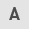Pyruvate kinase type M2 contributes to the development of pancreatic ductal adenocarcinoma by regulating the production of metabolites and reactive oxygen species
- Authors:
- Published online on: January 30, 2018 https://doi.org/10.3892/ijo.2018.4258
- Pages: 881-891
Abstract
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is among the most aggressive of types of cancer, and has a 5-year survival rate of <6% (1). This poor prognosis is due to a typically late-stage clinical diagnosis and the absence of effective chemotherapeutic treatment regimens. Although the 5-year survival rate for early-stage PDAC is ~60%, only 2% of PDAC cases are diagnosed at an early stage (1). Therefore, in order to improve the prognosis of patients with PDAC, the development of novel technologies for the early detection of the disease and effective therapeutic strategies is required.
One characteristic of cancer cells is the ability to reprogram their glucose metabolism. Differentiated tissues (non-proliferating cells) tend to use oxidative phosphorylation and anaerobic glycolysis under aerobic and anaerobic conditions, respectively, as metabolic pathways (2). By contrast, tumor cells and proliferative tissue tend to consume large amounts of glucose and to produce lactate via glycolysis even in the presence of oxygen, a phenomenon known as anaerobic glycolysis, or the Warburg effect. Previous studies have revealed that the Warburg effect is controlled by the pyruvate kinase (PK) M2 isoform (PKM2) (3,4). PK regulates the final step of glycolysis by converting phosphoenolpyruvate and adenosine diphosphate (ADP) into pyruvate and adenosine triphosphate (ATP). PK has 4 isoforms (L, R, M1 and M2) with tissue-specific and developmentally regulated expression (4): PKL is expressed in the liver, PKR in red blood cells, PKM1 in most differentiated tissues (including muscle, heart and brain) and PKM2 in embryonic and tumor cells (5).
PKM2 is strongly expressed in various types of human cancer (6), including pancreatic cancer (7). PKM2 is likely to promote tumorigenesis by regulating the Warburg effect: The downregulation of PKM2 increases oxygen consumption and decreases glucose uptake and lactate production (3), and restoring PKM2 increases cell proliferation and tumor formation in immunodeficient mice (8). In addition to its roles as a PK enzyme, PKM2 also functions as a co-activator or a protein kinase (9). For example, PKM2 interacts with hypoxia-inducible factor (HIF)-1 (10) to reprogram glucose metabolism in cancer cells, and it regulates cyclin D1 and c-Myc expression by phosphorylating histone H3 at threonine 11 (H3-T11), leading to G1-S phase transition, chromosome segregation, cell-cycle progression and tumorigenesis (11). However, little is known about the biological role of PKM2 in PDAC. In this study, to determine whether PKM2 promotes the development of PDAC, we examined the expression and function of PKM2 in human PDAC.
Our results clearly demonstrate that PKM2 is upregulated in PDAC tissues, and that PKM2 knockdown reduces tumorigenesis by altering the expression of genes that drive the cell-cycle G1-S phase transition and the production of intercellular metabolites, particularly spermine, which induces transcription. These findings suggest that PKM2 regulates stable cell division in PDAC and is a potential therapeutic target for PDAC.
Materials and methods
Microdissection of tissue samples
A total of 10 PDAC tissue samples and non-tumor tissue samples were obtained from patients who underwent surgical resection at the Miyagi Cancer Center Hospital, Natori, Japan during the period between 2010 and 2012. Each sample was examined histologically and diagnosed by two pathologists who were unaware of the present study. The non-tumor tissues obtained were adjacent to the tumor tissue samples. The tissues were embedded in paraffin and cut into 10-µm-thick sections, and ~10 sequential regions were microdissected from the same paraffin block using a Leica CIR MIC system (Leica Microsystems, Wetzlar, Germany) to obtain samples of cancer cells (n=10) and normal pancreatic duct cells (n=5). Total RNA was extracted using the Recover All™ Total Nucleic Acid Isolation kit (Ambion, Austin, TX, USA) following the manufacturer's instructions. This study was approved by the Ethics Committee of Miyagi Cancer Center (2010-039), and informed consent was obtained from each patient.
Cell culture
The PDAC cell lines, AsPC-1, BxPC-3, PANC-1 and MIAPaCa, were obtained from the American Type Culture Collection (Manassas, VA, USA), maintained in Dulbecco's modified Eagle's medium (DMEM; Wako Pure Chemical Industries, Osaka, Japan) containing 10% inactivated fetal bovine serum (FBS; EuroClone, Milano, Italy) with 100 U/ml penicillin and 100 µg/ml streptomycin (Nacalai Tesque, Kyoto, Japan) and cultured in a humidified incubator at 37°C, 5% CO2.
RNA interference
PKM2 was transiently knocked down by introducing siRNA against PKM2 (Nippongene, Tokyo, Japan) at a final concentration of 100 nM; Universal Negative Control siRNA (Nippongene) was used as a negative control. The siRNA was introduced using Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) with Opti-MEM I (Thermo Fisher Scientific) according to the manufacturer's instructions. For stable PKM2 knockdown, we used Knockout™ RNAi systems (Clontech Laboratories, Inc., Mountain View, CA, USA) following the manufacturer's instructions. Complementary shRNA oligonucleotides were annealed and ligated into the pSIREN vector. Subsequently, Plat-A packaging cells were transfected with shPKM2 or empty pSIREN vector (control) to produce recombinant retroviruses. PDAC cells stably infected with the recombinant retroviruses were selected with puromycin. The PKM2 sequences targeted by siRNA (si27, si155 and si156) and shRNA (sh27) are described elsewhere (12).
Reverse-transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the PDAC cell lines using the RNeasy Mini kit (Qiagen, Tokyo, Japan). First-Strand cDNA was generated from 300–500 ng of total RNA using the PrimeScript® 1st Strand cDNA Synthesis kit (Takara Bio, Shiga, Japan). PCR was performed at 95°C for 10 min, followed by 45 cycles of 95°C for 10 sec and 60°C for 25 sec. For RT-qPCR, we used a LightCycler® 480, a LightCycler® 480 probes master kit, TaqMan probes from the Universal Probe Library (Roche, Basel, Switzerland) and the following primers: PKM1 forward, cagccaaaggggactatcct and reverse, gaggctcgcacaagttcttc; PKM2 forward, ctatcctctggaggctgtgc and reverse, gtggggtcgctggtaatg; and β-actin forward, ccaaccgcgagaagatga and reverse, tccatcacgatgccagtg.
Cell proliferation assay
Cell proliferation rates were assayed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The cells were seeded at 1×103–1×104 per well in 96-well plates in normal cell growth medium. MTT solution (5 mg/m; Dojindo, Kumamoto, Japan) was added after 24, 48, 72 or 96 h, and the cells were incubated for a further 3 h at 37°C, after which MTT-formazan production was estimated by measuring the absorbance at 570 nm using a VersaMax microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA).
Cell migration assay
Cell migration was evaluated by a wound healing scratch assay. Confluent PDAC cells in 6-well plates were incubated with mitomycin C (Wako Pure Chemical Industries) for 2 h to inhibit cellular proliferation, after which the cells were scratched with the tip of a sterile P200 pipette. The medium was changed to a normal cell growth medium, and changes in the scratched area were measured following a 24-h incubation.
Western blot analysis
Cell lysates were extracted at 72 h after transfection with PKM2 siRNA or negative control. The cells were lysed with RIPA buffer supplemented with protease and phosphatase inhibitors (Roche). The total cell lysates were fractionated by 5–20% SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% non-fat milk in Tris-buffered saline (TBS) containing 0.1% Tween-20. The membranes were incubated overnight at 4°C with primary antibodies (anti-PKM2 and anti-α-tubulin antibodies and the details are described below), washed with TBS 3 times, and incubated with the horseradish peroxidase (HRP) linked secondary antibodies. Reactive bands were detected using Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). Anti-PKM2 rabbit polyclonal antibodies used as primary antibody were originally generated by our group against synthetic peptides. The amino acid sequences of antigen peptides were described previously (3). The α-tubulin (sc-23948; Santa Cruz Biotechnology, Santa Cruz, CA, USA) primary antibody was used as the internal standard. Anti-rat HRP-linked antibody (#7077) and anti-mouse IgG HRP-linked antibody (#7076) (both from Cell Signaling Technology, Danvers, MA, USA) were used as secondary antibodies. The dilution ratio of all the antibodies was 1:1,000.
Cell cycle analysis
Cell cycle regulation was analyzed by fluorescence-activated cell sorting (FACS). The cells were fixed with 70% ethanol, stored overnight at −30°C, and washed twice with FACS buffer (PBS, 0.5% BSA and 0.1% NaN3). The cells were incubated with an anti-Ki-67 antibody (BioLegend, San Diego, CA, USA) at a dilution of 1:60 for 1.5 h at room temperature and then with 20 µg/ml of RNase for 1 h at 37°C. The samples were stained with 5 mg/ml propidium iodide (PI) and analyzed with a FACSCanto™ II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Glucose consumption assay
Extracellular glucose was measured using a Glucose Assay kit (BioVision Technologies, Chester Springs, PA, USA). The cells were incubated in DMEM without FBS for 12 h prior to the assay, and glucose levels were quantified using a VersaMaxPLUS ROM plate reader (Molecular Devices LLC) according to the manufacturer's instructions.
Extracellular flux analyzer
The oxygen consumption rate (OCR, pmoles/min) and extracellular acidification rate (ECAR, mpH/min) were measured using Seahorse XFe96 extracellular flux analyzers (Seahorse Bioscience, North Billerica, MA, USA) according to the manufacturer's instructions. The cells in which PKM2 was knocked down or the control cells were seeded in an XF96 cell-culture microplate at 24 h prior to the assay. On the day of the experiment, the initial culture medium was changed to Agilent Seahorse XF base medium (102353-100; Agilent Technologies, Santa Clara, CA, USA) supplemented with GlutaMAX™ (Thermo Fisher Scientific) with or without 25 mM glucose, and the cells were cultured for 1 h in a CO2-free 37°C incubator and then subjected to a Seahorse XF Cell Mito Stress Test (103015-100) or Seahorse XF Glycolysis Stress Test (103020-100) (both from Agilent Technologies). The following drugs at the indicated concentrations were used for the tests: 3 µM oligomycin A, 3 µM FCCP, 2.78 µM antimycin and 2.78 µM rotenone A for the glycolysis test (all drugs were included in the Seahorse XF Glycolysis Stress Test; 103020-100; Agilent Technologies), and 10 mM glucose, 3 µM oligomycin A and 100 mM 2-DG for the Mito test (all drugs were included in the Seahorse XF Cell Mito Stress Test; 103015-100; Agilent Technologies). Measurements were recorded at the intervals prescribed by the test protocols.
Metabolome analysis
Intracellular metabolites were extracted from the MIAPaCa-2 cells (2×106) in which PKM2 was knocked down or the MIAPaCa-2 control cells. The culture medium was aspirated from the dish, and the cells were washed twice with a 5% mannitol solution. The cells were lysed with 1,300 µl of 60% methanol containing 10 µM commercial Internal Standard Solution 1 (Solution ID: H3304-1002; Human Metabolome Technologies, Inc., Tokyo, Japan) to inactivate enzymes and then harvested with a cell scraper. Tissue samples (50 mg) were plunged into 1,500 µl of a 50/50 mixture of acetonitrile and Milli-Q water containing the internal standards, 50 µM each of methionine sulfone (MetSul) and CSA, homogenized at 9,000 × g at 4°C for 30 sec twice with zirconia beads using a Precellys-24 Homogenizer (Bertin Technologies, Washington, DC, USA), and centrifuged again at 2,300 × g for 5 min. The upper aqueous layer was passed through a filter with a 5-kDa cut-off (UFC3LCCNB-HMT; Human Metabolome Technologies Inc.) to remove the proteins. The filtrate was dried under reduced pressure and suspended in 50 µl of Milli-Q water. The extracted metabolites were measured by a capillary electrophoresis time-of-flight mass spectrometer (CE-TOF/MS) with a commercial electrophoresis buffer (Solution ID H3302-1011and H3302-1021; Human Metabolome Technologies Inc.).
Microarray analysis
To identify the molecular target of PKM2 in the PDAC cells, comprehensive gene expression profiles were analyzed in the PDAC ells (MIAPaCa-2, PANC-1 and AsPC1) in which PKM2 was knocked down and the control (empty vector-transfected) cells using the Agilent Gene Expression Hybridization kit (Agilent Technologies). RNA concentration and purity were determined spectrophotometrically using the NanoDrop ND-1000 (Thermo Fisher Scientific) for all samples. Targets were prepared and the microarrays processed according to the manufacturer's instructions. Chips were scanned by the Agilent Feature Extraction 10.9 to assess the raw probe signal intensities.
Gene set enrichment analysis (GSEA)
GSEA was performed using the Broad Institute platform. Gene sets with a false discovery rate (FDR) value <0.25 after 1,000 permutations were considered significantly enriched.
Gene Expression Omnibus (GEO)
PKM expression was analyzed in PDAC tissue samples using the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) datasets (GDS4336 and GDS4103).
Xenograft tumor experiments
A total of 15 female NOD/Shi-scid-IL2Rγnull (NOG) mice aged 6–10 weeks (~20–25 g) were obtained from the Central Institute for Experimental Animals (CIEA, Kawasaki, Japan). The mice were housed in a cleanroom in the animal care facility at Miyagi Cancer Research Center, with standard temperature, humidity, and timed lighting conditions, and were fed mouse chow/water ad libitum. All animal experiments were approved by the Animal Welfare Committee of Miyagi Cancer Center (MCC-AE-2016-6). Tumor xenografts were generated by subcutaneously injecting 1×104–5 PDAC cells in 100 µl of PBS. Tumor volume was measured once a week for 9 weeks. Tumor volume was calculated using the following formula: [(long diameter) + (short diameter)2] ×1/2. The mice were sacrificed at 9–10 weeks after the injection of the PDAC cells. At that time, the mean weight of the mice was ~30 g. In addition, no mice developed multiple tumors.
Measurement of reactive oxygen species (ROS)
Hydrogen peroxide (H2O2) and ROS levels were measured with the ROS-Glo™ H2O2 assay (Promega, Madison, WI, USA) according to the manufacturer's instructions.
Statistical analysis
Significant differences between 2 groups were determined using the Student's t-test, and differences between >2 groups were evaluated by analysis of variance (ANOVA) and Dunnett's multiple comparisons test. A P-value <0.05 at the 95% confidence level was considered significant. All statistical analyses were performed using GraphPad Prism 6.0 software (GraphPad Software Inc., San Diego, CA, USA).
Results
PKM2 expression is enhanced in PDAC tissues and cell lines
We first analyzed the expression of whole PKM, including the M1 and M2 isoforms, in PDAC tissues using GEO data-sets (Fig. 1A and B), since the probe used in this dataset coded for the common sequence of PKM1 and PKM2, and found that PKM was significantly overexpressed in the PDAC compared with the non-tumorous pancreatic tissues (P<0.01). We then examined the expression of PKM1 and PKM2 by RT-qPCR using specific primers in the microdissected pancreatic lesions (Fig. 1C). As shown in Fig. 1D, the cancer cells expressed significantly more PKM2 than the normal pancreatic duct cells (P<0.01). On the other hand, there was no difference in PKM1 expression levels between the carcinoma and normal cells (Fig. 1E). PKM2 was predominantly expressed compared to PKM1 in the cultured PDAC cells (Fig. 1F and G). Taken together, these results indicated that PKM2, but not PKM1, was upregulated in the PDAC cells.
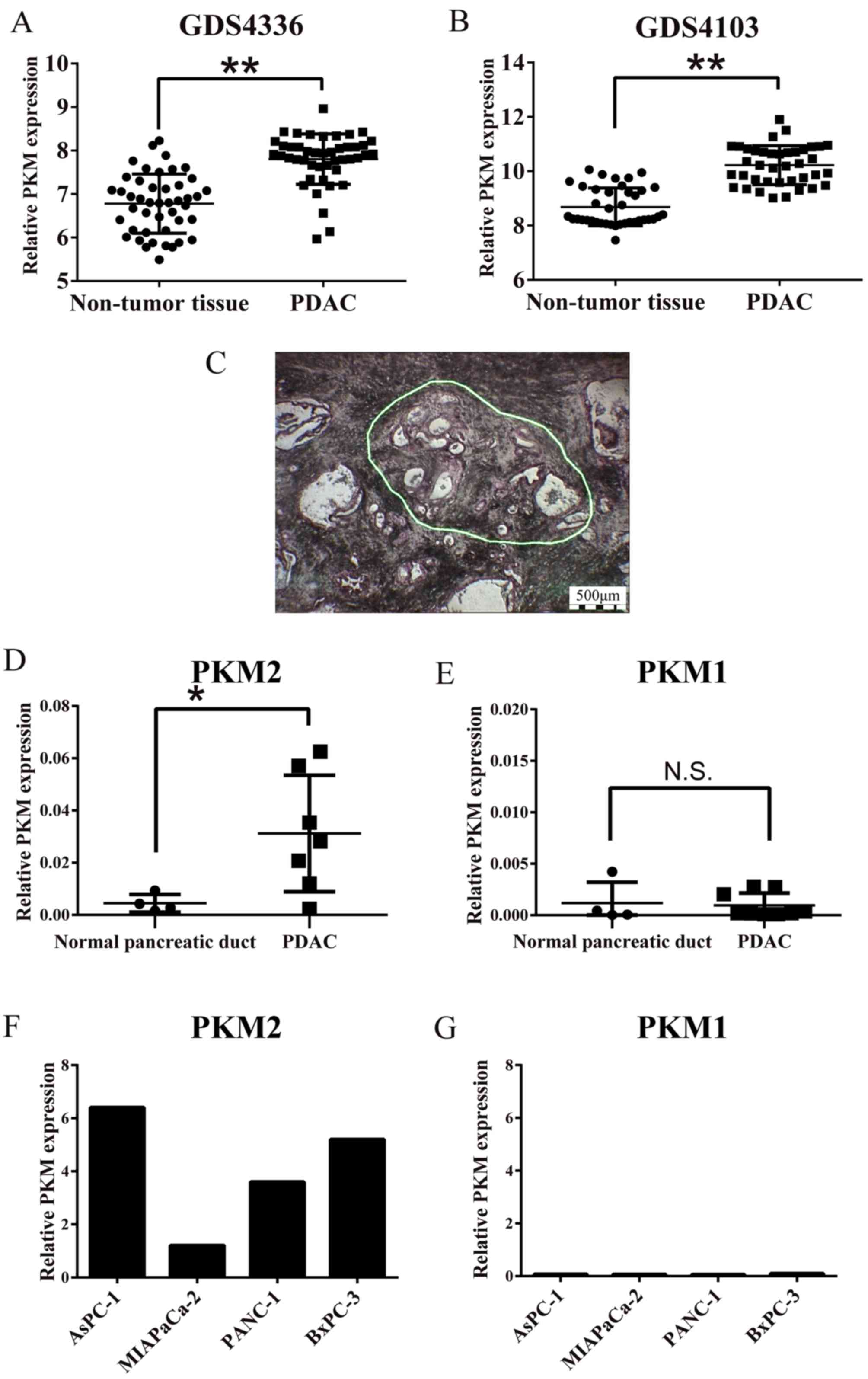
PKM2 knockdown in PDAC cells reduced cell growth, migration, and tumorigenecity
To clarify the role of PKM2 in PDAC, we introduced siRNA (in vitro) and shRNA (in vivo) into the PDAC cells to generate cells in which PKM2 was knocked down (PKM2-KD group). The results of western blot analysis confirmed the reduction of PKM2 expression in each PDAC cell line (Fig. 2A). The knockdown of PKM2 (PKM2-KD) also suppressed the growth and migration of the PDAC cells (Fig. 2B–D). Consistent with these results in vitro, PKM2-KD significantly decreased the growth of PDAC cell-derived tumors (PDAC cells were injected subcutaneously into NOG mice) (Fig. 2E). These data thus suggest that PKM2 enhances cellular growth, migration and tumorigenesis in PDAC. The xenograft and the following experiments were performed using sh27 or si27 RNA which downregulated PKM2 expression most effectively.
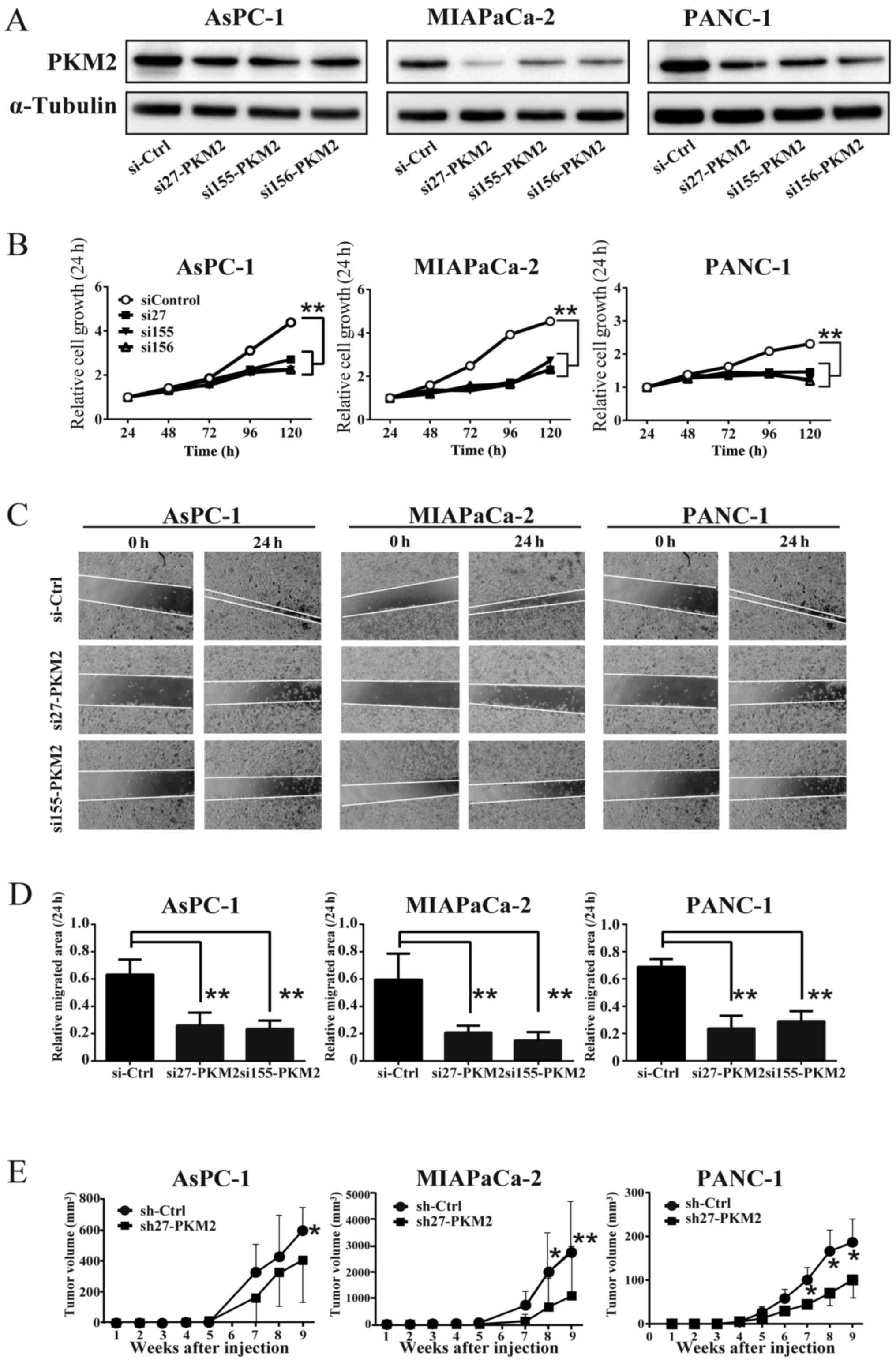
The expression of cell cycle-related genes is downregulated and the cell cycle is arrested in the PDAC cells in which PKM2 is downregulated
To determine whether the PKM2 expression level in PDAC was associated with the expression of molecules involved in tumor promotion, we used GSEA to identify gene expression signatures associated with PKM2 expression, and identified 146 gene sets that were significantly enriched in the PDA
C control cells compared to the PDAC cells in which PKM2 was knocked down (PKM2-KD group), with a P-value of <0.01 and a FDR of <25%. Among these gene sets, the expression levels of cell cycle signature genes were markedly downregulated in the siRNA-transfected PDAC cells compared to the PDAC cells in the PKM2-KD group, as shown by the enrichment plots for the activated genes related to the different phases of the cell cycle (Fig. 3A–D). In particular, the expression of MCM2-6, the gene that encodes Mcm2-6 (which makes up part of the hexameric Mcm2-7 complex) (13), was downregulated in the PDAC cells in the PKM2-KD group. The expression levels of ORC1 and ORC6, which encode proteins in the origin recognition complex (ORC), a multi-subunit DNA binding complex that binds to origins of replication, were also downregulated. Consistent with these affected genes, the cell cycle was arrested in the PKM2-deficient PDAC cells (Fig. 3E).
Glycolysis and the production of some metabolites are attenuated in PDAC cells in which PKM2 is downregulated
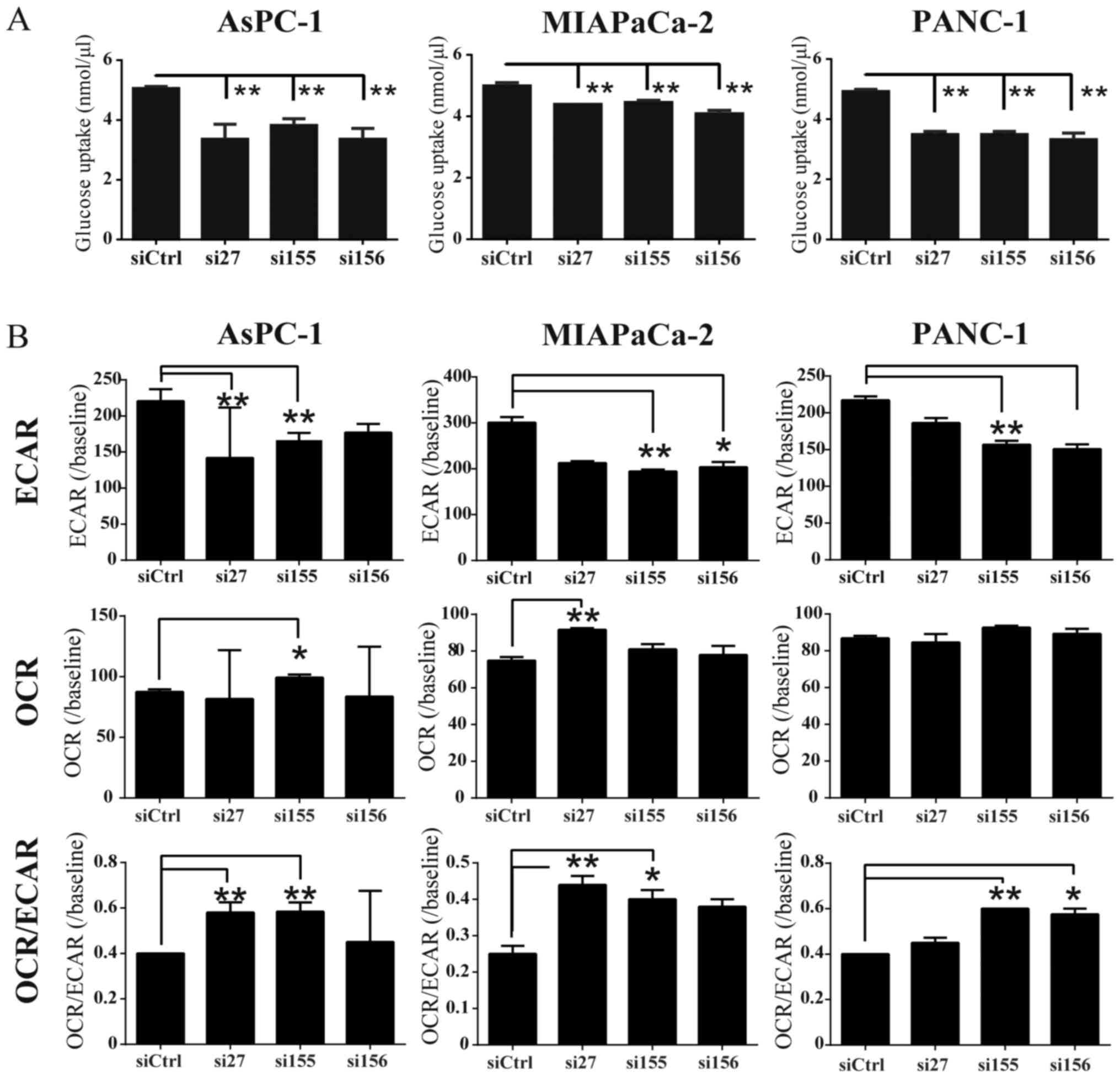
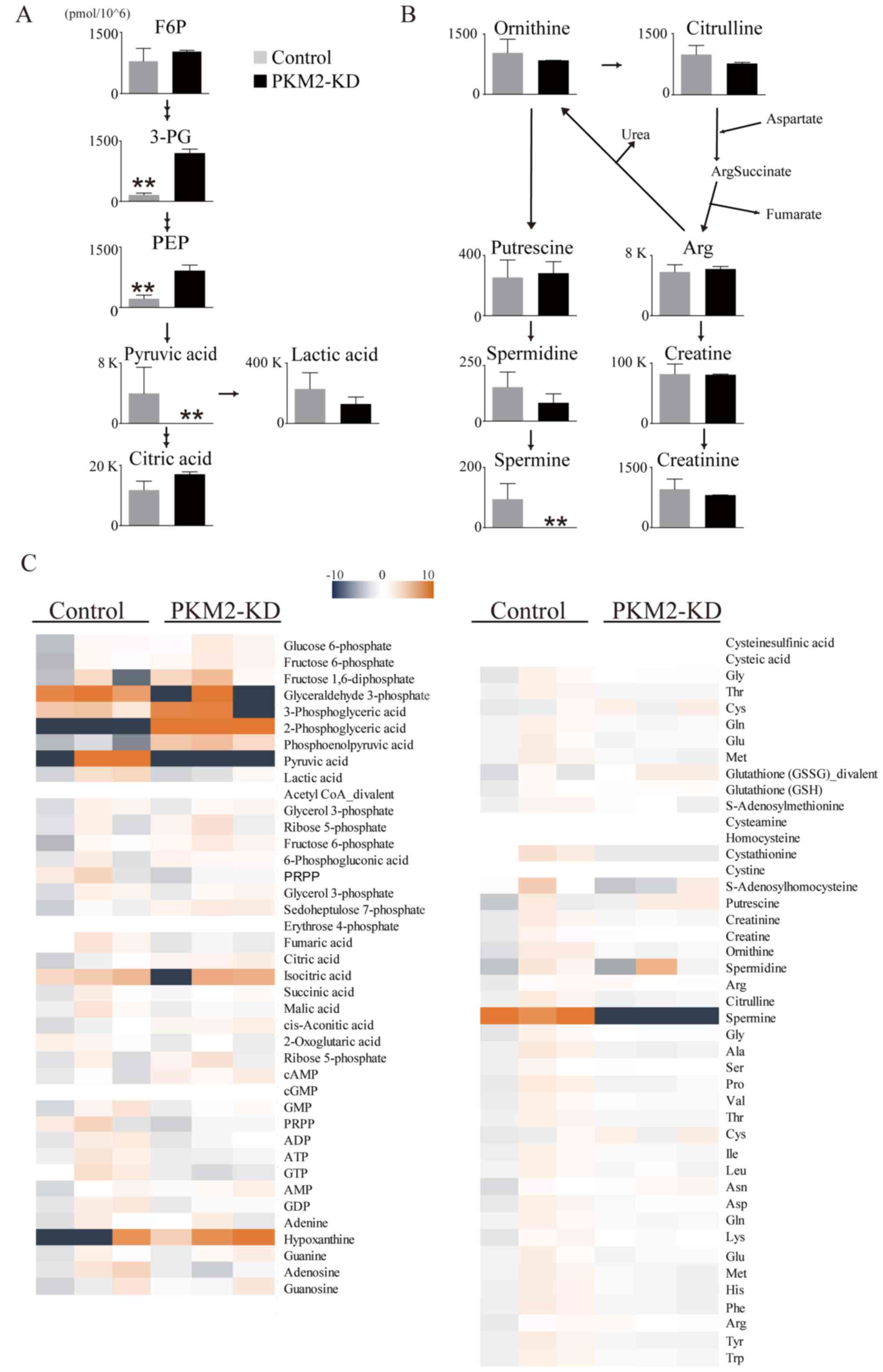
To determine whether PKM2 KD influences glucose uptake, we examined glucose consumption in PDAC cell lines, and found that it was decreased in the siPKM2-transfected PDAC cells (Fig. 4A). To examine whether PKM2 expression was associated with glycolysis and oxidative phosphorylation, we used extracellular flux analyzers to monitor ECAR and OCR, which indicate the glycolytic pathway and mitochondrial respiration, respectively. ECAR was significantly reduced in the cells in the PKM2-KD group compared to the controls (Fig. 4B). PKM2 deficiency had little effect on OCR; however, the OCR/ECAR ratio was increased in the cells in the PKM2-KD group, indicating that their energy was mainly generated by mitochondrial respiration. We then evaluated changes in PDAC intercellular metabolites following PKM2 knockdown (Fig. 5A and B). CE-TOF/MS revealed that the levels of some intracellular metabolites were decreased in the cells in the PKM2-KD group, particularly pyruvate and spermine (Fig. 5C). These data thus indicate that PKM2 plays an important role in maintaining glycolysis in PDAC cells.
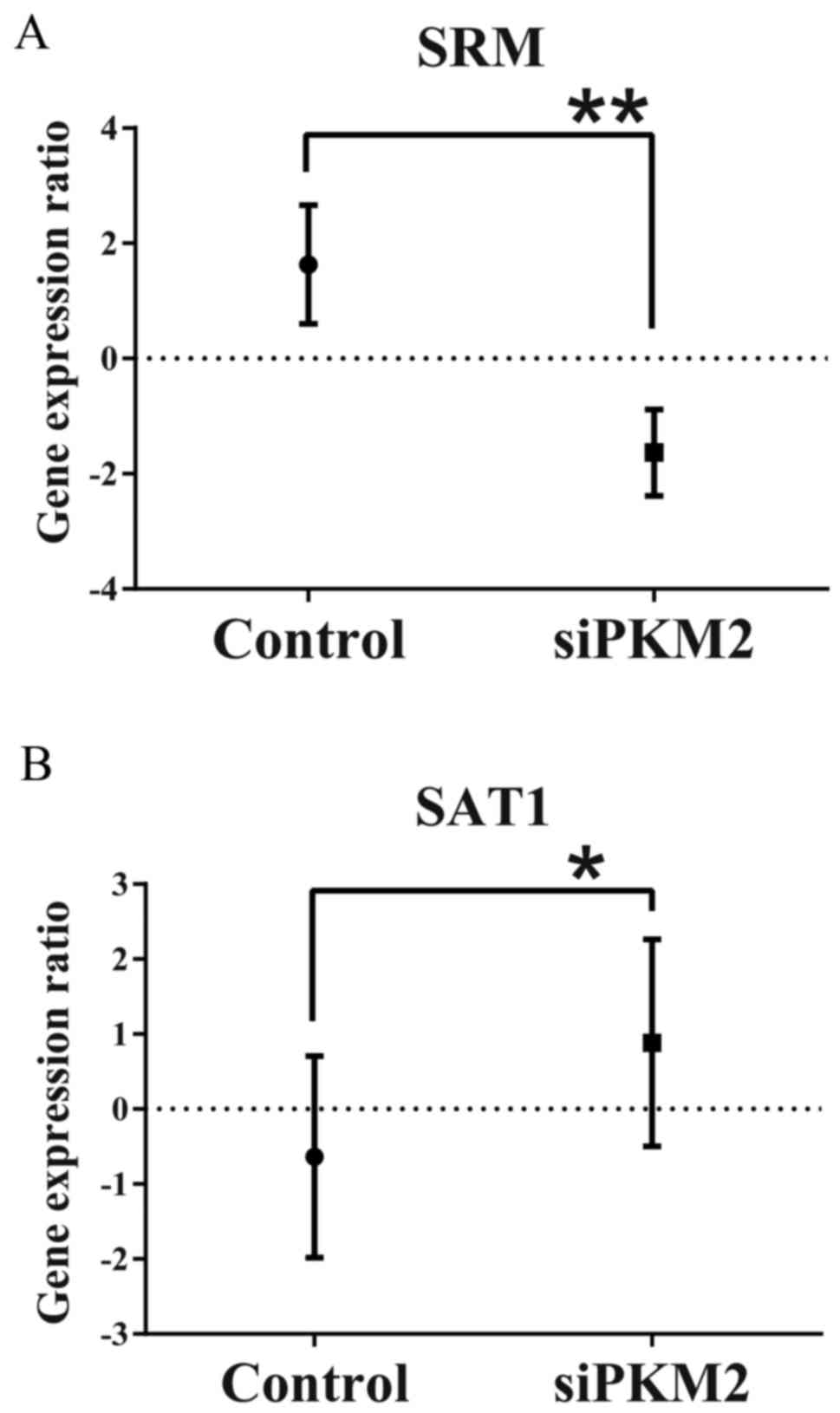
PKM2 knockdown results in the downregulation of spermidine synthase (SRM) and the upregulation of spermidine N1-acetyltransferase 1 (SAT1)
To assess the molecular mechanisms responsible for the decrease in spermine production in the PDAC cells in the PKM2-KD group, we evaluated the differences in gene expression profiles between the PDAC control cells and those in the PKM2-KD and group. We found a significant decrease in SRM levels in the PDAC cells in the PKM2-KD group (Fig. 6A). In addition, the expression of SAT1, which catalyzes the acetylation of spermidine and spermine to lead to rapid depletion of spermidine and spermine (14) was significantly increased in the PDAC cells in the PKM2-KD group (Fig. 6B). In this context, the attenuated production of spermine was due to the alteration of the expression of these enzymes by PKM2 suppression in the PDAC cells.
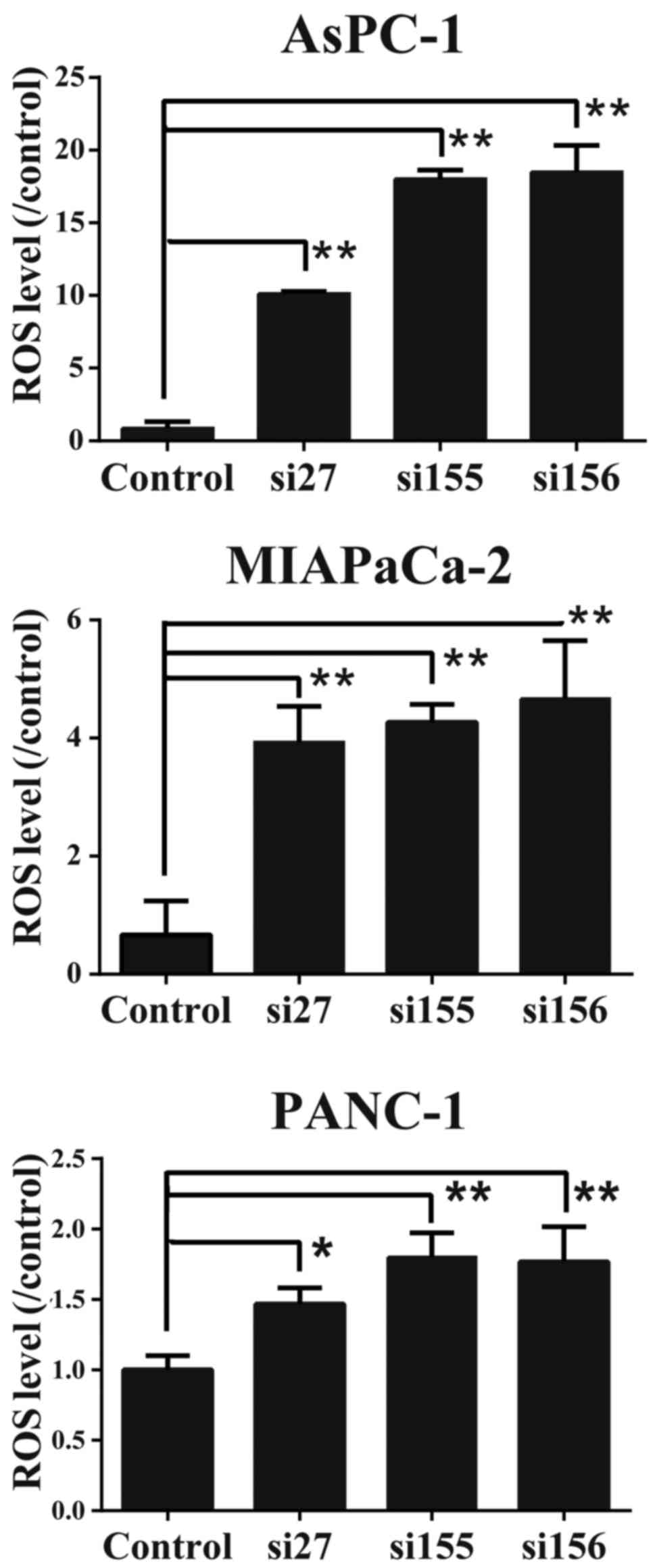
ROS levels increase in PDAC cells in which PKM2 is knocked down
To determine whether PKM2 downregulation affects the oxidative stress level, we examined the intercellular ROS concentrations in the PDAC cells. As shown in Fig. 7, ROS concentration was elevated in the PDAC cells in which PKM2 was knocked down. These data suggest that the presence of PKM2 regulates ROS production or concentration in PDAC cells.
Discussion
In this study, we demonstrated that PKM2, but not PKM1 expression was upregulated in PDAC cells compared to non-tumorous pancreatic tissue, and decreasing the PKM2 expression in PDAC cells suppressed cell proliferation, migration and tumor growth, and attenuated the Warburg effect, indicating that PKM2 contributes to carcinogenesis and development of PDAC. We also showed that when PKM2 expression was downregulated in the PDAC cells, the expression levels of genes that drive the cell cycle were also downregulated, leading to cell cycle arrest. In addition, PKM2 knockdown impeded the production of intercellular metabolites, particularly spermine, and increased the ROS concentration.
Cancer metabolism has been extensively researched, and some studies have suggested that PKM2 contributes to tumorigenesis in various types of cancer by regulating cancer metabolism (4). The PKM2 level is associated with overall survival in PDAC (15), and a decreased PKM2 expression suppresses cancer cell survival and invasion by altering the Warburg effect (7). Similar results were observed in this study, suggesting that PKM2 regulates the Warburg effect in the development of PDAC. In addition, our microarray analysis revealed that suppression of PKM2 expression resulted in the change in gene sets of glycolysis and glucose metabolism (data not shown), indicating that PKM2 is involved in more metabolic pathway facilitating cancer development other than expected.
Spermine is one of the polyamines, which are small basic molecules that are essential to normal cell growth and DNA stability (16–18). The polyamine content in the cell is highly regulated, and its metabolic alteration is implicated in many diseases. Studies have indicated correlations between polyamines and tumor progression in breast, colon, prostate, and skin cancers (19). Furthermore, depleting polyamines inhibits skin-tumor growth (20) and breast-cancer migration (21). The spermine concentration is increased in the urine of humans with pancreatic cancer (22). Spermine also accelerates hypoxia-initiated cancer-cell migration (23). In this study, we found that spermine was decreased in the cells in the PKM2-KD group compared to the control PDAC cells. To elucidate the molecular mechanisms responsible for the alteration of spermine production, we examined the changes in the expression of polyamine-related genes between the PDAC cells in the PKM2-KD group and the control PDAC cells by microarray analysis and found the SRM reduction and SAT1 induction, both of which cause the attenuation of spermine production, in the PDAC cells in the PKM2-KD group. Taken together, these data suggest the possibility that PKM2 facilitates PDAC cell growth and migration by enhancing spermine production.
We also found that the ROS levels were elevated in the cells in the PKM2-KD group. Increased ROS levels cause oxidative stress and promote tumorigenesis and tumor migration (24,25). On the other hand, excessive increases in ROS levels have been shown to induce cell cycle arrest and cell death (26,27). In breast cancer, cancer stem cells contain lower ROS concentrations than those found in corresponding non-tumorigenic cells (28). Taken together, these findings suggest that elevated ROS in levels in PDAC cells in which PKM2 is knocked down may inhibit the cell cycle and cell survival, along with tumor migration and growth.
In conclusion, in this study, we demonstrated that PKM2 was upregulated in PDAC cells compared to non-cancerous cells and that the knockdown of PKM2 decreased the of cell growth and migratory abilities that contribute to cancer cell development via the alteration of cancer-specific metabolism and the acceleration of ROS production, suggesting that PKM2 may be a biological marker and an effective therapeutic target in PDAC.
Abbreviations:
|
ADP |
adenosine diphosphate |
|
ATP |
adenosine triphosphate |
|
cDNA |
complementary DNA |
|
CE-TOF/MS |
capillary electrophoresis time-of-flight mass spectrometry |
|
DMEM |
Dulbecco's modified Eagle's medium |
|
FBS |
fetal bovine serum |
|
siRNA |
small interfering ribonucleic acid |
|
shRNA |
short hairpin ribonucleic acid |
|
PBS |
phosphate-buffered saline |
|
PVDF |
polyvinylidene difluoride |
|
RT-PCR |
reverse-transcription polymerase chain reaction |
Acknowledgments
Not applicable.
Notes
[1] Funding
This study was supported by Grants-in-Aid for Scientific Research (KAKENHI) (nos. 15K19080 and 15K09055) to MY and KS, respectively.
[2] Availability of data and materials
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
[3] Authors' contributions
M.Y., N.T., K.T. and K.S. designed the study, interpreted the results, analyzed the data and wrote and edited the manuscript. M.A., K.Y., K.M. and I.S. provided the surgical specimens and provided the histopathological findings. M.Y., R.S. and T.S. performed the in vivo experiments involving animals. M.Y., K.Y. and K.T performed the in vitro experiments. All authors are aware of the content in this manuscript and have read and edited the manuscript.
[4] Ethics approval and consent to participate
For the use of human samples, this study was approved by the Ethics Committee of Miyagi Cancer Center (2010-039), and informed consent was obtained from each patient. In addition, all animal experiments were approved by the Animal Welfare Committee of Miyagi Cancer Center (MCC-AE-2016-6).
[5] Consent for publication
Not applicable.
[6] Competing interests
The authors declare that they have no competing interests.
References
|
Ma J, Siegel R and Jemal A: Pancreatic cancer death rates by race among US men and women, 1970–2009. J Natl Cancer Inst. 105:1694–1700. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Vander Heiden MG, Cantley LC and Thompson CB: Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and Cantley LC: The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 452:230–233. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Mazurek S: Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 43:969–980. 2011. View Article : Google Scholar | |
|
Marie J, Levin MJ, Simon MP and Kahn A: Genetic and epigenetic control of the pyruvate kinase isozymes in mammals. Isozymes Curr Top Biol Med Res. 7:221–240. 1983.PubMed/NCBI | |
|
Dong G, Mao Q, Xia W, Xu Y, Wang J, Xu L and Jiang F: PKM2 and cancer: The function of PKM2 beyond glycolysis. Oncol Lett. 11:1980–1986. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Li C, Zhao Z, Zhou Z and Liu R: PKM2 promotes cell survival and invasion under metabolic stress by enhancing Warburg effect in pancreatic ductal adenocarcinoma. Dig Dis Sci. 61:767–773. 2016. View Article : Google Scholar | |
|
Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang L, You MJ, Koh MY, Cote G, Aldape K, et al: EGFR-induced and PKCε monoubiquitylation-dependent NF-κB activation upregulates PKM2 expression and promotes tumorigenesis. Mol Cell. 48:771–784. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Wong N, Ojo D, Yan J and Tang D: PKM2 contributes to cancer metabolism. Cancer Lett. 356:184–191. 2015. View Article : Google Scholar | |
|
Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, Cole RN, Pandey A and Semenza GL: Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 145:732–744. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK and Lu Z: PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 150:685–696. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Goldberg MS and Sharp PA: Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J Exp Med. 209:217–224. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Bell SP and Dutta A: DNA replication in eukaryotic cells. Annu Rev Biochem. 71:333–374. 2002. View Article : Google Scholar : PubMed/NCBI | |
|
Pegg AE: Spermidine/spermine-N(1)-acetyltransferase: A key metabolic regulator. Am J Physiol Endocrinol Metab. 294:E995–E1010. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
Lockney NA, Zhang M, Lu Y, Sopha SC, Washington MK, Merchant N, Zhao Z, Shyr Y, Chakravarthy AB and Xia F: Pyruvate kinase muscle isoenzyme 2 (PKM2) expression is associated with overall survival in pancreatic ductal adenocarcinoma. J Gastrointest Cancer. 46:390–398. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Pegg AE: Polyamine metabolism and its importance in neoplastic growth and a target for chemotherapy. Cancer Res. 48:759–774. 1988.PubMed/NCBI | |
|
Pegg AE: Mammalian polyamine metabolism and function. IUBMB Life. 61:880–894. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Childs AC, Mehta DJ and Gerner EW: Polyamine-dependent gene expression. Cell Mol Life Sci. 60:1394–1406. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Nowotarski SL, Woster PM and Casero RA Jr: Polyamines and cancer: Implications for chemotherapy and chemoprevention. Expert Rev Mol Med. 15:e32013. View Article : Google Scholar : PubMed/NCBI | |
|
Takigawa M, Verma AK, Simsiman RC and Boutwell RK: Inhibition of mouse skin tumor promotion and of promoter-stimulated epidermal polyamine biosynthesis by alpha-difluoromethylornithine. Cancer Res. 43:3732–3738. 1983.PubMed/NCBI | |
|
Gupta ED, Pachauri M, Ghosh PC and Rajam MV: Targeting polyamine biosynthetic pathway through RNAi causes the abrogation of MCF 7 breast cancer cell line. Tumour Biol. 37:1159–1171. 2016. View Article : Google Scholar | |
|
Russell DH: Increased polyamine concentrations in the urine of human cancer patients. Nat New Biol. 233:144–145. 1971. View Article : Google Scholar : PubMed/NCBI | |
|
Krüger A, Vowinckel J, Mülleder M, Grote P, Capuano F, Bluemlein K and Ralser M: Tpo1-mediated spermine and spermidine export controls cell cycle delay and times antioxidant protein expression during the oxidative stress response. EMBO Rep. 14:1113–1119. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, McCord JM and Harman D: Oxygen radicals and human disease. Ann Intern Med. 107:526–545. 1987. View Article : Google Scholar : PubMed/NCBI | |
|
Chio IIC and Tuveson DA: ROS in cancer: The burning question. Trends Mol Med. 23:411–429. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Liou GY and Storz P: Reactive oxygen species in cancer. Free Radic Res. 44:479–496. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Galadari S, Rahman A, Pallichankandy S and Thayyullathil F: Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic Biol Med. 104:144–164. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al: Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 458:780–783. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Zheng B, Peng J, Mollayup A, Bakri A, Guo L, Zheng J and Xu H: Construction of a prognostic prediction system for pancreatic ductal adenocarcinoma to investigate the key prognostic genes. Mol Med Rep. 17:216–224. 2018. | |
|
Badea L, Herlea V, Dima SO, Dumitrascu T and Popescu I: Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology. 55:2016–2027. 2008. |