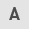p53 deficiency augments nucleolar instability after ionizing irradiation
- Authors:
- Published online on: September 27, 2019 https://doi.org/10.3892/or.2019.7341
- Pages: 2293-2302
-
Copyright: © Kakoti et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
The nucleolus is an intra-nuclear organelle that contains ribosomal DNA (rDNA) that encodes ribosomal RNA (rRNA), which is an essential component of ribosomes. The rRNA and a variety of ribosomal proteins form ribosomes, which serve as the site of biological protein synthesis. Previous studies have demonstrated that ribosome biogenesis is associated with multiple cellular activities. For example, limiting ribosome biogenesis extends the lifespan, whereas increased ribosome biogenesis has been reported as a hallmark of premature aging (1). In the context of cancer, alterations in ribosome biogenesis have been suggested to be involved in tumorigenesis by downregulating tumor suppressors (2,3). In addition, rDNA rearrangements have been observed in over half of adult solid tumors (4). This observation suggests that rDNA restructuring is one of the most common chromosomal alterations in adult solid tumors and may be involved in tumorigenesis or malignancy. Conversely, for cancer treatment, an inhibitor targeting RNA polymerase I, which is required for rDNA transcription, induces apoptosis in cancer cells by activating the ATM/ATR pathway (5).
Ionizing radiation (IR) generates multiple types of DNA damage, such as DNA double-strand breaks (DSBs), single-strand breaks (SSBs), and base damage (6). Among the various types of DNA damage, DSBs are considered to be a critical factor causing severe genome instability, such as deletion, duplication, inversion and translocation (7). Linear energy transfer (LET) is a parameter used to describe the pattern of energy deposition. X-rays or γ-rays are known as low LET IR, whereas heavy charged particle IR is high LET radiation. High LET particle irradiation causes complex DNA lesions, defined as DNA damage containing both DSBs and SSBs, as well as base damage, within 1–2 helical turns (<3–4 nm) (8).
Another feature of high LET particle irradiation-dependent DNA damage was recently identified, observed as clustered γH2AX foci along the particle track (9–11). A combination of complex DNA lesions within clustered DSBs is not efficiently repaired and is associated with a high cell-killing effect (9,12). IR is a critical risk factor for human health, particularly for astronauts during periods of space travel or when radiation accidents occur, whereas it is significantly beneficial for cancer treatment (13).
In human cells, ~150–200 copies of rDNA encoding rRNA is located on chromosomes 13, 14, 15, 21 and 22, and each chromosome contains repetitive rDNA sequences (14). These repetitive sequences are genetically unstable and highly sensitive when cells are exposed to exogenous DNA stress (15). Classical studies have demonstrated that morphological changes in the nucleolus have been observed following IR under a phase-contrast microscope (16). However, the stability of the nucleolus and the transcriptional activity of rDNA following IR in the context of DSBs and its signaling have not been thoroughly investigated. Moreover, although irradiation-induced dose-dependent increase in polysomal aggregates has been reported (17), the irradiation-dependent morphological changes of individual ribosomes have not been investigated.
The aim of the present study was to examine the alterations in the amount of nucleolin, a marker of the nucleolus, and its morphological changes following IR, in order to investigate nucleolar stability in response to DSBs. We also investigated the role of p53 in preventing nucleolar instability following IR and protecting the transcriptional activity of rDNA in irradiated cells.
Materials and methods
Cell culture and irradiation
HCT116 (human colorectal carcinoma, wild-type (WT) p53+/+ and isogenic p53−/− null derivative, provided by Dr Vogelstein of Johns Hopkins University) and U2OS (human osteosarcoma) cells were cultured in Eagle's minimum essential medium (MEM) with 10% fetal calf serum (FCS) (Sigma-Aldrich; Merck KGaA). 1BR (normal human dermal fibroblast strain) hTERT cells were cultured in the alpha modification of MEM (Wako Pure Chemical Industries, Ltd.) with 10% FCS. X-ray irradiation was performed using a Faxitron RX-650 at 100 kVp and 20 mA with a dose rate of 1.14 Gy/min (Faxitron Bioptics). Carbon ion irradiation was performed at Gunma University Heavy Ion Medical Center using a spread-out Bragg-peak (SOBP) beam (290 MeV/nucleon; mean LET at the center of a 6-cm SOBP, 50 keV/µm).
siRNA knockdown and p53 overexpression
In U2OS cells, p53 siRNA transfection was performed using HiPerFect (Qiagen GmbH), as previously described (18). The efficiency of the knockdown was confirmed by immunoblotting for the detection of p53. The sequence of the p53 siRNA oligonucleotide used was 5′-GUGCAGCUGUGGGUUGAUUdTdT-3′. To examine the effect of p53 overexpression on nucleolar stability after IR, Flag-p53 WT was transfected in HCT116 cells using the Neon® Transfection System (Thermo Fisher Scientific, Inc.). Transfection efficiency was confirmed by immunoblotting using p53 antibodies (1:500 anti-p53 antibody, sc-6243, Santa Cruz Biotechnology, Inc.) and flag (1:1,000 anti-flag antibody, F3165, Sigma-Aldrich; Merck KGaA). Cells were incubated in fresh MEM for 24 h and then irradiated and processed for immunofluorescence analysis.
Immunofluorescence staining
Cells were seeded on Fisherbrand microscope cover glasses (12-545-80 12CIR-1, Thermo Fisher Scientific, Inc.) 48 h prior to irradiation. At the indicated time points after irradiation, cells were fixed with 3% paraformaldehyde-2% sucrose for 10 min. After washing with phosphate-buffered saline (PBS), permeabilization was carried out by treatment with 0.2% TritonX-100-PBS (Sigma-Aldrich; Merck KGaA) for 2 min and 30 sec. Cells were then washed twice with PBS and incubated with the primary antibody in 2% bovine serum albumin (BSA)-PBS (1:1,000 anti-nucleolin antibody, ab22758, Abcam) at 37°C for 30 min. The cells were washed twice with PBS before being incubated with the appropriate secondary antibody conjugated to Alexa-Fluor-555 (1:500 in 2% BSA-PBS) at 37°C for 30 min. The cells were again washed twice with PBS and mounted onto slides using ProLong Gold antifade mountant (Thermo Fisher Scientific, Inc.).
Morphological evaluation of nucleoli
Immunofluorescence slides were examined with a Nikon microscope (ECLIPSE Ni-U with DS-Qi2 camera and NIS-Elements D) using a 60× magnification lens. From a representative area of each sample, at least 100 consecutive cells were evaluated by direct visualization. First, each cell was examined simultaneously for nuclear staining and nucleolar morphology, and then categorized into one of the seven nucleolar types. The number of nucleoli per cell was noted for all cells, excluding type 7 cells (cells undergoing apoptosis, mitosis, or mitotic catastrophe). Nucleolin signal within micronuclei (types 3 and 6) were also counted individually, and the total number was a combination of the signal within the main nuclei and micronuclei.
Quantification of 47S rRNA expression levels using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
Cells were seeded and incubated for 24 h prior to irradiation. At 72 h of irradiation, total RNA was extracted from the cells using NucleoSpin RNA (Macherey-Nagel GmbH). PrimeScript RT Reagent Kit (Perfect Real Time; Takara) was used to reverse-transcribe cDNA from total RNA, according to the manufacturer's instructions, and qPCR was performed using StepOnePlus (Thermo Fisher Scientific, Inc.). Reactions (20 µl each) were prepared in duplicate in a MicroAmp Fast Optical 96-Well Reaction Plate (Applied Biosystems; Thermo Fisher Scientific, Inc.). Each reaction contained 0.5 µM of each primer, 0.2 µM probe, 10 µl of TaqMan Universal PCR Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.), and cDNA as a template. The expression levels were normalized to GAPDH and were calculated using the 2−ΔΔCq method (19). The qPCR settings were as follows: Initial denaturation at 95°C for 10 min, followed by 45 cycles of denaturation at 95°C for 15 sec, and annealing and extension at 60°C for 1 min. The primers and probes used for the qPCR are listed below:
47S rRNA forward: 5′-GAACGGTGGTGTGTCGTTC-3′
47S rRNA reverse: 5′-GCGTCTCGTCTCGTCTCACT-3′
47S rRNA probe: 5′-CGGTGTGGGGTTCGAGGCGGTTT-3′
GAPDH forward: 5′-CTCCTCTGACTTCAACAGCGA-3′
GAPDH reverse: 5′-CCAAATTCGTTGTCATACCAGGA-3′
GAPDH probe: 5′-ATGCCAGCCCCAGCGTCAAAGGT-3′
Statistical analysis
Box plots were constructed using SigmaPlot 12.3 (Systat Software, Inc.) from the combined data of three independent experiments. The statistical significance was analyzed using the Student's two-tailed t-test or Mann-Whitney U test. For datasets where multiple comparisons were performed using single group, to avoid the high rate of familywise error, Bonferroni's correction was applied. The level of significance was determined using the α-value obtained by Bonferroni's correction and is specified in each individual figure legend.
Results
X-rays cause nucleolar fragmentation in human cancer cell lines
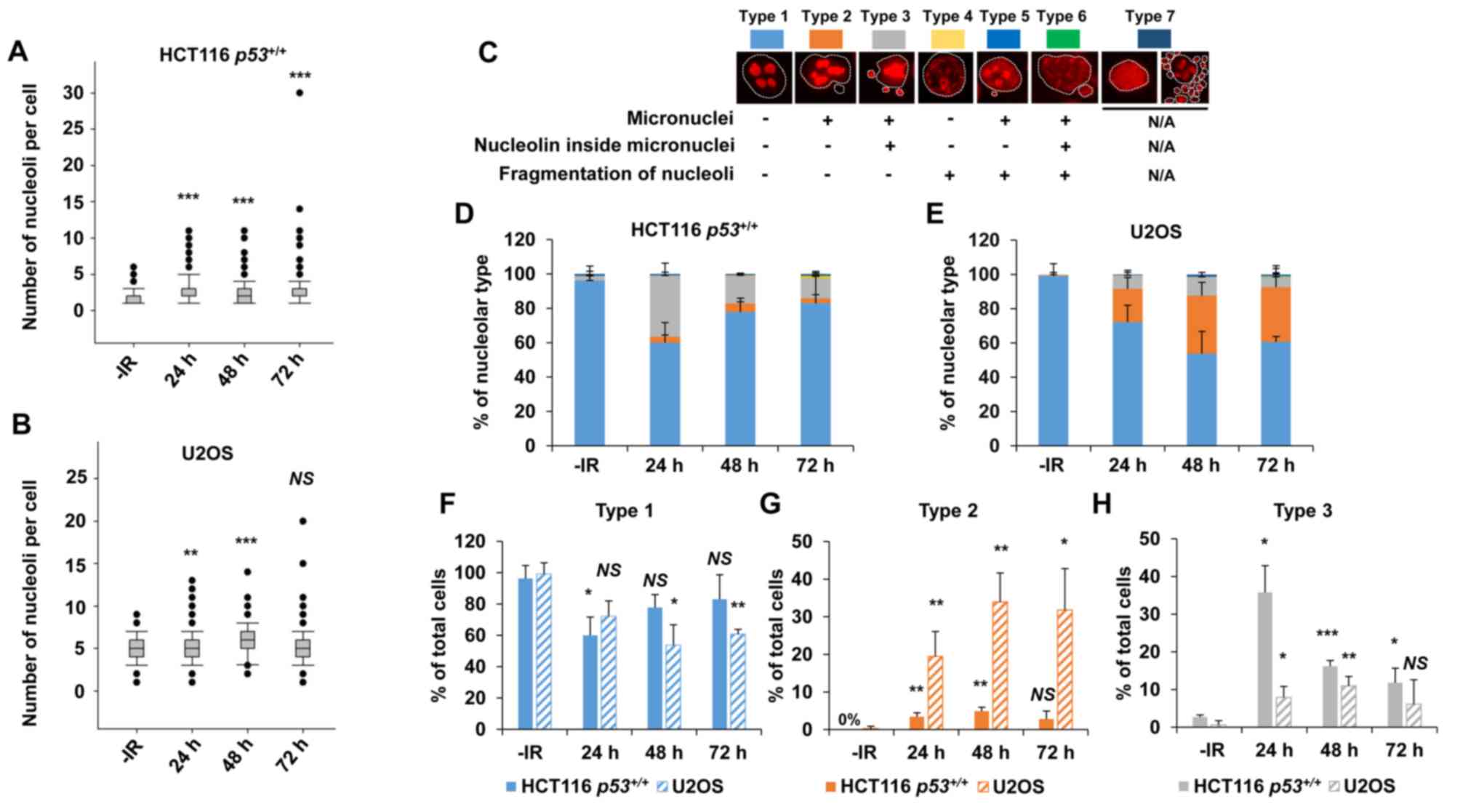
To investigate the nucleolar stability in response to IR in cancer cells, the number of nucleoli per cell and its morphological changes were examined following X-ray irradiation (n.b., nucleolin is localized within the nucleolus; therefore, it is used as a marker of nucleolus). In HCT116 p53+/+ and U2OS cell lines, there was an increase in the number of nucleoli per cell over time after 2 Gy of X-ray irradiation (Fig. 1A and B). Next, the morphological patterns were categorized to investigate the morphological changes after IR (Fig. 1C). Interestingly, a marked increase in type 3 HCT116 p53+/+ cells was observed following IR (Fig. 1D). Type 3 represents a cell containing micronuclei with a nucleolin signal. The peak of type 3 among HCT116 p53+/+ cells was observed at 24 h following IR. By contrast, U2OS cells exhibited an increase in types 2 and 3 following X-ray irradiation (Fig. 1E). The majority of the nucleolar aberrations in U2OS cells were type 2 (cell containing micronuclei without nucleolin signal). These data suggest that nucleolar instability is caused in cancer cells following IR, and the type of morphological changes is cell type-dependent (Fig. 1F-H, supplementary information Fig. S1A). Furthermore, the levels of nucleolar instability were examined after different doses of X-ray irradiation. Importantly, the incidence of nucleolar fragmentation and type 3 morphology increased with the dose of X-ray irradiation until it peaked at 2–4 Gy, followed by a modest decline at >8 Gy at all the evaluated time points (Fig. 2A and B).
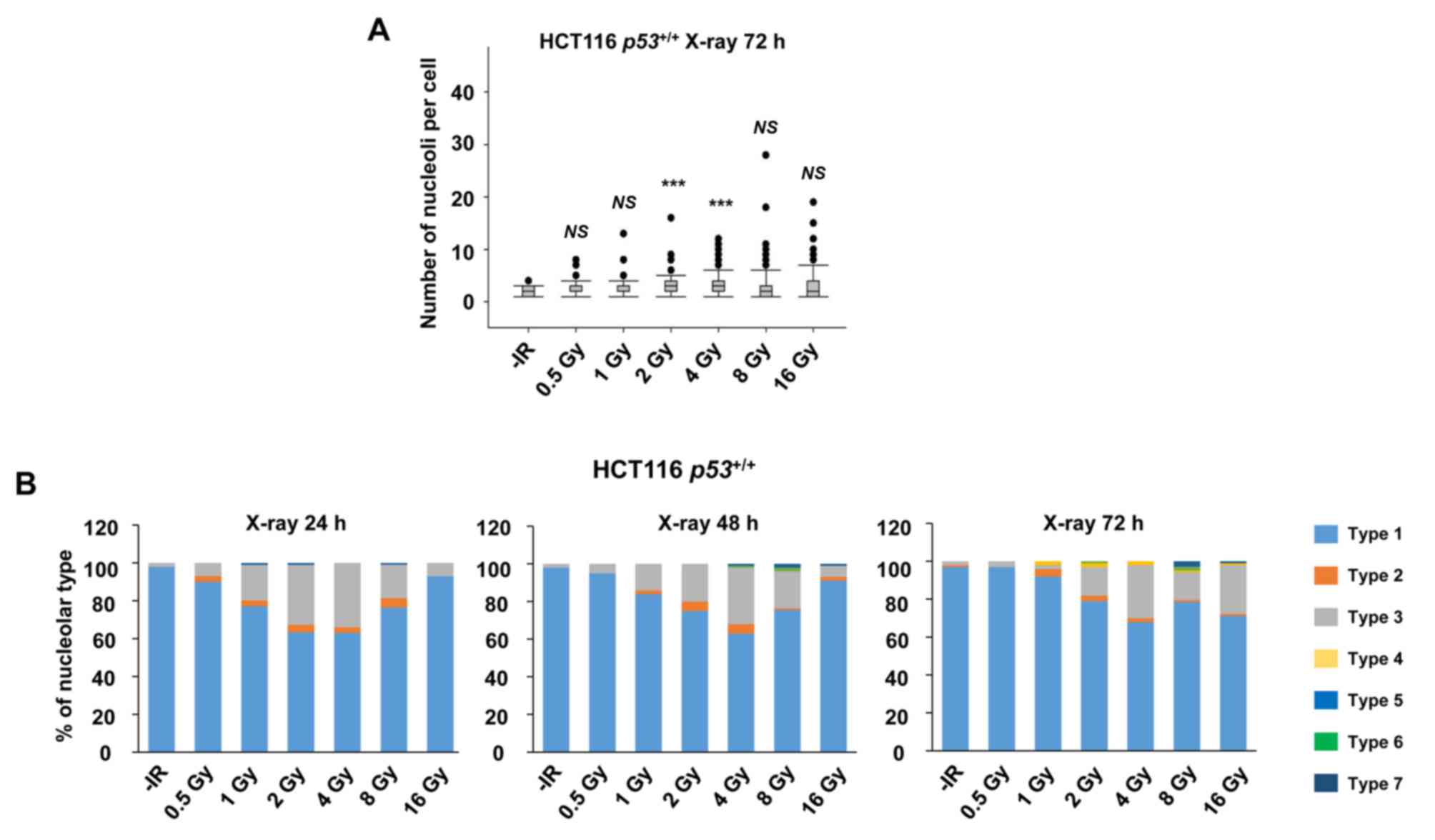
Next, the number of nucleoli in normal human fibroblasts was examined following IR to investigate the nucleolar stability in normal cells. In contrast to the observation of the increase in the number of nucleoli in cancer cell lines, X-ray irradiation did not increase the number of nucleoli in normal human fibroblasts, even at markedly higher radiation doses (Fig. 3).
p53 deficiency augments nucleolar fragmentation following exposure to X-rays
Furthermore, to address whether the genetic status involving genome stability affects nucleolar fragmentation after IR, the number of nucleoli per cell and any morphological changes were examined in cancer cells with p53 deficiency. Then, we compared the nucleolin signal in HCT116 p53+/+ and HCT116 p53−/− cells to investigate the impact of the p53 gene status on nucleolar stability. Importantly, a statistically significant (P=0.003) increase in nucleolar number was observed in HCT116 p53−/− cells at 72 h following exposure to X-rays (Fig. 4A). Furthermore, a dose-dependent increase in the nucleolar number of HCT116 p53−/− cells up to 8 Gy was found at 72 h (Fig. 4B). To investigate the effect of p53 on U2OS cells, siRNA knockdown targeting p53 was performed in U2OS cells. Similar to the results in HCT116 cells, there was a significant increase in the number of nucleoli in p53-depleted U2OS cells (Fig. 4C), with this increase being evident at later time points of 48–72 h after X-ray irradiation.
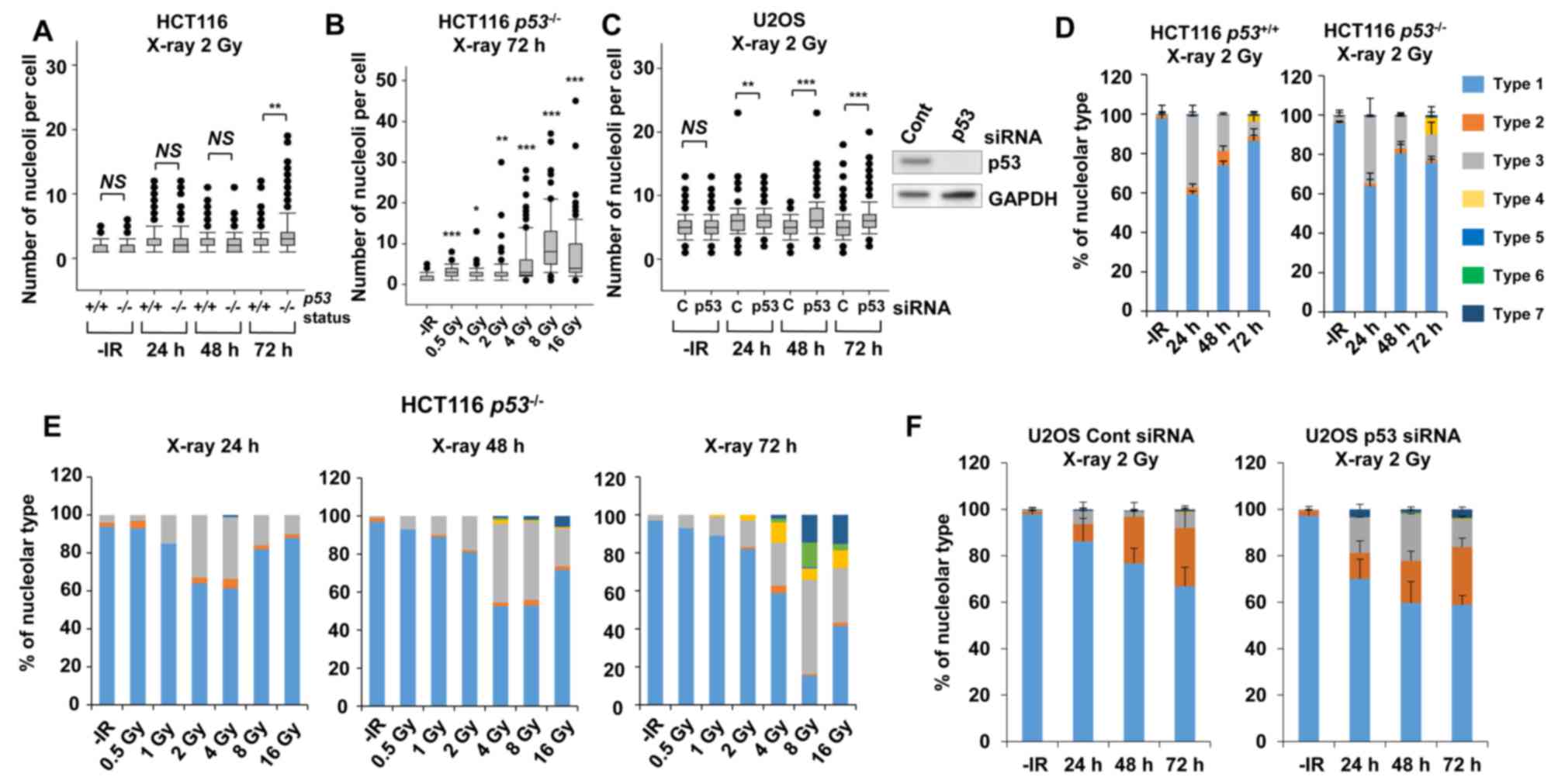
Next, morphological changes following the categorization shown in Fig. 1C in p53-defective cells were analyzed following IR. A morphological categorization analysis revealed the appearance of type 4 morphology (multiple nucleolar fragmentations without micronuclei) in HCT116 p53−/− cells at 72 h (Fig. 4D, supplementary information Figs. S1B and S2A-E). In addition, dose-dependent analysis confirmed more prominent type 4, type 6 and type 7 characteristics in HCT116 p53−/− cells (Fig. 4E). Alternatively, the morphological analysis in p53-depleted U2OS cells revealed a change in the nucleolar structure from type 2 to types 3 and 7 (Fig. 4F, supplementary information Figs. S1B and S2F-J). These results suggest that p53 is an important gene for suppressing nucleolar fragmentation following X-rays.
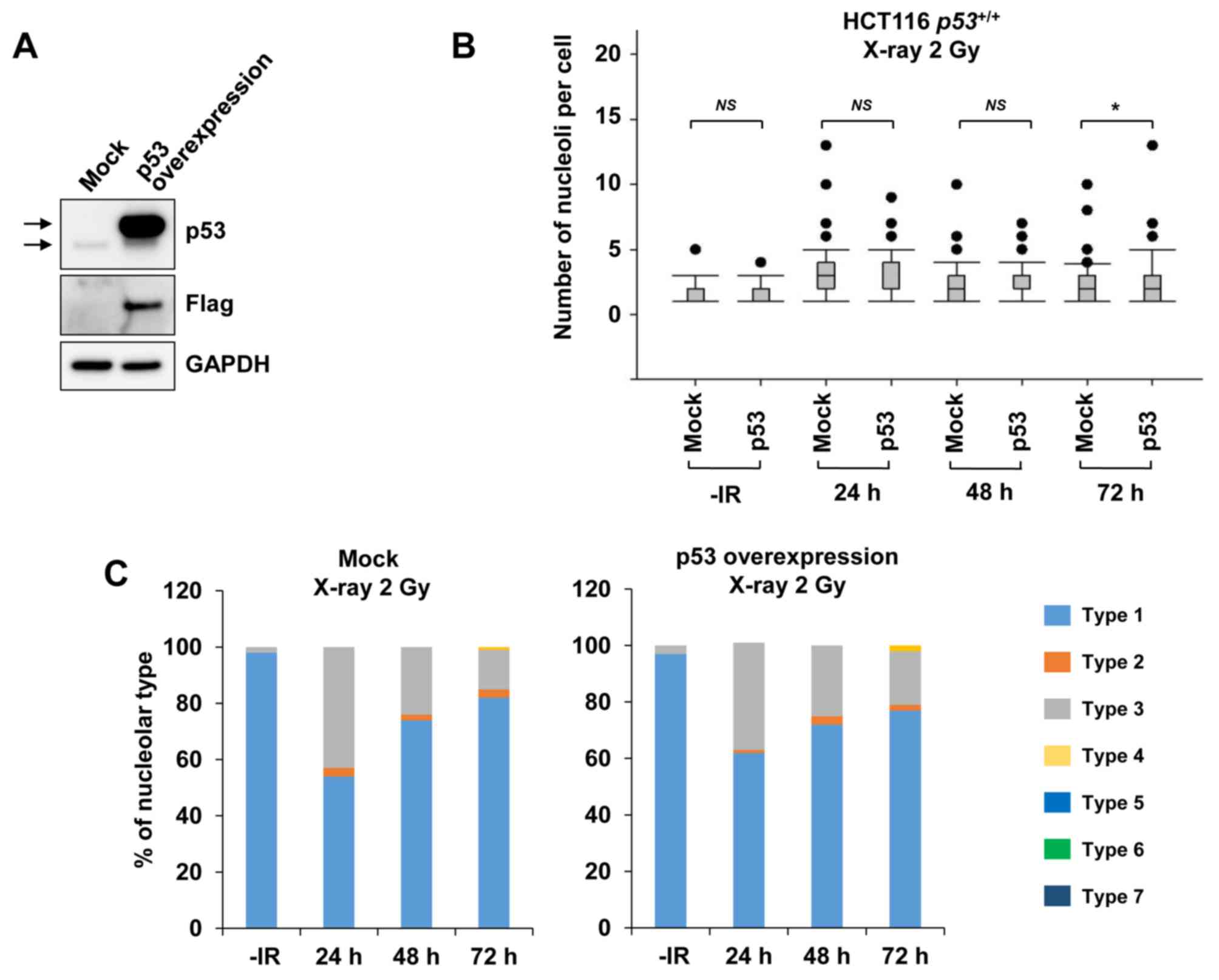
By contrast, when examining nucleolar fragmentation levels in HCT116 p53+/+ cells exogenously overexpressing p53 (Fig. 5A), it was observed that, compared with mock cells, the HCT116 p53+/+ cells overexpressing p53 exhibited a marginal increase in nucleolar fragmentation at 72 h after irradiation with 2 Gy of X-rays (Fig. 5B and C).
Carbon ions induce moderately greater nucleolar fragmentation compared with X-rays
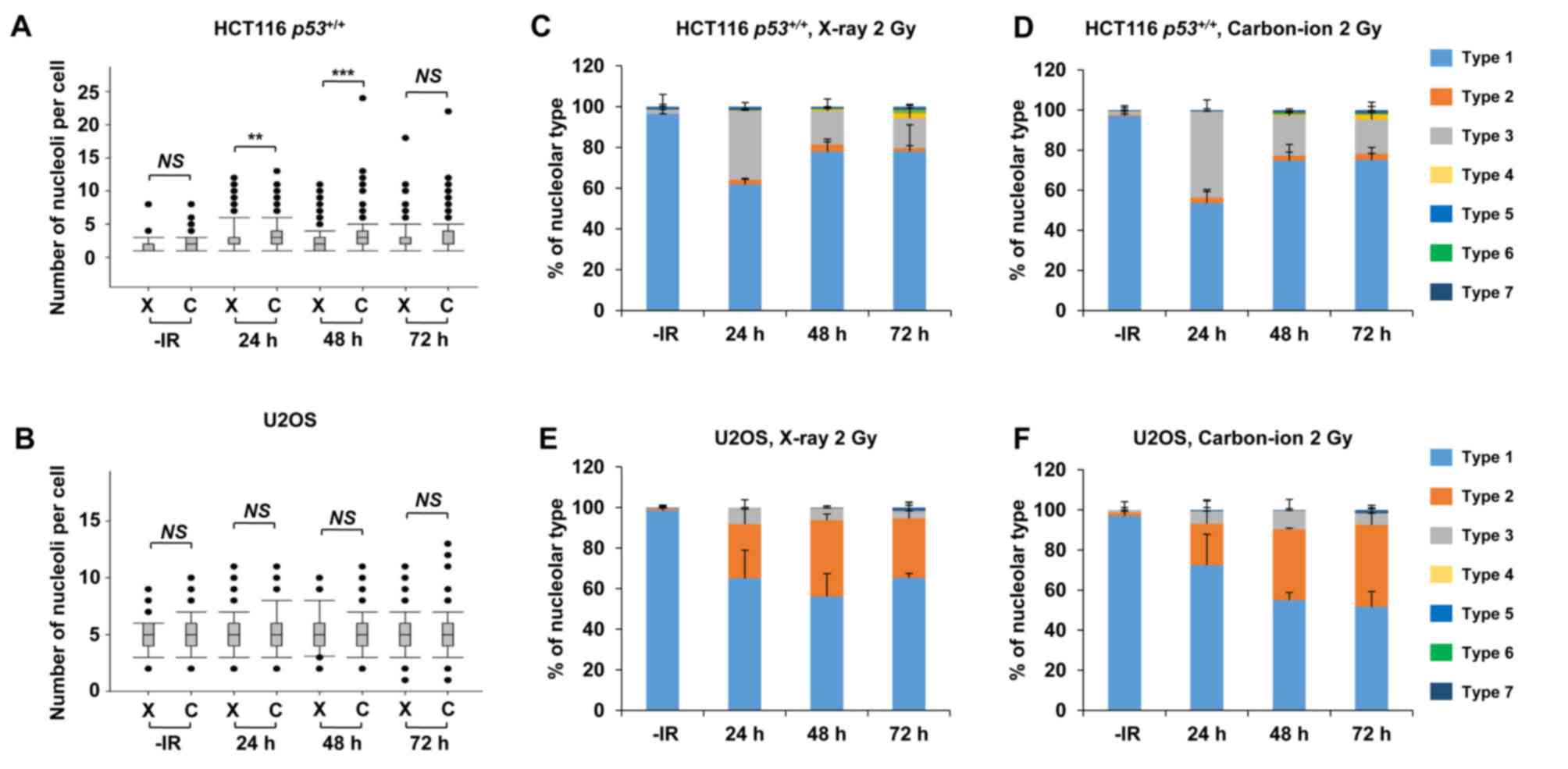
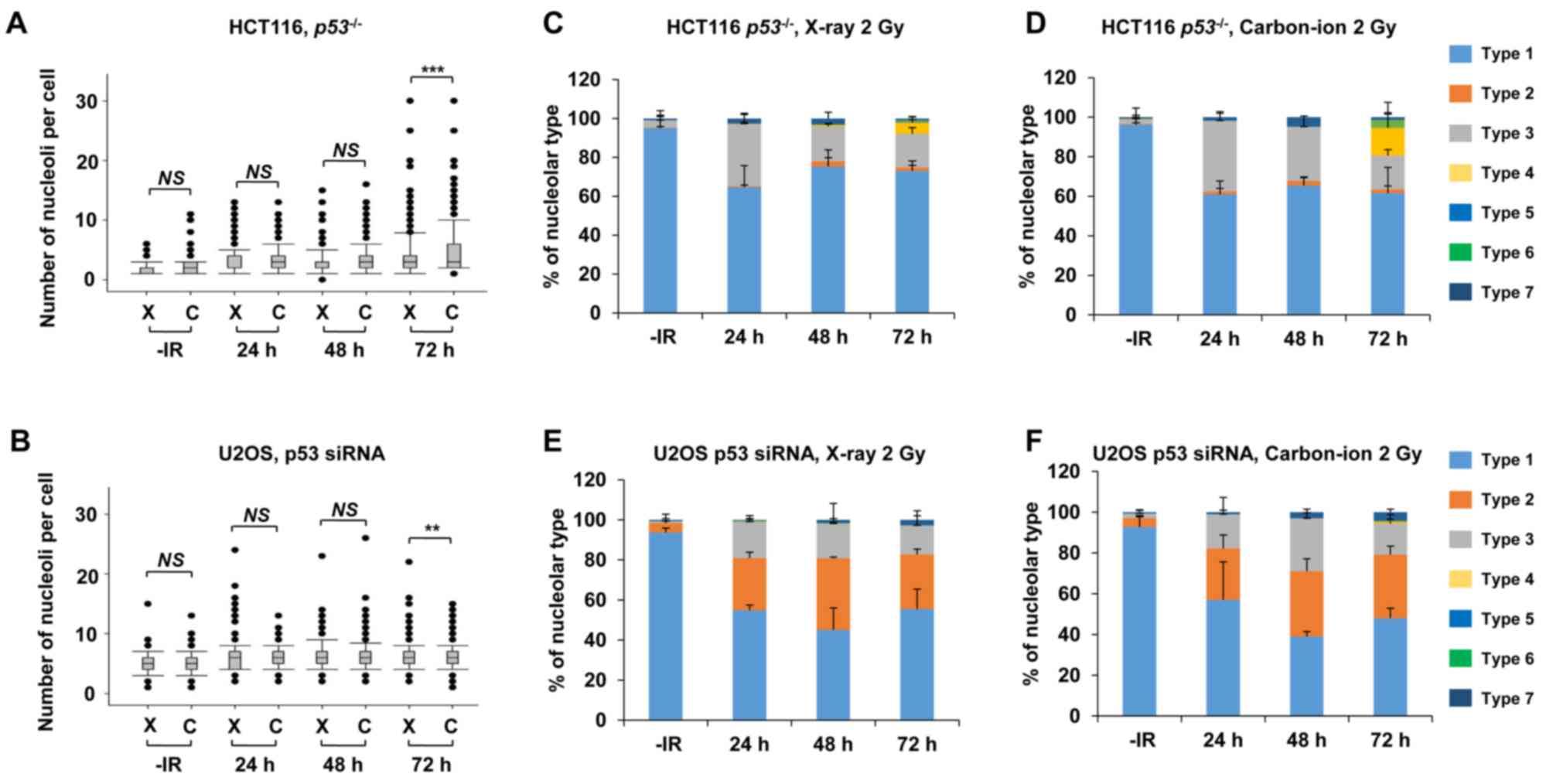
High LET carbon ion irradiation is one of the most promising next-generation radiotherapies (20). Following high LET carbon ion irradiation, critical genome instability causes greater cell-killing effect due to the formation of complex DNA lesions and clustered DSBs, leading to large deletions or chromosomal translocations (21,22). To evaluate the biological effects of high LET carbon ion irradiation on nucleolar stability, the number of nucleoli per cell and the morphology were examined after 2 Gy of X-ray or carbon ion irradiation. Similar to the results following X-ray irradiation (Fig. 1), the number of nucleoli per cell increased after carbon ion irradiation in HCT116 p53+/+ cells and U2OS cells (Fig. 6A and B). The increase in the number of nucleoli after carbon ion irradiation was modestly but statistically significantly higher at 24 h (P=0.001) and at 48 h (P<0.001) compared with that after X-ray irradiation in HCT116 p53+/+ cells. Compared with HCT116 p53+/+ cells, U2OS cells exhibited a modest increase in the number of nucleoli following carbon ion irradiation compared with that after X-ray irradiation; however, it was not statistically significant. Although the type distribution was similar between X-rays and carbon ion irradiation, the morphological changes in HCT116 p53+/+ and U2OS cells were also observed after carbon ion irradiation (Fig. 6C-F, supplementary information Figs. S1C and S3A-H). Next, we examined the number of nucleoli per cell and the morphological changes under a p53 defective background following carbon ion irradiation. At 24–48 h after IR, there was no significant alteration in the number of nucleoli due to p53 deficiency in either HCT116 or U2OS cells, as observed after X-ray exposure (Fig. 7A and B). However, a higher percentage of type 4 morphology was found in HCT116 p53−/− cells at 72 h after carbon ion irradiation, compared with after X-rays or HCT116 p53+/+ cells after carbon ion irradiation (Fig. 7C and D, supplementary information Figs. S1D and S4D). Type 6, which was rarely observed in other conditions, was detected in HCT116 p53−/− cells after carbon ion irradiation (Fig. 7D, supplementary information Fig. S4F). By contrast, a similar type of morphological change was observed in U2OS cells irrespective of the type of IR (Fig. 7E and F, supplementary information Fig. S4G-K). In the normal human fibroblast, similar to X-ray irradiation, carbon ion irradiation did not increase the number of nucleoli (supplementary information Fig. S5).
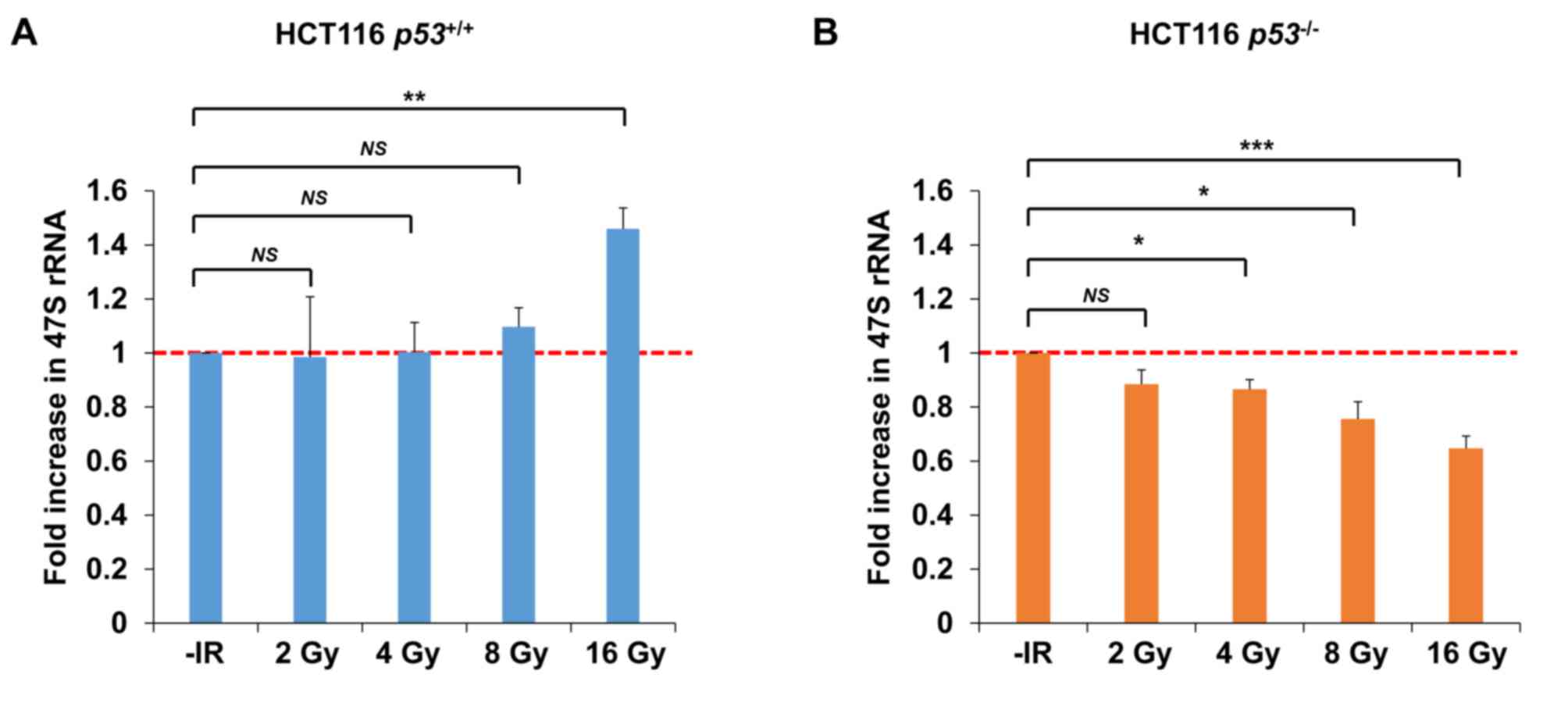
Transcriptional activity of rDNA is reduced under conditions of p53 deficiency following IR
To address the question whether nucleolar fragmentation after IR affects transcriptional activity of rDNA, qPCR was performed to measure the level of 47S rRNA, an immediate transcript of rDNA, which ultimately yields mature rRNA after RNA processing. Importantly, consistent with the results of nucleolar fragmentation, a significant decrease in rRNA synthesis was observed in HCT116 p53−/− cells 72 h after irradiation with 4–16 Gy of X-rays; however, X-ray irradiation did not change rRNA synthesis in HCT116 p53+/+ cells after 2, 4 or 8 Gy (Fig. 8A and B). Instead, an increase was observed in HCT116 p53+/+ cells after 16 Gy.
Discussion
In the present study, it was observed that IR causes nucleolar fragmentation, suggesting that DNA damage disrupts the nucleolar structure. Furthermore, it was observed that cancer cells lacking p53 or subjected to p53 siRNA display increased nucleolar fragmentation following IR. Compared with X-ray irradiation, carbon ion irradiation, generating complex DNA lesions and clustered DSBs, caused only slightly greater nucleolar fragmentation. In addition, the analysis of 47S rRNA using qPCR revealed a significant reduction of ribosomal RNA expression in p53-defective cells following IR. These results suggest that IR causes nucleolar instability, and p53 plays a key role in its prevention.
A chromosomal rearrangement, such as a large deletion, duplication, inversion or translocation, is a characteristic manifestation of dynamic genome instability following IR. The nucleolar fragmentation detected in the present study likely occurs during chromosomal rearrangements. In human cells, ~150–200 copies of rDNA encoding rRNA are located on five distinct chromosomes. Each chromosome contains a repetitive rDNA sequence. As rRNA forms secondary structures, such as hairpins, stem-loops, or cruciform structures (23), such loop structures are also expected to be formed in rDNA when DNA becomes single-stranded (e.g., during DNA replication or transcription). In the present study, however, it may be inferred that DSBs at rDNA regions are unlikely to be induced by IR, since the percentage of rDNA in the total genome is <1%. For example, 2 Gy of X-rays induce ~60 DSBs per cell in G1 phase cells and ~120 DSBs in G2 phase cells (24). Therefore, only 1 or <1 DSB per cell are induced at the rDNA region after 2–8 Gy of X-rays. Since such isolated DSBs are unlikely to be the cause of nucleolar fragmentation, it may be inferred that additional steps are required to induce nucleolar fragmentation following IR. One possibility is that DNA replication-associated DSBs in S phase cells may be the cause of nucleolar fragmentation. IR causes multiple types of DNA damage, such as DSBs, base damage, SSBs and cross-link damage. The amount of base damage and SSBs is estimated to be ~1,000-3,000 per cell per Gy (25). The number of direct DSBs after 1 Gy X-ray is ~30 per G1 cell (24). However, if a DNA replication fork encounters unrepaired base damage or SSBs, the damage is converted into DSBs, namely replication-associated DSBs (i.e., several thousand DSBs may be formed at replication sites) (26). The generation of DNA replication-associated DSBs may be augmented under p53 deficiency. Since p53 plays an important role in the G1/S checkpoint arrest following IR (27), the lack of a p53-dependent G1 arrest leads to the enhancement of the replication-associated DSBs. This model is supported by the findings of the present study, showing that nucleolar fragmentation following IR is augmented in p53-defective cells.
At the beginning of this study, it was expected that carbon ion irradiation may cause substantially greater nucleolar fragmentation, as it is associated with a higher frequency of chromosomal rearrangements compared with that observed after X-rays; however, the present analysis uncovered only a subtle enhancement of nucleolar fragmentation following carbon ion exposure. Replication-associated DSBs from base damage and SSBs may be the primary source of nucleolar fragmentation, as carbon ion-specific DSBs may not be implicated in the enhancement of nucleolar fragmentation. Consistent with this model, it may be inferred that p53 deficiency, which causes failure of G1/S checkpoint arrest (i.e., increase in S phase entry), led to nucleolar fragmentation. Additionally, G2/M checkpoint and mis-segregation during mitosis may be a major factor inducing nucleolar fragmentation. On morphological analysis, an increase in the formation of micronuclei with nucleolin signals was observed following IR, particularly in HCT116 cells and p53-depleted U2OS cells. As micronuclei are formed following mis-segregation in mitosis (28,29) the sensitivity of the G2/M checkpoint arrest is associated with nucleolar fragmentation. Although the role of p53 in G2/M checkpoint arrest following IR may be less significant, the lack of p53 may promote the formation of micronuclei with nucleolin signals through mis-segregation in mitosis following inefficient G2/M checkpoint arrest, particularly in U2OS cells. Additionally, it was observed that the primary type of fragmented nucleoli following IR in HCT116 cells was type 3, whereas in U2OS cells it was type 2. Although the precise mechanism underlying such a distinct type of formation is unclear, different responses between HCT116 and U2OS cells against the G1/S and G2/M checkpoint arrest may be involved. In contrast to the results observed in cancer cell lines, the normal human fibroblasts (1BR hTERT) did not exhibit an increase in the number of nucleolar fragments. Since normal fibroblasts display an intact G1/S and G2/M checkpoint arrest, nucleolar fragmentation and formation of micronuclei may not be efficiently induced via DNA replication and mitosis, respectively. In addition, following persistent checkpoint arrest, fibroblast cells tend to undergo cellular senescence rather than apoptosis (30); these distinct characteristics may explain the different levels of nucleolar fragmentation following IR. By contrast, cancer cells, which normally exhibit the predominant modes of cell death after irradiation, preferentially undergo apoptosis and mitotic catastrophe under p53 intact and deficient conditions, respectively (12). Thus, it appears that the checkpoint machinery involving G1/S and G2/M checkpoint arrest and apoptosis under the regulation of p53 plays a critical role in preventing nucleolar fragmentation following IR (Fig. 9).
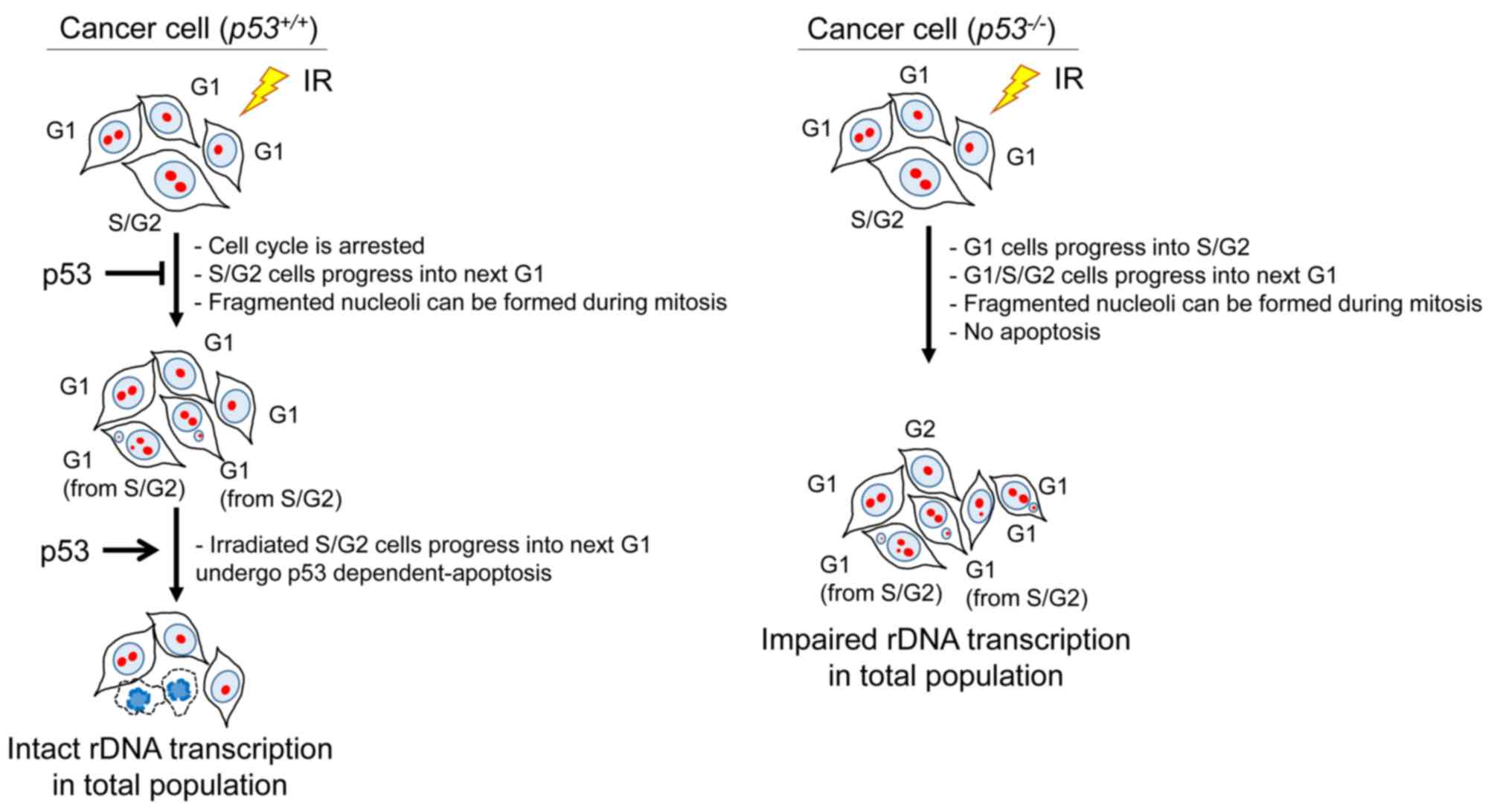
In addition to nucleolar fragmentation, rRNA production was found to be reduced in p53-defective cells following IR. Since rRNA is an essential component of the ribosome, which is required for all protein synthesis, the reduction of ribosomal biogenesis would impair cellular activity and viability. In contrast to p53-defective cells, rRNA production in p53-proficient cells was not reduced, but rather enhanced, following high-dose IR. As described above, a proportion of cells undergo mitosis due to a checkpoint release following a prolonged G2/M checkpoint arrest, which results in micronuclei formation. Following mitosis, cells with micronuclei may undergo apoptosis in the next G1 phase if the p53-dependent apoptosis pathway is intact. Thus, in p53-proficient cells, normal levels of rDNA transcription may be sustained in the remaining cells with intact nucleoli, as cells with fragmented nucleoli are eliminated by apoptosis. On the other hand, in p53-deficient cancer cells, the remaining cells exhibit inefficient rDNA transcriptional activity due to the nucleolar fragmentation, as cells displaying fragmented nucleoli with micronuclei cannot efficiently undergo apoptosis.
Consistently with the present findings, evidence supporting the involvement of p53 in nucleolar biogenesis has also been provided by previous studies (31,32). The downregulation of ribosome biogenesis leads to the release of ribosomal proteins, including Arf, from the nucleolus to the nucleoplasm, which promotes the binding between Murine Double Minute 2 (MDM2) and released ribosomal proteins (31). Thus, the prevention of MDM2-dependent p53 degradation results in p53 activation. In addition, nucleolin and ribosomal protein L26 (RPL26) bind to the 5′ untranslated region of p53 mRNA after DNA damage, thereby controlling the translation of p53 (32). In conclusion, the present study demonstrated that p53 plays an important role in maintaining the transcriptional activity of rDNA in response to DNA damage. The downregulation of the rDNA-rRNA axis (i.e., the pathway of ribosomal biogenesis) under conditions of p53 deficiency may be involved in cancer development via the impairment of cellular homeostasis, particularly after IR or genotoxic stress. These pathways play a significant role in the maintenance of ribosomal biogenesis as one of the mechanisms of cancer prevention, as the downregulation of the rDNA-rRNA axis may be caused by the two key functions of p53, including G1/S and G2/M checkpoint arrest and apoptosis.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Yoshimi Omi, Akiko Shibata, Yoko Hayashi, Shiho Nakanishi, Yukihiko Yoshimatsu, Itaru Sato and Yuka Hirota for assisting with the laboratory work.
Funding
The present study was supported by Grants-in-Aid from the Japan Society for the Promotion of Science for KAKENHI (JP17H04713 to AS), the Takeda Science Foundation, the Uehara Memorial Foundation, and the Program of the network-type Joint Usage/Research Center for Radiation Disaster Medical Science of Hiroshima University, Nagasaki University and Fukushima Medical University. The present study was also supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan for programs for Leading Graduate Schools, Cultivating Global Leaders in Heavy Ion Therapeutics and Engineering.
Availability of materials and data
All the datasets generated and analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
AS designed the experiments and wrote the paper with SK. The experiments including immunofluorescence, immunoblotting and qPCR were performed by SK, YH and AS. Acquired data were analyzed and interpreted by SK, WG, YH, TO, HS, MY, RK, TY and AS. The manuscript was reviewed by TO, HS, KDH, MY and TY. Administrative, technical, or material support was provided by TO, SL and TN. The study was supervised by AS.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
References
|
Buchwalter A and Hetzer MW: Nucleolar expansion and elevated protein translation in premature aging. Nat Commun. 8:3282017. View Article : Google Scholar : PubMed/NCBI | |
|
Ruggero D: Revisiting the nucleolus: From marker to dynamic integrator of cancer signaling. Sci Signal. 5:pe382012. View Article : Google Scholar : PubMed/NCBI | |
|
Montanaro L, Trere D and Derenzini M: Changes in ribosome biogenesis may induce cancer by down-regulating the cell tumor suppressor potential. Biochim Biophysica Acta. 1825:101–110. 2012. | |
|
Stults DM, Killen MW, Williamson EP, Hourigan JS, Vargas HD, Arnold SM, Moscow JA and Pierce AJ: Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 69:9096–9104. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Negi SS and Brown P: rRNA synthesis inhibitor, CX-5461, activates ATM/ATR pathway in acute lymphoblastic leukemia, arrests cells in G2 phase and induces apoptosis. Oncotarget. 6:18094–18104. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Shibata A and Jeggo P: A historical reflection on our understanding of radiation-induced DNA double strand break repair in somatic mammalian cells; interfacing the past with the present. Int J Radiat Biol. 95:945–956. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
van Gent DC, Hoeijmakers JH and Kanaar R: Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2:196–206. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Hada M and Georgakilas AG: Formation of clustered DNA damage after high-LET irradiation: A review. J Radiat Res. 49:203–210. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Nakajima NI, Brunton H, Watanabe R, Shrikhande A, Hirayama R, Matsufuji N, Fujimori A, Murakami T, Okayasu R, Jeggo P and Shibata A: Visualisation of gammaH2AX foci caused by heavy ion particle traversal; distinction between core track versus non-track damage. PLoS One. 8:e701072013. View Article : Google Scholar : PubMed/NCBI | |
|
Hagiwara Y, Niimi A, Isono M, Yamauchi M, Yasuhara T, Limsirichaikul S, Oike T, Sato H, Held KD, Nakano T and Shibata A: 3D-structured illumination microscopy reveals clustered DNA double-strand break formation in widespread γH2AX foci after high LET heavy-ion particle radiation. Oncotarget. 8:109370–109381. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Hagiwara Y, Oike T, Niimi A, Yamauchi M, Sato H, Limsirichaikul S, Held KD, Nakano T and Shibata A: Clustered DNA double-strand break formation and the repair pathway following heavy-ion irradiation. J Radiat Res. 60:69–79. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Amornwichet N, Oike T, Shibata A, Ogiwara H, Tsuchiya N, Yamauchi M, Saitoh Y, Sekine R, Isono M, Yoshida Y, et al: Carbon-ion beam irradiation kills X-ray-resistant p53-null cancer cells by inducing mitotic catastrophe. PLoS One. 9:e1151212014. View Article : Google Scholar : PubMed/NCBI | |
|
Durante M and Cucinotta FA: Heavy ion carcinogenesis and human space exploration. Nat Rev Cancer. 8:465–472. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Henderson AS, Warburton D and Atwood KC: Location of ribosomal DNA in the human chromosome complement. Proc Natl Acad Sci USA. 69:3394–3398. 1972. View Article : Google Scholar : PubMed/NCBI | |
|
Durkin SG and Glover TW: Chromosome fragile sites. Annu Rev Genet. 41:169–192. 2007. View Article : Google Scholar : PubMed/NCBI | |
|
Montgomery PO, Karney D, Reynolds RC and McClendon D: Cellular and subcellular effects of ionizing radiations. Am J Pathol. 44:727–746. 1964.PubMed/NCBI | |
|
Hidvegi EJ, Holland J, Boloni E, Lonai P, Antoni F and Varteresz V: The effect of whole-body x-irradiation of guinea pigs on liver ribosomes. Biochem J. 109:495–505. 1968. View Article : Google Scholar : PubMed/NCBI | |
|
Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Löbrich M and Jeggo PA: Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 30:1079–1092. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Ohno T: Particle radiotherapy with carbon ion beams. EPMA J. 4:92013. View Article : Google Scholar : PubMed/NCBI | |
|
Oike T, Niimi A, Okonogi N, Murata K, Matsumura A, Noda SE, Kobayashi D, Iwanaga M, Tsuchida K, Kanai T, et al: Visualization of complex DNA double-strand breaks in a tumor treated with carbon ion radiotherapy. Sci Rep. 6:222752016. View Article : Google Scholar : PubMed/NCBI | |
|
Niimi A, Yamauchi M, Limsirichaikul S, Sekine R, Oike T, Sato H, Suzuki K, Held KD, Nakano T and Shibata A: Identification of DNA double strand breaks at chromosome boundaries along the track of particle irradiation. Genes Chromosomes Cancer. 55:650–660. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Petrov AS, Bernier CR, Gulen B, Waterbury CC, Hershkovits E, Hsiao C, Harvey SC, Hud NV, Fox GE, Wartell RM and Williams LD: Secondary structures of rRNAs from all three domains of life. PLoS One. 9:e882222014. View Article : Google Scholar : PubMed/NCBI | |
|
Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O and Jeggo PA: gammaH2AX foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimization. Cell Cycle. 9:662–669. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Powell S and McMillan TJ: DNA damage and repair following treatment with ionizing radiation. Radiother Oncol. 19:95–108. 1990. View Article : Google Scholar : PubMed/NCBI | |
|
Vilenchik MM and Knudson AG: Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci USA. 100:12871–12876. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Amundson SA, Myers TG and Fornace AJ Jr: Roles for p53 in growth arrest and apoptosis: Putting on the brakes after genotoxic stress. Oncogene. 17:3287–3299. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, Norppa H, Eastmond DA, Tucker JD and Thomas P: Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 26:125–132. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Huang H, Fletcher L, Beeharry N, Daniel R, Kao G, Yen TJ and Muschel RJ: Abnormal cytokinesis after x-irradiation in tumor cells that override the G2 DNA damage checkpoint. Cancer Res. 68:3724–3732. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Johmura Y, Shimada M, Misaki T, Naiki-Ito A, Miyoshi H, Motoyama N, Ohtani N, Hara E, Nakamura M, Morita A, et al: Necessary and sufficient role for a mitosis skip in senescence induction. Mol Cell. 55:73–84. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
James A, Wang Y, Raje H, Rosby R and DiMario P: Nucleolar stress with and without p53. Nucleus. 5:402–426. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Takagi M, Absalon MJ, McLure KG and Kastan MB: Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 123:49–63. 2005. View Article : Google Scholar : PubMed/NCBI |