Introduction
Pulmonary arterial hypertension (PAH) is a common
clinical condition with a range of underlying etiologies
characterized by pathological changes in the pulmonary arteries,
which lead to significant increases in the pulmonary arterial
pressure (PAP) and in the right ventricular hypertrophy (RVH).
There is no cure for this disease and medical regimens are limited.
All forms of PAH share a common pathogenesis characterized by
vasoconstriction, pulmonary vascular remodeling and thrombosis
in situ (1). Vascular
remodeling is majorly caused by pulmonary artery smooth muscle cell
(PASMCs) proliferation and migration, which is critical to the
pathogenesis of PAH, however, the mechanism of pulmonary vascular
remodeling is incompletely understood. Therefore, preventing or
treating the development of PAH via the inhibition of pulmonary
vascular remodeling is an important strategy.
Peroxisome proliferator-activated receptor γ (PPARγ)
is a family member of nuclear receptors, which consists of PPARα,
PPARβ (also known as PPARδ) and PPARγ, and previous studies have
suggested that PPARγ has broad protective effects on the
cardiovascular system that surpass the regulation of adipogenesis
and glucose metabolism (2–5). Activation of PPARγ plays an important
role in anti-inflammation, anti-proliferation and anti-fibrosis,
suggesting that activation of PPARγ may benefit a variety of
diseases (6–8). Evidence has been accumulating that
activation of PPARγ may be a novel PAH therapeutic target. Studies
have indicated that activation of PPARγ by rosiglitazone (Rosi),
one of the most potent and selective synthetic agonists of PPARγ
receptors, protects against PAH development in a series of
experimental models (9). However, the
mechanisms underlying the inhibition of proliferation of PASMCs by
activation of PPARγ have not been completely understood; therefore
hemodynamic, histology and immunoblotting were performed in the
present study to test whether the activation of PPARγ inhibits
monocrotaline (MCT)-induced PAH in rats.
Materials and methods
Animals
Male Sprague-Dawley (SD) rats (250–350 g) were
purchased from the animal center of the Xi'an Jiaotong University
Medical College (Xi'an, China). The protocol of the study was
approved by the Institutional Ethics Committee of Xi'an Jiaotong
University Health Science Center and complied with the Declaration
of the National Institutes of Health Guide for Care and Use of
Laboratory Animals (publication no. 85–23, revised 1985). All the
animals were housed in climate-controlled conditions with a 12-h
light:12-h dark cycle and had free access to chow and water.
Experimental protocols
The 18 SD (7-week-old) rats were randomly divided
into three groups (n=6 in each group). Models of PAH rats were
induced by subcutaneous injection of MCT (60 mg/kg; Sigma, St.
Louis, MO, USA) on day 1, and another group model of PAH rats were
orally administered Rosi (5 mg/kg/day; GlaxoSmithKline, London, UK)
for 4 weeks until the rats were sacrificed. The control rats were
subcutaneously injected with normal saline and orally administered
the same volume of saline.
Measurement of the right ventricular
systolic pressure (RVSP) and RVH
At day 28, rats were anesthetized with 10% chloral
hydrate (0.3 ml/kg, intraperitoneal injection). Following stable
anesthesia, the right jugular vein was exposed and a polyethylene
catheter (PE50) was inserted into the right atrium and right
ventricle (RV) and the RVSP was monitored using a Grass polygraph
(PowerLab; AD Instruments, Sydney, Australia). RVSP was assumed to
be equal to the PAP in the presence of a normal pulmonary valve.
The analog signals of pressure were digitized with a sampling
frequency of 1,000 Hz and expressed in millimeters of mercury. The
RVH was routinely checked to observe the establishment of PAH in
the MCT-treated rats, which was assessed by the ratio of the weight
of the right ventricle to that of the left ventricle plus septum:
RV/(LV + S).
Tissue preparation and morphometric
analysis of the pulmonary vessels
The rats were sacrificed following homodynamic
measurements and the lungs were harvested. The right lungs were
fixed in 4% paraformaldehyde overnight, sectioned at 5-µm and
stained with hematoxylin and eosin (H&E). Explants of
intrapulmonary arteries were isolated from the left lungs and
stored at −80°C for future use.
Pulmonary vascular remodeling was evaluated by
determining the percentage medial wall thickness (%MT) and wall
area (%WA) of vessels (diameter, 50–200 µm) according to the method
by Ochoa et al (10) and Wang
et al (11). The medial
thickness of the vessel wall is defined as the distance between
inner and outer elastic lamina. Vessel external diameter (ED),
lumen internal area (IA) and average total vessel area (TA) were
examined. The %MT was calculated as (2MT/ED) × 100 and %WA was
calculated as (TA - IA)/TA × 100. All the analyses were performed
in a blinded manner.
Western blotting
Lung tissues were lysed in radioimmunoprecipitation
assay lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP40, 0.5%
sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 1 mM
phenyl-methylsulfonyl fluoride, 1 mM Na3VO4,
1mM NaF and proteinase inhibitors]. Lysates were centrifuged at
13,000 rpm at 4°C for 15 min and the supernatant was collected as
total protein. The protein concentrations were measured using
Pierce BCA protein assay kit (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Protein was separated on SDS-PAGE gel and
transferred onto a Trans-Blot nitrocellulose membrane (Bio-Rad,
Hercules, CA, USA). Polyclonal antibodies against phosphatase and
tensin homologue deleted on chromosome ten (PTEN; 9188S), p-Akt
(13038S), total Akt (8596S; all Cell Signaling Technologies,
Danvers, MA, USA) and rabbit anti-rat GAPDH (ABS16; Chemicon
International, Inc., Billerica, MA, USA) (1:1,000 dilution) were
used according to the manufacturer's instructions. Subsequently,
the membrane was incubated with horseradish peroxidase conjugated
with goat anti-rabbit IgG antibody (A0545; Sigma) (1:5,000
dilution), and reactions were developed with SuperSignal West Pico
Chemiluminescent Substrate (Pierce Biotechnology, Inc., Rockford,
IL, USA) and exposed to autoradiographic film. Signaling was
quantified from scanned films using Quality One software
(Bio-Rad).
Statistics analysis
Values are presented as mean ± standard error of the
mean. SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA) processing
was used for statistical analysis. Data were analyzed using one-way
analysis of variance followed by Tukey post hoc test analysis of
variance. P<0.05 was considered to indicate a statistically
significant difference between groups.
Results
Activation of PPARγ inhibits
MCT-induced PAH
To examine whether activation of PPARγ suppresses
the development of PAH in rats induced by MCT, RVSP and the weight
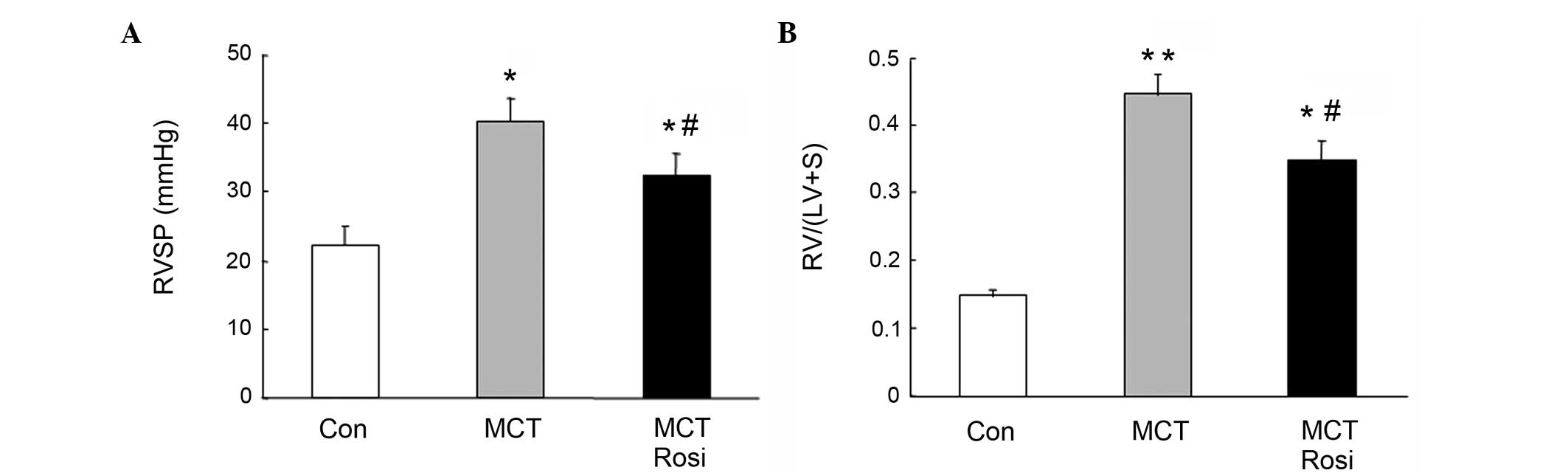
ratio of RV/(LV + S) were examined. As shown in Fig. 1, RVSP was significantly increased in
MCT-treated rats (40.32±3.44 mmHg) compared with the control rats
(22.34±2.55 mmHg; P<0.05), and this was accompanied with the
increased ratio of RV/(LV + S). The ratio of RV/(LV + S) in the
model of PAH (0.45±0.03) was significantly increased compared with
that in the control rats (0.15±0.02; P<0.01). However, treatment
of the PAH model with the PPARγ agonist rosiglitazone for 4 weeks
reduced the RVSP (32.44±3.12 mmHg; P<0.05 vs. MCT group) and
ameliorated the ratio of RV/(LV + S) (0.34±0.03; P<0.05 vs. MCT
group), suggesting that activation of PPARγ effectively suppressed
the development of PAH.
Effects of PPARγ activation on
MCT-induced pulmonary arterial remodeling
To determine the mechanisms underlying activation of
PPARγ inhibition of PAH, remodeling of the pulmonary artery was
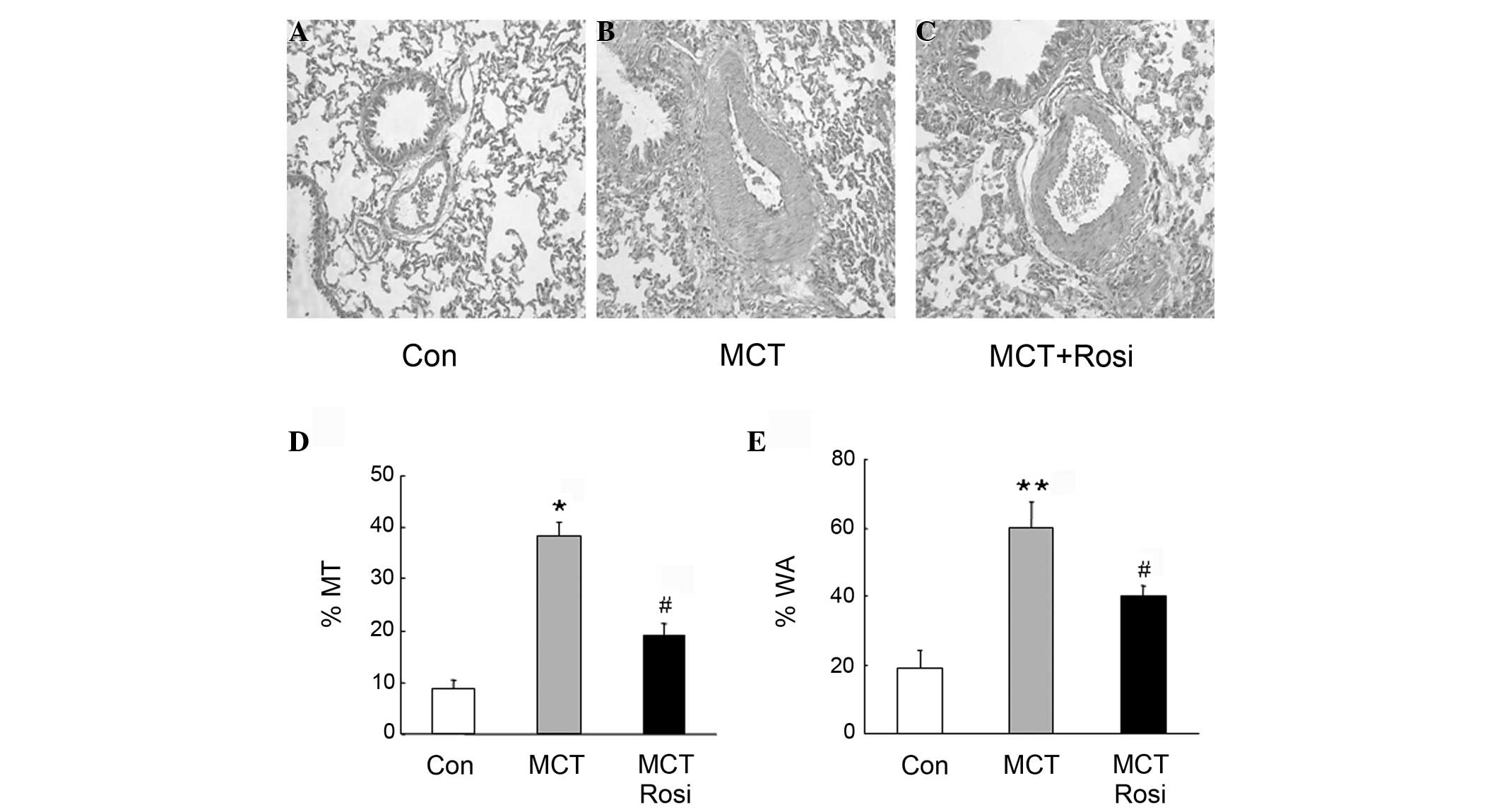
examined using histological analysis. Fig.
2A and B show that medial MT and WA were significantly elevated
in small pulmonary arterioles in MCT-treated rats compared with
that of the control rats. This was accompanied with the increase in
cell number of PASMCs in the medial layer of the small pulmonary
artery, whereas treatment with rosiglitazone suppressed these
changes in the pulmonary artery in MCT-treated rats (Fig. 2C), indicating that rosiglitazone
inhibited pulmonary artery remodeling by suppressing the
proliferation of PASMCs. Fig. 2D and E
further indicated that %MT and %WA in rats of MCT-induced PAH were
significantly increased, while the presence of rosiglitazone
reduced these parameters in the PAH model (MCT + Rosi vs. MCT
group, P<0.05), suggesting that activation of PPARγ inhibited
the pulmonary vascular remodeling in MCT-treated rats.
Mechanisms of activation of PPARγ
inhibiting PASMCs proliferation
Activation of the PI3K/Akt signaling pathway is
associated with a variety of types of cells proliferation,
including PASMCs. Therefore, it is important to know whether
activation of PPARγ by rosiglitazone inhibits PASMCs proliferation
through negative regulation of the PI3K/Akt pathway in
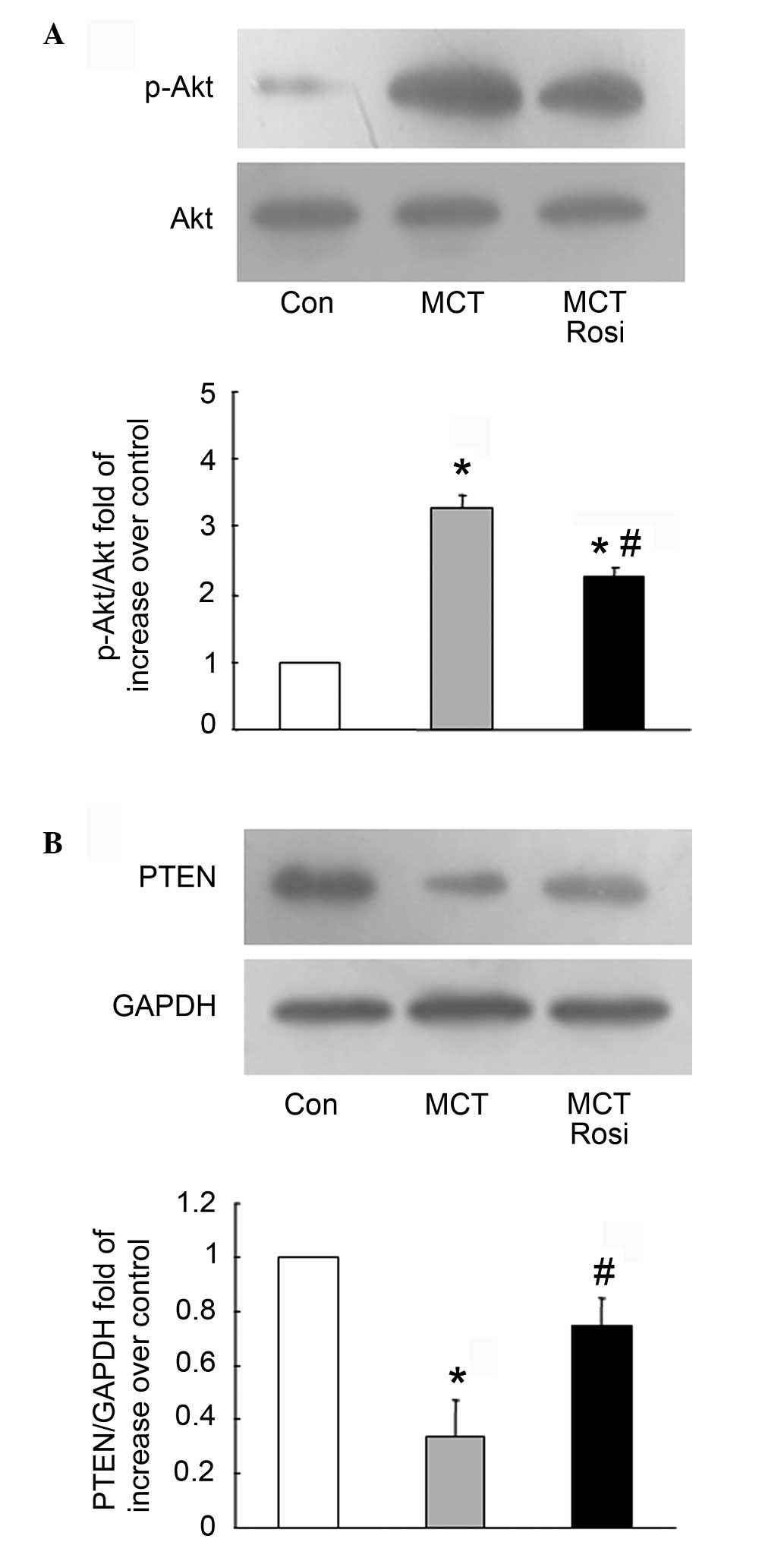
intrapulmonary arteries. Fig. 3A shows
that the phosphorylation of Akt was increased with a 3.29-fold
compared with the control in MCT-treated rats, while the level of
Akt phosphorylation decreased from a 3.29-fold to 2.27-fold
increase over the control in the rosiglitazone-treated model of PAH
(MCT + Rosi vs. MCT group, P<0.05), suggesting that activation
of PPARγ suppresses the proliferation of PASMCs by modulating
PI3K/Akt signaling.
To further clarify the mechanisms of activation of
PPARγ negatively regulating PI3K/Akt cascade, the intrinsic
negative regulator of PI3K, PTEN, was examined. As shown in
Fig. 3B, the endogenous level of PTEN
was lower than the control in the MCT-induced model of PAH (MCT vs.
control, P<0.05), while the co-treatment with rosiglitazone
reversed the reduction of the PTEN protein level compared to the
model of PAH (MCT + Rosi vs. MCT group, P<0.05), indicating
activation of PPARγ regulation of the PI3K/Akt pathway by
upregulation of PTEN.
Discussion
The major finding of the present study is that
activation of PPARγ by rosiglitazone significantly induced the
expression of PTEN, which in turn inhibits the PI3K/Akt signaling
pathway, mediates the proliferation of PASMCs and subsequently
suppresses pulmonary vascular remodeling and the development of
PAH. The study provides a novel molecular basis whereby activation
of PPARγ modulates vascular remodeling and treats PAH.
Vascular remodeling is believed to be the major
contributor during PAH, which is characterized by enhanced
proliferation and migration of PASMCs. Several signaling pathways
are associated with the growth and apoptosis of PASMCs during PAH
(12,13). For example, stimulation of human PASMCs
with platelet-derived growth factor induced PI3K-dependent
activation of Akt, p70 S6 kinase and ribosomal protein S6,
suggesting that PI3K signaling is necessary to mediate human PASMCs
proliferation and the activation of PI3K may play an important role
in vascular remodeling (14). In the
present study, the level of p-Akt significantly increased in rats
with MCT-induced PAH, along with the elevation of RVSP and right
ventricle hypertrophy. Furthermore, the H&E staining showed
that there was a significant increase of the proliferation of
PASMCs and the vascular remodeling of pulmonary arteries in the
MCT-treated group. Taken together, these results suggest that
PI3K/Akt signaling pathway is responsible for the proliferation of
PASMCs, pulmonary vascular remodeling and the development of
PAH.
The PTEN tumor-suppressor gene is frequently mutated
in numerous types of human cancer, such as pancreatic cancer,
glioblastoma and lung cancer (15–17).
Previous studies have suggested that PTEN not only regulates
cardiomyocyte hypertrophy and survival, but also regulates
pulmonary smooth muscle cell proliferation and cell survival
(18,19). Phosphatidylinositol 3-kinases (PI3Ks)
are a subfamily of lipid kinases, which catalyze the formation of
phosphatidylinositol-3,4,5-trisphosphate (PIP3) from
phosphatidylinositol-4,5-bisphosphate (PIP2). The serine-threonine
kinase Akt (also known as PKB) is a centrally important downstream
effector of PIP3, which regulates proliferation, growth and
survival (20,21). Previous studies have shown that PTEN
plays an important role in the pathogenesis of diabetes and
numerous cancer diseases, and further study has indicated that
induction of PTEN by activation of PPARγ inhibits the PI3K/Akt
signaling pathway-mediated proliferation of tumor cells (22–25). The
present study showed that the expression of PTEN was significantly
decreased in the MCT-induced group, along with the activation of
PI3K/Akt pathway, and the accelerated process of PASMCs
proliferation and pulmonary vascular remodeling.
PPARγ is one of the ligand-activated nuclear hormone
receptors of the steroid receptor, and it has been shown to be
involved in lipid metabolism and glucose homeostasis (26). Previous studies indicate that PPARγ
also regulates cellular proliferation and differentiation and
anti-angiogenesis and anti-inflammatory functions (27–29). PTEN
contains two PPARγ responsive elements within its promoter region,
and PPARγ can bind to its putative PPAR response elements and
regulate the expression of PTEN. Increasing evidence supports that
the PPARγ agonists of the TZD class (rosiglitazone and
troglitazone) inhibit PAH and pulmonary vascular remodeling in
several experimental models of PAH (30–33). By
contrast, targeted genetic deletion of PPARγ from endothelial or
vascular smooth muscle cells is associated with the development of
spontaneous PAH in mice (34–36). Furthermore, recent studies showed that
activation of PPARγ reduces PAH through regulation of the ET-1
receptors or alteration in miR-27a and ET-1 levels (37,38). The
results of the present study found that rosiglitazone treatment
decreased RVSP and the ratio of RV/(LV + S) in MCT-induced PAH in
rats, and %MT and %WA induced by MCT were also similarly decreased
in MCT rats, which was associated with the induction of PTEN and
suppression of PI3K/Akt signaling cascades. Taken together, the
present results identify that the activation of PPARγ suppresses
MCT-induced PAH in rats, at least partly, by the regulation of the
PTEN/PI3K/Akt signaling pathway, suggesting that the therapeutic
potential of strategies targeting PPARγ will be beneficial to treat
PAH.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81070045) and the
Key Clinical Project for Affiliated Hospital of Ministry of Public
Health of China (grant no. 111).
References
|
1
|
Humbert M, Sitbon O and Simonneau G:
Treatment of pulmonary arterial hypertension. N Engl J Med.
351:1425–1436. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Martens FM, Visseren FL, Lemay J, de
Koning EJ and Rabelink TJ: Metabolic and additional vascular
effects of thiazolidinediones. Drugs. 62:1463–1480. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Meng Y, Chen C, Tian C, Du J and Li HH:
Angiotensin II-induced Egr-1 expression is suppressed by peroxisome
proliferator-activated receptor-γ ligand 15d-PGJ2 in macrophages.
Cell Physiol Biochem. 35:689–698. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gao D, Hao G, Meng Z, et al: Rosiglitzone
Suppresses Angiotensin II-Induced Production of KLF5 and Cell
Proliferation in Rat Vascular Smooth Muscle Cells. PLoS One.
10:e01237242015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nicola T, Ambalavanan N, Zhang W, et al:
Hypoxia-induced inhibition of lung development is attenuated by the
peroxisome proliferator-activated receptor-γ agonist rosiglitazone.
Am J Physiol Lung Cell Mol Physiol. 301:L125–134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang C, Ting AT and Seed B: PPAR-γ
agonists inhibit production of monocyte inflammatory cytokines.
Nature. 391:82–86. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morales-Garcia JA, Luna-Medina R,
Alfaro-Cervello C, Cortes-Canteli M, Santos A, Garcia-Verdugo JM
and Perez-Castillo A: Peroxisome proliferator-activated receptor γ
ligands regulate neural stem cell proliferation and differentiation
in vitro and in vivo. Glia. 59:293–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kulkarni AA, Thatcher TH, Olsen KC,
Maggirwar SB, Phipps RP and Sime PJ: PPAR-γ ligands repress
TGFβ-induced myofibroblast differentiation by targeting the
PI3K/Akt pathway: Implications for therapy of fibrosis. PLoS One.
6:e159092011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nisbet RE, Bland JM, Kleinhenz DJ,
Mitchell PO, Walp ER, Sutliff RL and Hart CM: Rosiglitazone
attenuates chronic hypoxia-induced pulmonary hypertension in a
mouse model. Am J Respir Cell Mol Biol. 412:482–490. 2010.
View Article : Google Scholar
|
|
10
|
Ochoa CD, Yu L, Al-Ansari E, Hales CA and
Quinn DA: Thrombospondin-1 null mice are resistant to
hypoxia-induced pulmonary hypertension. J Cardiothorac Surg.
5:322010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang LX, Sun Y, Chen C, Huang XY, Lin Q,
Qian GQ, Dong W and Chen YF: Effects and mechanism of oridonin on
pulmonary hypertension induced by chronic hypoxia-hypercapnia in
rats. Chin Med J (Engl). 122:1380–1387. 2009.PubMed/NCBI
|
|
12
|
Ismail S, Sturrock A, Wu P, Cahill B,
Norman K, Huecksteadt T, Sanders K, Kennedy T and Hoidal J: NOX4
mediates hypoxia-induced proliferation of human pulmonary artery
smooth muscle cells: The role of autocrine production of
transforming growth factor-{β}1 and insulin-like growth factor
binding protein-3. Am J Physiol Lung Cell Mol Physiol.
296:L489–L499. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang D, Ma C, Li S, Ran Y, Chen J, Lu P,
Shi S and Zhu D: Effect of Mitofusin 2 on smooth muscle cells
proliferation in hypoxic pulmonary hypertension. Microvasc Res.
84:286–296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goncharova EA, Ammit AJ, Irani C, Carroll
RG, Eszterhas AJ, Panettieri RA and Krymskaya VP: PI3K is required
for proliferation and migration of human pulmonary vascular smooth
muscle cells. Am J Physiol Lung Cell Mol Physiol. 283:L354–L363.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fenton TR, Nathanson D, Ponte de
Albuquerque C, Kuga D, Iwanami A, Dang J, Yang H, Tanaka K,
Oba-Shinjo SM, Uno M, et al: Resistance to EGF receptor inhibitors
in glioblastoma mediated by phosphorylation of the PTEN tumor
suppressor at tyrosine 240. Proc Natl Acad Sci USA.
109:14164–14169. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ying H, Elpek KG, Vinjamoori A, Zimmerman
SM, Chu GC, Yan H, Fletcher-Sananikone E, Zhang H, Liu Y, Wang W,
et al: PTEN is a major tumor suppressor in pancreatic ductal
adenocarcinoma and regulates an NF-κB-cytokine network. Cancer
Discov. 1:158–169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K
and Yang GH: MicroRNA-21 (miR-21) represses tumor suppressor PTEN
and promotes growth and invasion in non-small cell lung cancer
(NSCLC). Clin Chim Acta. 411:846–852. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oudit GY and Penninger JM: Cardiac
regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc Res.
82:250–260. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oudit GY, Sun H, Kerfant BG, Crackower MA,
Penninger JM and Backx PH: The role of phosphoinositide-3 kinase
and PTEN in cardiovascular physiology and disease. J Mol Cell
Cardiol. 37:449–471. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen L, Monti S, Juszczynski P, Ouyang J,
Chapuy B, Neuberg D, Doench JG, Bogusz AM, Habermann TM, Dogan A,
et al: SYK inhibition modulates distinct PI3K/AKT- dependent
survival pathways and cholesterol biosynthesis in diffuse large B
cell lymphomas. Cancer Cell. 23:826–838. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
De Luca A, Maiello MR, D'Alessio A,
Pergameno M and Normanno N: The RAS/RAF/MEK/ERK and the PI3K/AKT
signalling pathways: Role in cancer pathogenesis and implications
for therapeutic approaches. Expert Opin Ther Targets. 16:(Suppl 2).
S17–S27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Moon SH, Kim DK, Cha Y, Jeon I, Song J and
Park KS: PI3K/Akt and Stat3 signaling regulated by PTEN control of
the cancer stem cell population, proliferation and senescence in a
glioblastoma cell line. Int J Oncol. 42:921–928. 2013.PubMed/NCBI
|
|
23
|
Conley-LaComb MK, Saliganan A, Kandagatla
P, Chen YQ, Cher ML and Chinni SR: PTEN loss mediated Akt
activation promotes prostate tumor growth and metastasis via
CXCL12/CXCR4 signaling. Mol Cancer. 12:852013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Akca H, Demiray A, Aslan M, Acikbas I and
Tokgun O: Tumour suppressor PTEN enhanced enzyme activity of GPx,
SOD and catalase by suppression of PI3K/AKT pathway in non-small
cell lung cancer cell lines. J Enzyme Inhib Med Chem. 28:539–544.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kitagishi Y and Matsuda S: Redox
regulation of tumor suppressor PTEN in cancer and aging (Review).
Int J Mol Med. 31:511–515. 2013.PubMed/NCBI
|
|
26
|
Monsalve FA, Pyarasani RD, Delgado-Lopez F
and Moore-Carrasco R: Peroxisome proliferator-activated receptor
targets for the treatment of metabolic diseases. Mediators Inflamm.
2013:5496272013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nickkho-Amiry M, McVey R and Holland C:
Peroxisome proliferator-activated receptors modulate proliferation
and angiogenesis in human endometrial carcinoma. Mol Cancer Res.
10:441–453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
O'Sullivan SE and Kendall DA: Cannabinoid
activation of peroxisome proliferator-activated receptors:
Potential for modulation of inflammatory disease. Immunobiology.
215:611–616. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Robinson E and Grieve DJ: Significance of
peroxisome proliferator-activated receptors in the cardiovascular
system in health and disease. Pharmacol Ther. 122:246–263. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stephen J, Delvecchio C, Spitale N,
Giesler A, Radford K, Bilan P, Cox PG, Capone JP and Nair P: PPAR
ligands decrease human airway smooth muscle cell migration and
extracellular matrix synthesis. Eur Respir J. 41:425–432. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang H, Wu Q, Liu Z, Luo X, Fan Y, Liu Y,
Zhang Y, Hua S, Fu Q, Zhao M, et al: Downregulation of FAP
suppresses cell proliferation and metastasis through PTEN/PI3K/AKT
and Ras-ERK signaling in oral squamous cell carcinoma. Cell Death
Dis. 5:e11552014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Crossno JT Jr, Garat CV, Reusch JE, Morris
KG, Dempsey EC, McMurtry IF, Stenmark KR and Klemm DJ:
Rosiglitazone attenuates hypoxia-induced pulmonary arterial
remodeling. Am J Physiol Lung Cell Mol Physiol. 292:L885–L897.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang D, Wang G, Han D, Zhang Y, Xu J, Lu
J, Li S, Xie X, Liu L, Dong L, et al: Activation of PPAR-γ
ameliorates pulmonary arterial hypertension via inducing heme
oxygenase-1 and p21(WAF1): An in vivo study in rats. Life Sci.
98:39–43. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nicol CJ, Adachi M, Akiyama TE and
Gonzalez FJ: PPARgamma in endothelial cells influences high fat
diet-induced hypertension. Am J Hypertens. 18:549–556. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hansmann G, de Jesus Perez VA, Alastalo
TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T,
Wang L, Morrell NW, et al: An antiproliferative
BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in
pulmonary hypertension. J Clin Invest. 118:1846–1857. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu X, Bijli KM, Ramirez A, Murphy TC,
Kleinhenz J and Hart CM: Hypoxia downregulates PPARγ via an
ERK1/2-NF-κB-Nox4-dependent mechanism in human pulmonary artery
smooth muscle cells. Free Radic Biol Med. 63:151–160. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu Y, Tian XY, Huang Y and Wang N:
Rosiglitazone Attenuated Endothelin-1-Induced Vasoconstriction of
Pulmonary Arteries in the Rat Model of Pulmonary Arterial
Hypertension via Differential Regulation of ET-1 Receptors. PPAR
Res. 2014:3740752014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kang BY, Park KK, Green DE, Bijli KM,
Searles CD, Sutliff RL and Hart CM: Hypoxia mediates mutual
repression between microRNA-27a and PPARγ in the pulmonary
vasculature. PLoS One. 8:e795032013. View Article : Google Scholar : PubMed/NCBI
|