Introduction
Malignant melanoma (MM), a tumor arising from the
malignant transformation of melanocytes, is characterized by high
tumorigenic potential. Over the past 30 years, the global incidence
of metastatic melanoma has rapidly increased, resulting in a
notable increase in mortality (1).
Although the incidence of melanoma in China is relatively low,
~20,000 new cases are observed annually (2). Although early-stage melanoma is
curable via wide local excision (3), it can invade the dermis within
months, becoming life-threatening upon metastasis. Notably,
approximately one-third of patients with advanced melanoma present
with metastases to the lungs, liver or brain at the time of
diagnosis (4). Overall, the 5-year
survival rate is as high as 99% for patients with localized
melanoma, but it decreases to 27.3% for those with distant
metastases (5); therefore,
metastatic melanoma is generally associated with a poor
prognosis.
Melanoma pathogenesis involves a complex interplay
among ultraviolet (UV)-induced DNA damage, genetic mutations (such
as BRAF and NRAS) and dysregulated melanogenesis. UV
radiation, particularly UVB, induces thymine dimers and reactive
oxygen species, leading to oxidative DNA damage and activation of
oncogenic pathways, such as the p53 and melanocyte-inducing
transcription factor-dependent melanin synthesis pathways (6-8).
Although melanin protects against UV radiation, its intermediates
can leak from melanosomes under pathological conditions, promoting
tumor progression by enhancing hypoxia-inducible factor 1α-driven
angiogenesis, metabolic reprogramming and immunosuppression
(9,10). This dual role of melanogenesis
underscores its potential as a therapeutic target. Therefore,
systematic investigation of the pathogenesis and progression
mechanisms of melanoma using various experimental approaches has
marked theoretical and clinical value. Specifically, bioinformatics
enables the analysis of melanoma-related genes and signaling
pathways from large-scale data, revealing key molecular networks
involved in pathogenesis and progression and providing precise
targets for subsequent experimental research. Therefore, the
present study aimed to integrate public transcriptomic datasets
from Gene Expression Omnibus and The Cancer Genome Atlas (TCGA) to
identify high-confidence differentially expressed genes (DEGs) and
prioritize candidates associated with melanoma prognosis, validate
the expression and prognostic significance of targeting protein for
Xklp2 (TPX2) in independent cohorts, and functionally characterize
the role of TPX2 in melanoma cell proliferation and migration in
vitro and define its regulatory relationship with aurora kinase
A (AURKA) to evaluate TPX2 as a potential prognostic biomarker and
therapeutic target.
Materials and methods
Bioinformatics analysis
MM-related gene expression data were retrieved from
the Gene Expression Omnibus database of the National Center for
Biotechnology Information (https://www.ncbi.nlm.nih.gov/geo/) using ‘melanoma’ as
the search term. The GSE98394(11)
dataset (platform GPL16791) containing 27 commonly acquired nevus
(normal control) and 51 primary melanoma samples was obtained and
used for analysis. Differential analysis was conducted using the R
package ‘limma’ v3.50.0 (https://bioinf.wehi.edu.au/limma/). Statistical
significance of the differences between normal and tumor samples
was analyzed via unpaired t-test (for normally distributed data) or
Wilcoxon rank-sum test (for non-normally distributed data), with
adjusted P<1x10-10 and |fold change (FC)|>4 as
thresholds. The R package ‘pheatmap’ v1.0.12 (https://CRAN.R-project.org/package=pheatmap) was used
to generate a heatmap with hierarchical clustering, and a scatter
plot was constructed to observe the expression levels and trends of
DEGs. DEGs identified by limma were subjected to functional
enrichment analysis to identify over-represented Gene Ontology
categories (biological processes, cellular components and molecular
functions) and Kyoto Encyclopedia of Genes and Genomes pathways
(12,13). Enrichment results were summarized
and visualized using the R package ‘ggplot2’ v4.0.1 (https://CRAN.R-project.org/package=ggplot2).
The mRNA-sequencing and gene mutation data of 469
skin cutaneous melanoma (SKCM) and 558 normal tissue samples were
obtained from TCGA-SKCM dataset (https://tcga-data.nci.nih.gov/). Statistical analyses
were conducted using Gene Expression Profiling Interactive Analysis
2 (http://gepia2.cancer-pku.cn), with
adjusted P<1x10-8 and |FC|>2 as thresholds. Gene
expression values were presented as transcripts per million.
Significantly upregulated and downregulated genes in the GSE98394
and TCGA-SKCM datasets were separately intersected to obtain the
final lists of consistently upregulated and downregulated genes.
Kaplan-Meier survival curves were generated to compare survival
outcomes between groups, and statistical significance was assessed
using the log-rank (Mantel-Cox) test. Hazard ratios (HRs) and
corresponding 95% confidence intervals were calculated using Cox
proportional hazards regression models. Genes significantly
associated with patient survival were identified as candidates for
further investigation.
To validate TPX2 expression and its association with
melanoma progression, two independent datasets (GSE3189 and
GSE46517) were retrieved from the Gene Expression Omnibus database.
The GSE3189(14) and
GSE46517(15) datasets were used
to validate differential TPX2 expression between melanoma and nevus
tissues. Statistical significance was assessed via unpaired t-tests
for comparisons between two groups. For prognostic validation, the
GSE65904 dataset was used to analyze the association between gene
expression and disease-specific survival. Optimal cut-off points
for separating high and low expression groups were determined using
the survive_cutpoint function in the R package ‘survminer’.
Kaplan-Meier survival curves were generated, and differences were
assessed using the log-rank test. Additionally, multivariate Cox
proportional hazards regression models were constructed to evaluate
the independent prognostic value of TPX2 and AURKA, adjusting for
clinical covariates including age, sex and tumor stage.
Cell lines and culture
Human MM cells (A375 and C32) and immortalized human
melanocytes (PIG1) originally from LMAI were provided by another
laboratory at Guangdong Medical University (Zhanjiang, China). All
cells were cultured in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) with 10% FBS (Shanghai ExCell Biology, Inc.) and
1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.)
at 37˚C in a constant temperature incubator with 5%
CO2.
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was extracted from cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) and
reverse-transcribed into cDNA (incubation at 50˚C for 15 min
followed by 85˚C for 2 min) using HiScript II Q RT SuperMix for
qPCR (Vazyme Biotech Co., Ltd.). RT-qPCR was performed using ChamQ
Universal SYBR qPCR Master Mix (Vazyme Biotech Co., Ltd.) on the
Bio-Rad CFX Opus 96 system (Bio-Rad Laboratories, Inc.). The
thermocycling conditions were as follows: Initiation with a
hot-start activation at 95˚C for 30 sec, followed by 40 cycles of a
two-step amplification program consisting of denaturation at 95˚C
for 10 sec and a combined annealing/extension step at 60˚C for 30
sec. Fluorescence signal acquisition was performed at the end of
each 60˚C phase. To confirm the specificity of the amplified
products and the absence of primer-dimers, a melting curve analysis
was conducted immediately following the final cycle (60-95˚C).
Relative gene expression was calculated using the 2-ΔΔCq
method (16), with GAPDH as the
endogenous control for normalization. All experiments were
independently repeated three times. The following primer sequences
were used in the present study: TPX2 forward,
5'-GAGGGCCTTTCTGGTTCTCT-3'; TPX2 reverse,
5'-CTCCTGTAGTCTGGCCTCCT-3'; GAPDH forward,
5'-GTCTCCTCTGACTTCAACAGCG-3'; and GAPDH reverse,
5'-ACCACCCTGTTGCTGTAGCCAA-3'.
Western blotting
Proteins were extracted using RIPA buffer (Beyotime
Biotechnology) with a protease inhibitor (P6730; Beijing Solarbio
Science & Technology Co., Ltd.). The BCA assay (Beyotime
Biotechnology) was used for protein quantification, and 20 µg total
protein was loaded per lane. SDS-PAGE was used to separate
proteins, which were transferred to PVDF membranes
(MilliporeSigma). After blocking with 5% skimmed milk (Beyotime
Biotechnology) for 1 h at room temperature, the membranes were
incubated overnight with primary antibodies, including anti-TPX2
(dilution, 1:5,000; cat. no. 11741-1-AP; Proteintech Group, Inc.),
anti-AURKA (dilution, 1:2,000; cat. no. 66757-1-Ig; Proteintech
Group, Inc.) and anti-GAPDH (dilution, 1:50,000; cat. no.
60004-1-Ig; Proteintech Group, Inc.) antibodies, at 4˚C. The
membranes were further washed with 1X TBS with 0.05% Tween-20
(Beyotime Biotechnology) and incubated with goat anti-rabbit
secondary antibodies (dilution, 1:5,000; cat. no. RGAR001;
Proteintech Group, Inc.) or goat anti-mouse secondary antibodies
(dilution, 1:5,000; cat. no. RGAM001; Proteintech Group, Inc.) at
room temperature for 1 h. Protein bands were visualized using an
enhanced chemiluminescence detection reagent (Thermo Fisher
Scientific, Inc.). Band intensities were semi-quantified via
densitometric analysis using ImageJ software v1.53 (National
Institutes of Health). All experiments were independently repeated
three times.
MTT assay
Cells were seeded in a 96-well plate (Thermo Fisher
Scientific, Inc.) at a density of 1x103 cells/well and
cultured for 0, 12, 24, 36 and 48 h. An MTT solution (Beyotime
Biotechnology) was used to assess cell viability. Formazan crystals
were dissolved in DMSO (Thermo Fisher Scientific, Inc.), and the
absorbance [optical density (OD)] was measured at 490 nm using a
microplate reader. All experiments were independently repeated
three times.
Wound healing assay
The wound healing assay was performed on cells grown
to 90% confluence in a 6-well plate (Thermo Fisher Scientific,
Inc.). Cells were cultured in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) with 10% FBS (Shanghai ExCell Biology,
Inc.) and 1% penicillin/streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) at 37˚C in a constant temperature incubator with
5% CO2. To establish a uniform wound, the cells were
scraped with a 200-µl micropipette tip. After washing with
phosphate-buffered saline, the cells were cultured with 2% FBS
RPMI-1640 medium. Cell migration was observed using an inverted
fluorescence microscope at 0 and 24 h. Wound closure was
quantitatively assessed by measuring the wound area at 0 and 24 h
using ImageJ software v1.53 (National Institutes of Health). The
percentage of wound closure was calculated as follows: Healing
rates (%)=[(initial wound width-wound width at 24 h)/initial wound
width] x100. All measurements were performed in at least three
randomly selected fields per well, and the mean value was used for
statistical analysis. All experiments were independently repeated
three times.
Transwell assay
A serum-free cell suspension containing
1x105 cells was seeded in the upper chamber of a
Transwell system (8.0 µm; Corning, Inc.), and culture medium with
10% fetal bovine serum was added to the lower chamber. After
incubation at 37˚C in a constant temperature incubator with 5%
CO2 for 24 h, non-migrated cells on the upper surface of
the membrane were gently removed, and the cells that had migrated
to the lower surface of the filter were fixed with 4%
paraformaldehyde for 30 min at room temperature, stained with
crystal violet for 10 min at room temperature (Beyotime
Biotechnology), washed with phosphate-buffered saline and allowed
to dry. Then, the number of migrating cells was determined using an
inverted microscope. All experiments were independently repeated
three times.
Small interfering RNA (siRNA)
transfection
A total of 5x105 A375 and C32 cells were
seeded in a 6-well plate at a density of 70-90%. TPX2 siRNA
(sense, 5'-AUUAUUAGCCUUAGUAAUGUA-3' and antisense,
5'-UACAUUACUAAGGCUAAUAAU-3') and siNC (sense,
5'-UUCUCCGAACGUGUCACGU-3' and antisense, 5'-ACGUGACACGUUCGGAGAA-3')
were obtained from Changzhou Ruibo Bio-Technology Co., Ltd. siRNA
(50 pmol) was transfected into the cells using the
Lipofectamine® RNAiMAX reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). siRNA-lipid complexes were prepared in
serum-free medium and added to cells, which were then incubated at
37˚C in a humidified atmosphere with 5% CO2 for 6 h.
Subsequently, the transfection medium was replaced with complete
culture medium. Cells were harvested for RT-qPCR and western blot
analyses 48 h after transfection. Functional assays were performed
at the following intervals after transfection unless otherwise
stated: MTT assays at 0, 12, 24, 36 and 48 h, and wound healing and
Transwell migration assays at 24 h. Cells transfected with
non-targeting siNC were used as negative controls for all siRNA
transfection experiments. Untransfected parental A375 and C32 cells
were included as blank controls where indicated.
Statistical analyses
Data were analyzed using SPSS v22.0 (IBM, Corp.) and
are presented as the mean ± standard deviation. GraphPad Prism v8.0
(Dotmatics) was used for data visualization. Statistical analyses
of quantitative data were conducted using one-way ANOVA. Levene's
test and F-test were applied to test for unequal variances, and
Welch's ANOVA was used for analysis when variances were unequal.
Tukey's honestly significant difference test was used when Levene's
test indicated equal variances. If Levene's test indicated unequal
variances and Welch's ANOVA was applied, pairwise comparisons were
performed using the Games-Howell post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Differential expression and functional
enrichment analyses of the GSE98394 and TCGA-SKCM datasets
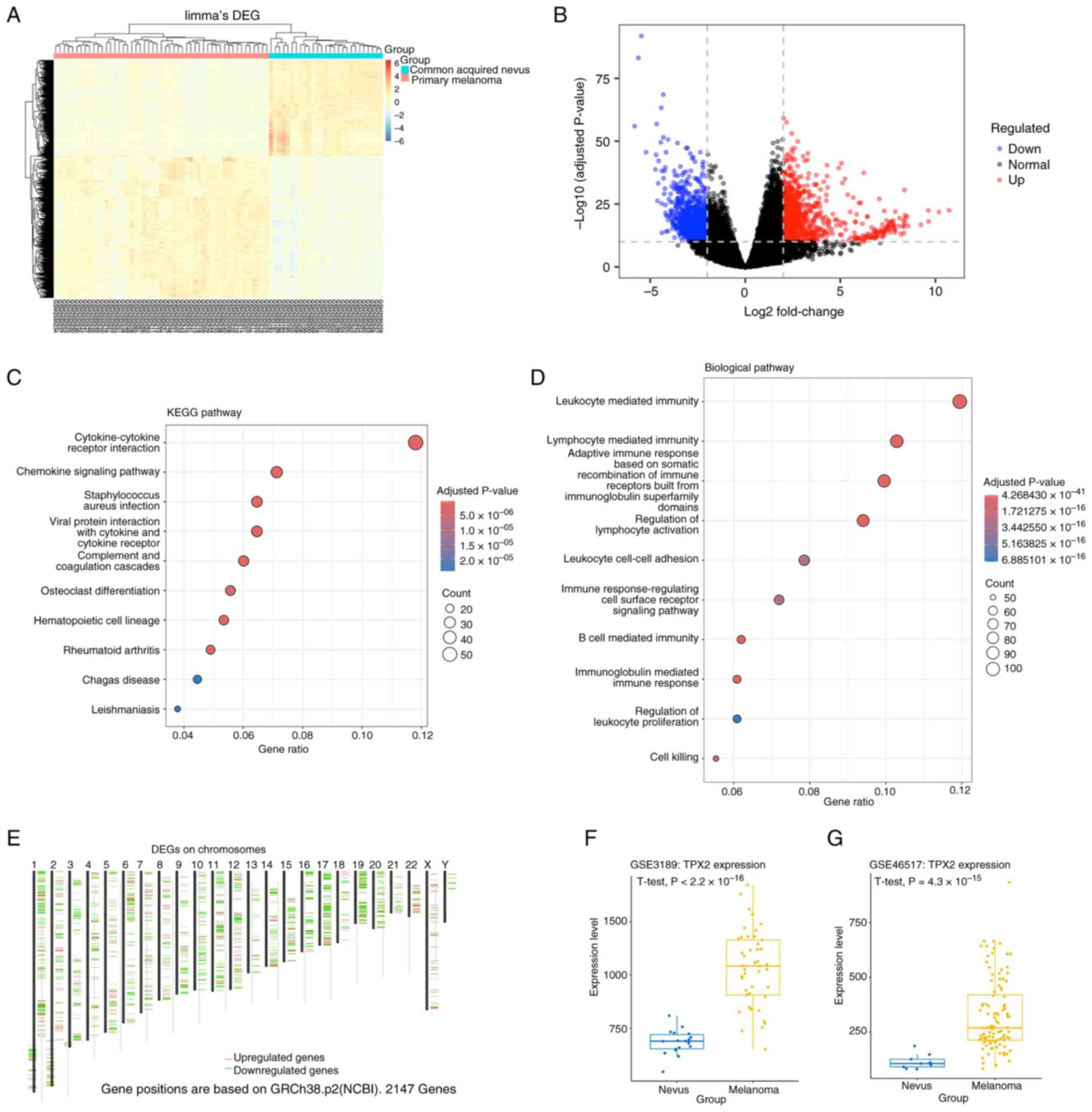
Differential expression analysis of the GSE98394
dataset identified 878 significantly downregulated and 812
significantly upregulated genes based on the thresholds of
|FC|>4 and adjusted P<1x10-¹0 (Fig. 1A and B). Functional enrichment analysis of
these DEGs revealed significant involvement in pathways such as the
‘cytokine-cytokine receptor interaction’ and ‘chemokine signaling
pathway’. The enriched biological processes included
‘leukocyte-mediated immunity’, ‘lymphocyte mediated immunity’ and
‘regulation of lymphocyte activation’ (Fig. 1C and D). Similarly, analysis of TCGA-SKCM
dataset using cut-offs of |(FC)|>2 and adjusted
P<1x10-8 identified 357 significantly downregulated
and 67 significantly upregulated genes (Fig. 1E). Intersecting the DEGs from both
datasets revealed eight potentially upregulated genes
[anti-silencing function 1B histone chaperone, TPX2,
transmembrane protein 132A, PDZ-binding kinase, von Willebrand
factor, protein kinase membrane-associated tyrosine/threonine 1
(PKMYT1), ubiquitin-conjugating enzyme E2 C (UBE2C)
and insulin-like growth factor-2] and five potentially
downregulated genes [adhesion G protein-coupled receptor V1,
phytanoyl-CoA 2-hydroxylase-interacting protein, complement factor
H (CFH), troponin C1 and family with sequence similarity 153
member B]. Moreover, TPX2 mRNA levels were significantly
higher in melanoma than in nevi (P<0.001) in the GSE3189 cohort
(Fig. 1F). These findings were
consistent with those obtained from the GSE46517 dataset, in which
TPX2 levels were also significantly upregulated in malignant
tissues (P<0.001; Fig. 1G).
Across both validation cohorts, TPX2 expression demonstrated an
~2.8-fold increase in melanoma compared to that in benign
lesions.
Impact of candidate functional genes
on patient overall survival (OS)
To evaluate the prognostic significance of candidate
genes in melanoma, Kaplan-Meier survival analyses were conducted
using OS data from TCGA melanoma cohort. Patients were divided into
high and low expression groups based on median expression levels.
As shown in Fig. 2A, patients with
low expression of TPX2 exhibited significantly longer OS compared
with those with high expression (log-rank P=0.0074; HR=1.4),
suggesting a potential oncogene function of TPX2 in melanoma. To
further validate the prognostic value of the identified candidate
genes, an independent survival analysis was performed using the
GSE65904 dataset. Patients were stratified into high and low
expression groups based on the optimal cut-off values for TPX2 and
AURKA. Kaplan-Meier survival analysis revealed that high expression
levels of both TPX2 and AURKA were significantly associated with
poor disease-specific survival (log-rank P<0.001 for both;
Fig. 2B and C). Specifically, patients in the high
TPX2 expression group exhibited significantly lower survival
probability than those in the low TPX2 expression group. Similarly,
elevated AURKA expression was an indicator of unfavorable
prognosis. The univariate and multivariate Cox regression results
are shown in Table I. These
results from an independent cohort further confirm that TPX2 and
AURKA are reliable biomarkers for predicting survival outcomes in
patients with melanoma.
 | Table IUnivariate and multivariate cox
regression analysis. |
Table I
Univariate and multivariate cox
regression analysis.
| A, TPX2 |
|---|
| Variable | Univariate HR (95%
CI) | Univariate
P-value | Multivariate HR
(95% CI) | Multivariate
P-value |
|---|
| High vs. low
expression | 1.938
(1.308-2.871) | <0.001 | 1.910
(1.273-2.866) | 0.002 |
| Age (continuous
variable) | 0.998
(0.985-1.012) | 0.797 | 1.002
(0.987-1.016) | 0.827 |
| Sex (male vs.
female) | 1.335
(0.885-2.016) | 0.169 | 1.170
(0.766-1.788) | 0.468 |
| Tumor stage
(in-transit, local, primary and regional vs. ‘general’) | 0.354
(0.204-0.613) | <0.001 | 0.374
(0.214-0.652) | <0.001 |
| B, AURKA |
| Variable | Univariate HR (95%
CI) | Univariate
P-value | Multivariate HR
(95% CI) | Multivariate
P-value |
| High vs. low
expression | 2.440
(1.460-4.079) | <0.001 | 2.174
(1.284-3.682) | 0.004 |
| Age (continuous
variable) | 0.998
(0.985-1.012) | 0.797 | 1.001
(0.986-1.015) | 0.932 |
| Sex (male vs.
female) | 1.335
(0.885-2.016) | 0.169 | 1.278
(0.841-1.941) | 0.250 |
| Tumor stage
(in-transit, local, primary and regional vs. ‘general’) | 0.354
(0.204-0.613) | <0.001 | 0.391
(0.223-0.685) | 0.001 |
TPX2 promotes the growth and migration
of A375 melanoma cells
To evaluate the differential expression of TPX2
between normal melanocytes and melanoma cells, TPX2 mRNA
levels were assessed in PIG1 and A375 cells. RT-qPCR analysis
revealed that TPX2 mRNA expression was significantly higher
in A375 cells than in PIG1 cells (P<0.05; Fig. 3A). This upregulation was further
confirmed at the protein level via western blotting, which showed
greater TPX2 protein expression in A375 cells compared with PIG1
cells (P<0.05; Fig. 3B). To
investigate whether elevated TPX2 expression is associated with
functional alterations in melanoma cells, the migration and
proliferation of A375 and PIG1 cells were evaluated. MTT
proliferation assays indicated that A375 cells exhibited
significantly higher OD values from 12 to 48 h, indicating greater
proliferation compared with PIG1 cells (P<0.05; Fig. 3C). Additionally, Transwell
migration assays demonstrated that A375 cells exhibited
significantly enhanced migration, as indicated by a higher number
of A375 cells traversing the membrane compared with PIG1 cells
(P<0.05; Fig. 3D).
Consistently, wound healing assays revealed that A375 cells
migrated more rapidly, with a noticeably smaller wound area at 24 h
(P<0.05; Fig. 3E).
Collectively, these findings suggested that TPX2 is highly
expressed in A375 melanoma cells and may be associated with
melanoma progression by promoting cell proliferation and
migration.
TPX2 knockdown inhibits AURKA
expression and attenuates the proliferation and migration of
melanoma cells
To further investigate the functional roles of TPX2
in melanoma cells, siRNA-mediated knockdown of TPX2 in
melanoma cells was performed. RT-qPCR analysis confirmed a
significant reduction in TPX2 mRNA levels after transfection
with TPX2-specific siRNA compared with those in control A375 cells
(P<0.05; Fig. 4A). Western
blotting analysis further validated the decrease in TPX2 protein
levels in A375-siTPX2 and C32-siTPX2 cells and revealed a
concomitant reduction in AURKA protein levels (P<0.05; Fig. 4B). Functionally, MTT assays
demonstrated that TPX2 knockdown significantly inhibited A375 cell
proliferation. The A375-siTPX2 group exhibited markedly lower OD
values than the control A375 group at all time points (P<0.05;
Fig. 4C). Transwell migration
assays showed that the number of migrated cells was significantly
reduced in the A375-siTPX2 and C32-siTPX2 groups, indicating
impaired migration (P<0.05; Fig.
4D). Consistently, wound healing assays revealed that the
scratch area remained largely open in TPX2-silenced cells
after 24 h, whereas A375 and C32 cells exhibited notable wound
closure (P<0.05 for A375 cells; Fig. 4E). Collectively, these results
suggested that TPX2 promotes melanoma cell proliferation and
migration, which could be related to AURKA upregulation.
 | Figure 4TPX2 knockdown reduces AURKA
expression and suppresses proliferation and migration in A375 and
C32 melanoma cells. (A) Reverse transcription-quantitative PCR
analysis of TPX2 mRNA levels in A375 and C32 cells transfected with
siTPX2 or non-targeting siNC. Data are presented separately for
each cell line. (B) Western blot analysis of TPX2 and AURKA protein
levels in parental A375 and C32 cells and in the corresponding
siTPX2- and siNC-transfected cells. GAPDH served as a loading
control. (C) Cell proliferation assessed using an MTT assay at the
indicated time points. Proliferation of siTPX2-transfected cells
and parental controls is shown. (D) Transwell migration assay of
A375 and C32 cells and their siTPX2 transfectants. Migrated cells
were stained and counted under a microscope (magnification, x200;
scale bar, 50 µm). siTPX2-transfected cells were compared with
parental cells within each cell line. (E) Wound healing assay
images at 0 and 24 h. Wound closure was quantified and siTPX2
transfectants were compared with parent cell controls
(magnification, x50; scale bar, 200 µm). Data are presented as the
mean ± SD. *P<0.05 vs. parental cells,
#P<0.05 vs. siNC, A375 or C32 as applicable. si,
small interfering; NC, negative control; AURKA, aurora kinase A;
TPX2, targeting protein for Xklp2; OD, optical density. |
Discussion
MM remains a challenging malignancy with limited
therapeutic options, particularly in advanced stages (17). Melanoma arises from melanocytes,
which are responsible for melanin production and predominantly
located in the skin but also found in mucosal tissues, eyes and
other organs (18). Globally, over
324,635 new melanoma cases and 57,043 deaths were reported in 2020,
with cutaneous melanoma being dominant in white populations
(>90%) and acral subtypes being more common in East Asian
populations (19). The present
study integrated bioinformatics analysis with functional
experiments to identify key molecular drivers of melanoma
progression, focusing on the oncogenic role of TPX2 and its
interplay with AURKA. The present findings revealed TPX2 as a
notable regulator of melanoma cell proliferation and migration,
highlighting it as a novel therapeutic target.
The intersection of DEGs from the GSE98394 and
TCGA-SKCM datasets identified TPX2, UBE2C and
PKMYT1 as consistently upregulated genes. These genes are
implicated in mitotic regulation and cell cycle progression,
processes frequently dysregulated in cancer (20-22).
Notably, TPX2, a microtubule-associated protein essential for
spindle assembly, drives genomic instability and metastasis in
various cancers (20).
Consistently, the present survival analysis revealed that high TPX2
expression was associated with shorter OS in patients with
melanoma. This aligns with previous reports linking TPX2 to
aggressive phenotypes in hepatocellular carcinoma (23) and breast cancer (24), suggesting a conserved oncogenic
role across malignancies.
Clinically, early-stage melanoma is curable with
surgery; however, metastatic disease has a poor 5-year survival
rate of 27.3% (5). Traditional
chemotherapy, such as dacarbazine and temozolomide, shows limited
efficacy (10-20% response rates) and marked toxicity (25-27).
Advances in targeted therapies (BRAF/MEK inhibitors) and
immunotherapies (anti-cytotoxic T-lymphocyte-associated protein-4
and anti-programmed death protein-1 antibodies, as well as
oncolytic viruses such as Talimogene laherparepvec) have improved
outcomes in patients with BRAF-mutant and advanced melanoma
(28-31).
However, drug resistance and immune-related adverse events remain
major obstacles. The identification of TPX2 as a driver of cell
proliferation and migration in the present study adds to the
existing molecular toolkit for addressing these challenges.
Functional validation revealed that TPX2
silencing significantly impaired melanoma cell proliferation and
migration. Notably, TPX2 knockdown concurrently reduced
AURKA expression at the protein level. AURKA, a serine/threonine
kinase critical for mitotic entry, is often co-amplified with TPX2
in cancer (32,33). TPX2 primarily stabilizes AURKA by
binding to it, recruiting it to microtubules and protecting it from
degradation, thereby indirectly contributing to high AURKA levels
by increasing its stability and activation (34,35).
Furthermore, AURKA is a driver of epithelial-mesenchymal transition
and metastasis, linking TPX2 to melanoma aggressiveness (36). This interaction may underpin the
mitotic defects and reduced migration observed in TPX2-depleted
cells. Although TPX2 is well characterized as a physical scaffold
that stabilizes AURKA and protects it from degradation, its
contribution to downstream cellular phenotypes remains to be fully
elucidated (37). The reduction in
transcript abundance does not necessarily imply a direct
transcriptional role of TPX2. Instead, it possibly reflects an
indirect consequence of G2/M phase or cell cycle arrest
typically observed following TPX2 depletion (38). Because AURKA expression is strictly
regulated by the cell cycle, peaking during the late G2
and early M phases, a shift in the cell cycle profile toward
G1/S possibly results in lower steady-state AURKA
mRNA levels (39). However, the
precise molecular association between TPX2 and AURKA warrants
further investigation, including chromatin immunoprecipitation,
promoter activity and pull-down assays, to determine the mechanisms
by which TPX2 regulates AURKA transcription and protein
translation.
To the best of our knowledge, the present study is
the first to demonstrate that TPX2 not only drives melanoma cell
proliferation but also directly enhances cell migration,
potentially via AURKA-dependent pathways. This dual functionality
positions TPX2 as a coordinator of melanoma progression, bridging
cell cycle dysregulation and metastatic potential. The association
between TPX2 overexpression and poor survival in TCGA-SKCM cohort
further underscores the clinical relevance of TPX2 as a prognostic
biomarker. However, this study has several limitations: First,
experimental validation focused solely on TPX2 in the A375 and C32
cell lines, necessitating further studies on other melanoma models
(such as BRAF/NRAS-mutant cell lines) and in vivo systems to
validate the findings. Second, the mechanism linking TPX2 to AURKA
regulation remains unclear. Lastly, although bioinformatics
analysis prioritized high-confidence DEGs, functional studies on
other candidates (PKMYT1 and UBE2C) are necessary to
determine their contributions to melanoma progression.
In summary, the present integrated approach
identified TPX2 as a key oncogene driving melanoma
proliferation and migration, potentially via AURKA upregulation.
The present findings suggested that TPX2 may be both a prognostic
biomarker and potential therapeutic target. Future studies should
evaluate TPX2 inhibition in preclinical models and investigate
combinatorial strategies targeting TPX2 and AURKA or melanogenesis
pathways to mitigate melanoma aggressiveness and enhance the
efficacy of existing immunotherapies and targeted therapies.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Guangdong
Province Medical Science Research Project (grant no. A2019103).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
FL conceptualized the study, designed the
methodology, performed bioinformatics analysis and the experiments,
and wrote the original draft of the manuscript. RY generated
figures, was involved in validation, and performed formal analysis,
bioinformatics analysis and the experiments. ZW conceptualized the
study, acquired funding and reviewed the manuscript. FL and RY
confirm the authenticity of all the raw data. All authors have read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Giaquinto AN and Jemal A:
Cancer statistics, 2024. CA Cancer J Clin. 74:12–49.
2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Xu L, Cheng Z, Cui C, Wu X, Yu H, Guo J
and Kong Y: Frequent genetic aberrations in the cell cycle related
genes in mucosal melanoma indicate the potential for targeted
therapy. J Transl Med. 17(245)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kozovska Z, Gabrisova V and Kucerova L:
Malignant melanoma: Diagnosis, treatment and cancer stem cells.
Neoplasma. 63:510–517. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Luke JJ, Flaherty KT, Ribas A and Long GV:
Targeted agents and immunotherapies: Optimizing outcomes in
melanoma. Nat Rev Clin Oncol. 14:463–482. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Herndon TM, Demko SG, Jiang X, He K,
Gootenberg JE, Cohen MH, Keegan P and Pazdur R: U.S. Food and drug
administration approval: Peginterferon-alfa-2b for the adjuvant
treatment of patients with melanoma. Oncologist. 17:1323–1328.
2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Slominski A, Tobin DJ, Shibahara S and
Wortsman J: Melanin pigmentation in mammalian skin and its hormonal
regulation. Physiol Rev. 84:1155–1228. 2004.PubMed/NCBI View Article : Google Scholar
|
|
7
|
dos Santos Videira IF, Moura DF and Magina
S: Mechanisms regulating melanogenesis. An Bras Dermatol. 88:76–83.
2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Garmyn M, Young AR and Miller SA:
Mechanisms of and variables affecting UVR photoadaptation in human
skin. Photochem Photobiol Sci. 17:1932–1940. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Slominski RM, Sarna T, Plonka PM, Raman C,
Brozyna AA and Slominski AT: Melanoma, melanin, and melanogenesis:
The Yin and Yang relationship. Front Oncol.
12(842496)2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Slominski AT, Zmijewski MA, Plonka PM,
Szaflarski JP and Paus R: How UV light touches the brain and
endocrine system through skin, and why. Endocrinology.
159:1992–2007. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Badal B, Solovyov A, Di Cecilia S, Chan
JM, Chang LW, Iqbal R, Aydin IT, Rajan GS, Chen C, Abbate F, et al:
Transcriptional dissection of melanoma identifies a high-risk
subtype underlying TP53 family genes and epigenome deregulation.
JCI Insight. 2(e92102)2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Talantov D, Mazumder A, Yu JX, Briggs T,
Jiang Y, Backus J, Atkins D and Wang Y: Novel genes associated with
malignant melanoma but not benign melanocytic lesions. Clin Cancer
Res. 11:7234–7242. 2005.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kabbarah O, Nogueira C, Feng B, Nazarian
RM, Bosenberg M, Wu M, Scott KL, Kwong LN, Xiao Y, Cordon-Cardo C,
et al: Integrative genome comparison of primary and metastatic
melanomas. PLoS One. 5(e10770)2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zavaleta-Monestel E, Quesada-Villaseñor R,
Barrantes-López M, Arguedas-Chacón S, Campos-Hernández J,
Rojas-Chinchilla C, García-Montero J, Castro-Ulloa J, Anchía-Alfaro
A and Montenegro-Chaves JR: Advancements in the treatment of
multiple myeloma. Cureus. 16(e74970)2024.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Jitian Mihulecea CR and Rotaru M: Review:
The key factors to melanomagenesis. Life (Basel).
13(181)2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Aguirre-Portoles C, Bird AW, Hyman A,
Canamero M, Perez de Castro I and Malumbres M: Tpx2 controls
spindle integrity, genome stability, and tumor development. Cancer
Res. 72:1518–1528. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang S, You X, Zheng Y, Shen Y, Xiong X
and Sun Y: The UBE2C/CDH1/DEPTOR axis is an oncogene and tumor
suppressor cascade in lung cancer cells. J Clin Invest.
133(e162434)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wang S, Xiong Y, Luo Y, Shen Y, Zhang F,
Lan H, Pang Y, Wang X, Li X, Zheng X, et al: Genome-wide CRISPR
screens identify PKMYT1 as a therapeutic target in pancreatic
ductal adenocarcinoma. EMBO Mol Med. 16:1115–1142. 2024.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang Y, Wang H, Yan Z, Li G, Hu G, Zhang
H, Huang D, Wang Y, Zhang X, Yan Y, et al: The critical role of
dysregulated Hh-FOXM1-TPX2 signaling in human hepatocellular
carcinoma cell proliferation. Cell Commun Signal.
18(116)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Marugán C, Sanz-Gómez N, Ortigosa B,
Monfort-Vengut A, Bertinetti C, Teijo A, González M, Alonso de la
Vega A, Lallena MJ, Moreno-Bueno G and de Cárcer G: TPX2
overexpression promotes sensitivity to dasatinib in breast cancer
by activating YAP transcriptional signaling. Mol Oncol.
18:1531–1551. 2024.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Jiang G, Li RH, Sun C, Liu YQ and Zheng
JN: Dacarbazine combined targeted therapy versus dacarbazine alone
in patients with malignant melanoma: a meta-analysis. PLoS One.
9(e111920)2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Middleton MR, Grob JJ, Aaronson N,
Fierlbeck G, Tilgen W, Seiter S, Gore M, Aamdal S, Cebon J, Coates
A, et al: Randomized phase III study of temozolomide versus
dacarbazine in the treatment of patients with advanced metastatic
malignant melanoma. J Clin Oncol. 18:158–166. 2000.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Guven K, Kittler H, Wolff K and
Pehamberger H: Cisplatin and carboplatin combination as second-line
chemotherapy in dacarbazine-resistant melanoma patients. Melanoma
Res. 11:411–415. 2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Larkin J, Del Vecchio M, Mandalá M, Gogas
H, Arance Fernandez AM, Dalle S, Cowey CL, Schenker M, Grob JJ,
Chiarion-Sileni V, et al: Adjuvant nivolumab versus ipilimumab in
resected stage III/IV melanoma: 5-year efficacy and biomarker
results from CheckMate 238. Clin Cancer Res. 29:3352–3361.
2023.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Eggermont AMM, Blank CU, Mandala M, Long
GV, Atkinson V, Dalle S, Haydon A, Lichinitser M, Khattak A,
Carlino MS, et al: Adjuvant pembrolizumab versus placebo in
resected stage III melanoma. N Engl J Med. 378:1789–1801.
2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Eggermont AM, Suciu S, Santinami M,
Testori A, Kruit WH, Marsden J, Punt CJ, Salès F, Gore M, MacKie R,
et al: Adjuvant therapy with pegylated interferon alfa-2b versus
observation alone in resected stage III melanoma: Final results of
EORTC 18991, a randomised phase III trial. Lancet. 372:117–126.
2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J,
Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al: An
immunogenic personal neoantigen vaccine for patients with melanoma.
Nature. 547:217–221. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Holder J, Miles JA, Batchelor M, Popple H,
Walko M, Yeung W, Kannan N, Wilson AJ, Bayliss R and Gergely F:
CEP192 localises mitotic Aurora-A activity by priming its
interaction with TPX2. EMBO J. 43:5381–5420. 2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Li H, Wang Y, Lin K, Venkadakrishnan VB,
Bakht M, Shi W, Meng C, Zhang J, Tremble K, Liang X, et al: CHD1
promotes sensitivity to aurora kinase inhibitors by suppressing
interaction of AURKA with its coactivator TPX2. Cancer Res.
82:3088–3101. 2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bayliss R, Sardon T, Vernos I and Conti E:
Structural basis of Aurora-A activation by TPX2 at the mitotic
spindle. Mol Cell. 12:851–862. 2003.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Eyers PA, Erikson E, Chen LG and Maller
JL: A novel mechanism for activation of the protein kinase Aurora
A. Curr Biol. 13:691–697. 2003.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Shen HM, Zhang D, Xiao P, Qu B and Sun YF:
E2F1-mediated KDM4A-AS1 up-regulation promotes EMT of
hepatocellular carcinoma cells by recruiting ILF3 to stabilize
AURKA mRNA. Cancer Gene Ther. 30:1007–1017. 2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Polverino F, Mastrangelo A and
Guarguaglini G: Contribution of AurkA/TPX2 overexpression to
chromosomal imbalances and cancer. Cells. 13(1397)2024.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Liu S, Cai J, Qian X, Zhang J, Zhang Y,
Meng X, Wang M, Gao P and Zhong X: TPX2 lactylation is required for
the cell cycle regulation and hepatocellular carcinoma progression.
Life Sci Alliance. 8(e202402978)2025.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Vats P, Saini C, Baweja B, Srivastava SK,
Kumar A, Kushwah AS and Nema R: Aurora kinases signaling in cancer:
From molecular perception to targeted therapies. Mol Cancer.
24(180)2025.PubMed/NCBI View Article : Google Scholar
|