Thalassemia is an inherited haemoglobin blood
disorder that is the result of an absence or insufficient
production of normal haemoglobin. Defects in α-like and β-like
globin genes result in defective production of haemoglobin leading
to α- and β-thalassemia, respectively (1). It has recently been estimated that
there are 270 million carriers of haemoglobinopathies worldwide
(2). The high rate of carriers of
pathogenic globin gene variants is attributed to the natural
selection and consanguinity. Heterozygotes, individuals that carry
one defective gene for ineffective Hb synthesis, are selected
naturally because of conferred protection against malaria.
Heterozygotes are less fit than normal individuals and produce
small corpuscles; however, these are less prone to attack by
Plasmodium falciparum. Therefore, the prevalence of
mutations is high in Greece, Sicily and Italy where malaria is more
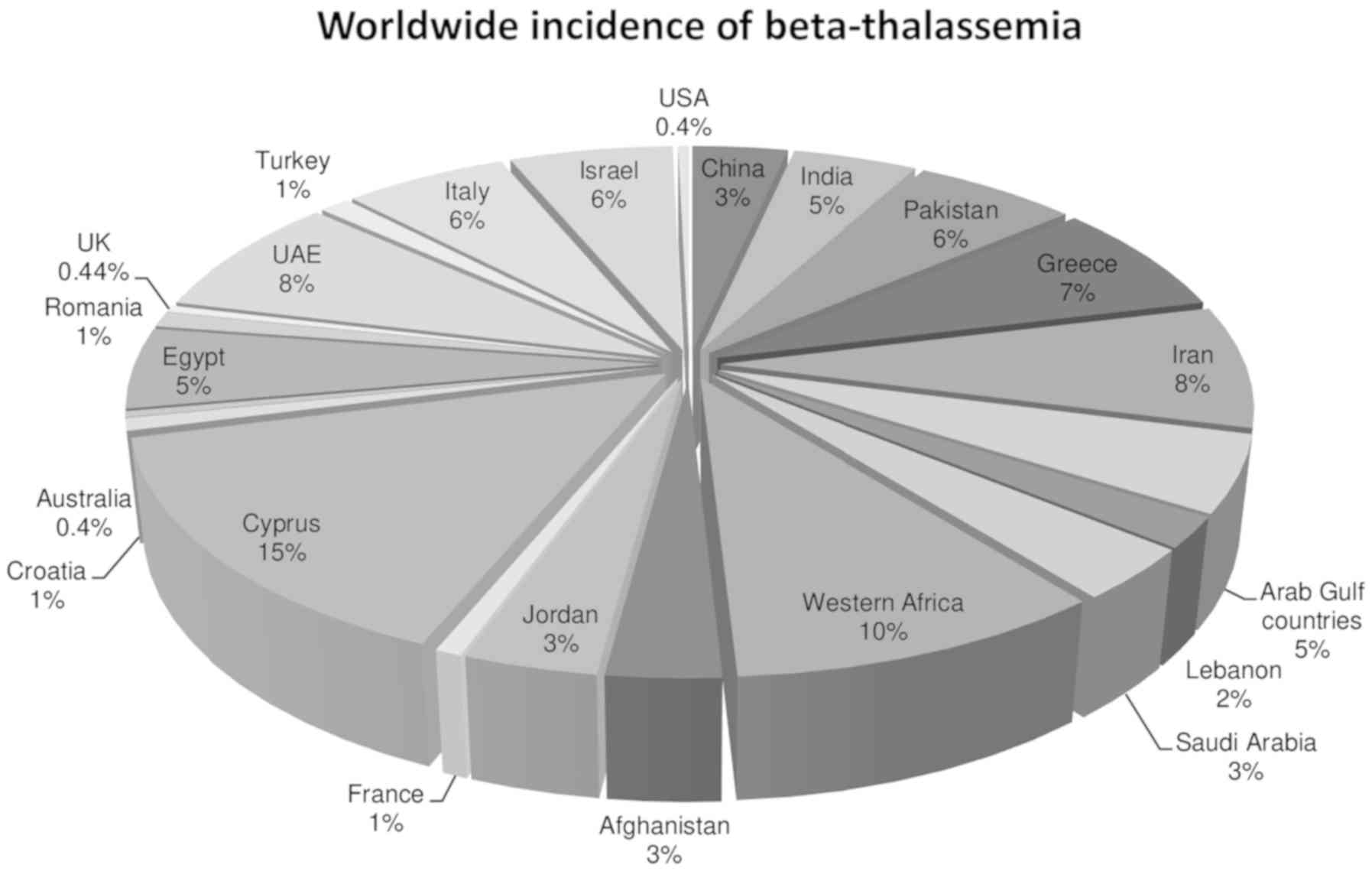
prevalent (Fig. 1) (3). Consanguineous marriages are also a
salient factor underlying the high incidence of thalassemia. In the
Eastern Mediterranean region, Pakistan is the country with the
highest number of infants born with β thalassemia each year
(4).
Adult haemoglobin is a tetramer composed of two α
and two β-globin chains that are bound to a haem prosthetic group.
α-thalassemia is a common haemoglobin disorder that is the result
of the absence or ineffective synthesis of α-globin chains. The
α-gene locus is present on chromosome 16(5). α-globin chains are constituents of
several haemoglobin types; HbA (α2β2), fetal
haemoglobin (HbF) (α2ϒ2) and HbA2
(α2δ2) (6).
Clinically, four α-thalassemia conditions are recognized. α+
thalassemia which is characterized by deletion in one of the
α-globin genes while αº thalassemia results from in-cis deletion in
two α-globin genes. The other two clinical forms are Hb Bart
hydrops fetalis syndrome (complete absence of functional α-globin
gene) and HbH disease (only one functional α gene) (7). α-thalassemia is usually the result of
deletion mutations and single nucleotide substitutions; insertions
and deletions are less frequently associated with α-thalassemia
(8). The severity of the
haemoglobinopathy can be reduced by increasing the concentration of
HbF. Borgio et al (9)
performed gene sequencing of HBA1 and HBA2 in the
Saudi Population, and found that 5.7% of the population carried a
novel convert of HBA2 termed α12. Individuals who
carried this gene convert exhibited reduced expression of
HBA2. The association between the variants of the α-globin
gene and iron concentration in the body was also determined. Using
multiplex PCR, α-globin gene deletions were identified. Female
β-thalassemia patients carrying α-globin wild type genotype
(αα/αα) had increased levels of iron. In contrast, female
β-thalassemia patients who co-inherited deletions in the α-globin
gene had normal iron levels. It was concluded that the serum iron
levels were significantly lowered when there was a co-inheritance
of a deleted α-globin gene in female patients with β-thalassemia
(10).
Homozygotes for β-thalassemia may either develop
thalassemia major (Cooley's anemia) or thalassemia intermedia. A
severe form of the disease, β-thalassemia major, is usually
diagnosed earlier in life and requires regular blood transfusions,
whereas thalassemia intermedia results in less severe anemia and
presents later in life. β-thalassemia intermedia patients require
less frequent blood transfusions compared with β-thalassemia major
patients (11). Individuals with
β-thalassemia minor are heterozygotes for a mutation in one of the
β-globin chains and do not present with any severe symptoms. These
individuals have mild anemia and their haemoglobin levels vary
between 9-11 g/dl (12).
β-thalassemia is the result of single nucleotide
substitutions, such as small insertions and deletions within the
HBB gene or its flanking sequence. Rarely, β-thalassemia may
occur due to large deletions (13).
Only a fraction of β-thalassemia cases are the result of deletions
in the HBB gene coding sequence. At present, 1,811
haemoglobin gene variants are known, of which, 404 mutations are
associated with β-thalassemia. These mutations include causative
mutations, mutations that modify disease presentation and neutral
polymorphisms (IthaGenes database; ithanet.eu/db/ithagenes). It has
been estimated that ~1.5% of the world's population are carriers of
β thalassemia trait (1). The
mutational spectrum of β-thalassemia in India, Pakistan,
Bangladesh, Saudi Arabia, Greece, and Italy is provided in Table I.
β-thalassemia patients, as well as their families,
face serious clinical, socio-economic and psychosocial challenges
throughout their life (14). In
Pakistan, there are ~100,000 patients that require treatment on a
regular basis, and each patient requires a minimum of 8,000
pkr/month (~$52) for treatment (15). Regular blood transfusion is required
for α-thalassemia major patients, which is not an easy or absolute
approach. Acquiring fully screened and compatible blood samples at
regular time intervals (every 2-4 weeks) is a difficult task for
the families of thalassaemic individuals (16).
The primary health issue for the survival of
thalassemia major patients is iron overload, which is a consequence
of multiple transfusions of blood (17). To overcome this problem, various iron
chelating drugs are given to the patients, to prevent iron
accumulation in the patients organs and tissues (1). Several complications are observed in
patients who have received multiple transfusions, including lethal
infectious diseases (18).
Additionally, in several countries, the patients and their families
do not come from financially stable backgrounds. Other challenges
faced by the patients include: Difficulty forming relationships due
to ignominy, discomfort, and a lack of acceptance by individuals.
Thalassemic individuals suffer from a lot of emotional and
environmental stresses which hinders their chances at marriage
(14). Improper neurosecretory
functioning is frequently observed in thalassemia major patients
due to the low production of growth hormones (19). Hypogonadism, frontal bossing,
improper skeletal maturation and stunted growth are often observed
as a result of insufficient growth hormone production (20).
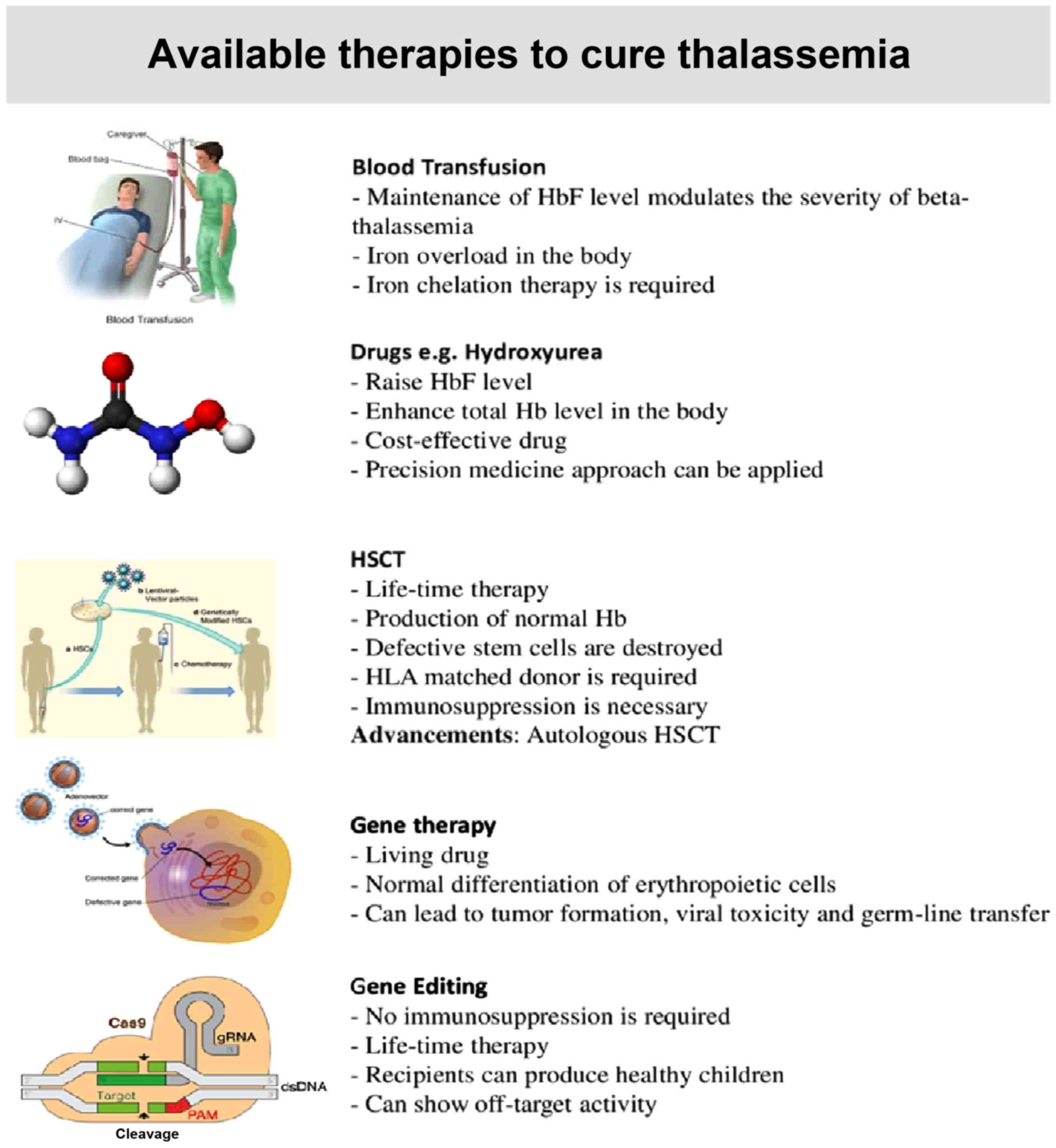
Possible therapies for Thalassemia include
conventional therapy, such as regular transfusions and
iron-chelating drugs, induction of γ-globin gene pharmaceutically,
allogenic transplantation or a single dose cure in the form of gene
therapy which does not require immunosuppression (21). A summary of these therapies is
presented in Fig. 2.
Regular transfusion leads to iron overload in the
body which has detrimental effects on the heart and liver (22,23).
Augmentation of HbF through the use of different pharmaceutical
drugs is used to modulate the severity of thalassemia. Among these
drugs, hydroxyurea is a good and cost-effective drug that
normalizes the activity of several signalling pathways resulting in
augmented HbF production, which ultimately results in reduction in
the frequency of blood transfusions required (24). Allogenic (hemopoietic cell)
transplantation is a promising cure for thalassemia; however, there
are several hurdles preventing its use: Limited availability of
Human Leukocyte Antigen (HLA)-identical donors, graft rejection in
certain cases and the role of iron toxicity in haematopoietic stem
cell transplantation (HSCT) rejection or failure (25). Lentiglobin gene therapy is the latest
method for cure without any mortality, graft rejection and clonal
dominance issues. However, delayed platelet engraftment has been
reported in certain patients (26).
Severe anemia is a result of insufficient production
of normal haemoglobin alongside a build-up of α-globin chains,
which ultimately results in ineffective erythropoiesis (27). Thalassemia major/intermedia patients
require blood transfusions at regular intervals, and the
transfusion frequency varies from one individual to another
(28,29). The life expectancy of patients is
increased by a few years by giving blood transfusions at regular
intervals, as the proper functioning of organs is preserved.
Perfect transfusion of blood also lessens lethargy, laziness and
fatigue (30). Additionally, blood
provides the strength to affected patients, allowing them to live
normally and to cope with stress and other situations confidently
(31).
Patients often get transfusion transmitted
infections when unscreened blood is transfused (32). Due to incompatible blood
transfusions, certain patients also suffer from allergic reactions
that may prove fatal (33). The
immunological interactions that occur between the patient and the
donor due to incompatible blood may be devastating for the
thalassemic individuals. The infections that most commonly occur
following the use of unscreened blood are Hepatitis B virus, dengue
virus, HIV, Hepatitis C virus, malaria and syphilis (34). The patients may also suffer due to
bacterial contamination in blood (35).
The most concerning and dangerous health issue faced
by thalassemia major patients during the blood transfusion phase of
treatment is iron overload (36).
Iron overload is the primary cause of the death in patients with
thalassemia, and it cannot be avoided when blood is transfused, as
extra iron is introduced in the patient's body, which proves to be
lethal as it accumulates around the organs, such as the liver,
kidneys and the heart, causing cardiovascular disorders and renal
failure (37). Repeated transfusions
also results in the formation of skin hemorrhages and spots of
blood (38).
Hydroxyurea activates the γ-globin gene and enhances
the production of HbF. Hydroxyurea is a cheap and cost effective
drug that is effective in certain patients with thalassemia for
reducing the frequency of blood transfusions required (39). Two α chains combine with the γ-globin
chains and form HbF that functions in place of the defective
haemoglobin (40). Hydroxyurea not
only augments the HbF levels, but also increases the levels of
total haemoglobin in the body. Its effectiveness is dependent on
the genetic makeup of the patient, and it has proven to be a
suitable treatment option for several thalassemia major patients
(24).
At present, HSCT is the only practically available
option which has a high curative rate. Donald Thomas performed the
first successful HSCT in an 18 months old thalassemia major child
using an HLA matched elder sibling as a donor 37 years ago
(41). Bone marrow transplant stands
on the following principles: i) Destroy defective stem cells to
stop them from proliferating; ii) suppress the immune system of the
host to ensure good engraftment; iii) infuse stem cells with normal
genes; and iv) prevent graft vs. host disease (GVHD). This
procedure requires progenitor stem cells to be administered in an
individual. The procedure is sub-categorized on the basis of the
source of progenitor cells (42) as
follows: i) Progenitor stem cells from the recipient (autologous
transplant); ii) stem cells from someone other than the recipient
(allogeneic transplant); or iii) umbilical cord blood
transplant.
In the case of allogeneic transplantation, the
traditional source of stem cells is the bone marrow. Children who
undergo HSCT before developing severe iron overload or viral
hepatitis and who also receive bone marrow from an HLA matched
donor exhibit a high likelihood of thalassemia remission (43). The selection of a suitable donor is
of significant importance regarding HSCT. Donors may be HLA matched
siblings (44). For patients without
any HLA matched siblings the foremost choice is alternative donors
including HLA matched unrelated donors, HLA mismatched related
donors and unrelated cord blood (45). The best results have been achieved
with HLA matched siblings; however, there is a <50% probability
of finding a histocompatible donor.
The outcome of HSCT is strongly influenced by
factors such as age at transplantation, irregular history of iron
chelation, histocompatibility and source of stem cells (21). In the 1980s, a prognostic scheme used
was developed to predict transplant outcomes. In a study by the
Pesaro group, young patients were assigned to three classes based
on a prognostic scheme underlying three variable factors: Quality
of chelation therapy before transplantation, hepatomegaly and
liver/portal fibrosis (46). Chances
of overall survival and thalassemia free survival varied in all of
the three classes based on the presence or absence of these risk
factors. Low-risk group (class I) had an 87-94% chance of survival,
whereas the intermediate-risk group (class II) had an 81-84% chance
of survival. In the high-risk group (class III) the chance of
overall survival was 70%, and the probability of thalassemia free
survival was 58% (46). This risk
classification does not apply to adults (individuals >17 years)
(47). When HSCT is performed,
active chronic hepatitis infection has a strong negative impact on
the overall survival of adults (48). Adult patients with long-term exposure
to iron overload exhibit characteristics similar to individuals
present in the high-risk group. Approaches to treat these
individuals through HSCT along with cyclophosphamide have thus far
proved unsuccessful (49).
HSCT failure is attributed either due to graft
rejection or GVHD. GVHD is considered to be a notable cause of
morbidity and mortality in patients who undergo allogeneic bone
marrow transplants; 10-50% of individuals with an HLA matched
related donor develop grade II-IV GVHD. It is also estimated that
15-40% of deaths in patients who receive HSCT occur due to GVHD
(50).
Veno-occlusive diseases (VODs) of the liver may
occur following bone marrow transplants. The incidence of VODs is
high in the Indian population, whereas in the Italian population
the risk is lower. This is likely due to differences in
pharmacogenetic variables and busulfan dosing between the two
populations. However, a low incidence of VODs is observed in
Pakistani children, who have undergone HSCT, with a similar genetic
background as that of Indians (51).
The acceptability of HSCT is limited due to GVHD,
transplant conditioning and graft failure. Complete myeloablation
can have toxic effects and can lead to infertility (52). Infertility is a common concern and
can occur in ~60% of the patients who undergo HSCT. The problem of
infertility may be addressed through fertility preservation
strategies, which include cryopreservation of ovarian tissue and
sperm banks (53-55).
β-thalassemia is a significant cause of early
mortalities worldwide. The available therapies only improve the
quality of life, but are associated with several side effects after
long-term use. Bone marrow transplant is considered a curative
therapy when an HLA matched donor is available. HSC gene transfer
may serve as a potential option once developed, but at present, the
survival advantage of corrected HSCs is low (56). Appropriate correction of globin gene
expression is required to correct erythrocyte defects (57).
Gene therapy has been hypothesized to serve as an
effective cure for monogenic blood disorders for several decades
(58). Gene therapy is a viral
vector-based therapy termed a living drug that is used to fix
errors in the defective genes (59).
Retroviral vectors are considered powerful tools for autoHSCs as
these have long terminal repeats with efficient and universal
enhancers; however, the resultant high expression of genes may
result in genotoxicity (60).
Additionally, retroviral vectors may integrate in or near
proto-oncogenes resulting in aberrant proliferation and
genotoxicity (61).
The first successful gene therapy trial for
thalassemia was performed using lentiviral vectors by transducing
autologous CD34+ HSCs which encode functional β-globin
and the patient did not require transfusions for the following 2
years (62). Development of
lentiviral vectors with self-inactivating capacity without any
pathogenic elements will be a significant milestone in the search
and development for the cure of thalassemia (63). Lentiviral vectors have an advantage
over retroviral vectors, as they are not involved in insertional
gene activation (64).
Globin-expressing lentiviral vectors (GLOBE LV) with transplanted
transduced HSCs corrected the pathological indications of
thalassemia major and intermedia (65) as well as restoring normal
differentiation of erythroid cells (66). However, in certain patients, GLOBE LV
resulted in alteration of transcriptional activity, premature
transcription and aberrant splicing, hence interfering with normal
gene regulation during gene therapy clinical trials in patients
with thalassemia major (62).
Additional clinical studies are required to
determine whether there are any interactions between the viral
vectors and the genome of the patient, and the effects of such
interactions. In β-thalassemia gene therapy, due to
erythroid-restricted activity of promoters, there is a very low
probability of genotoxicity caused by trans-activation (67). Thus, no toxicity or tumor production
has been reported in studies where lentiviral vectors were used in
mouse models. However, checks are required to ensure the vector has
not inserted, the toxicity of the vector and germline transfer
(67). A recent study showed that
the absence of transplant based mortality as well as replication of
competent lentiviruses resulted in other complications, such as
febrile neutropenia, epistaxis, stomatitis, pyrexia, irregular
menstruation and liver diseases in some patients (68). Furthermore, it is difficult to
contain all the required genetic elements required for gene
expression in the limited size of the vectors. Chromatin domain
insulators may be required to ensure the precise activity of
enhancers, and this may be suppressed by the chromatin structure at
the vector integration site. There is also the chance of altered
transgene expression or insertional oncogenesis (69). Alternatively, gene editing may be
used to modify mutated genes through the use of different
engineered nucleases (70), with the
advantage of retaining endogenous control of target gene expression
(71).
Gene therapy is a potential curative option in which
viruses are used as vectors that are permanently integrated into
the patient's genome. However, there are several hurdles in the
implementation of gene therapy for patients with thalassemia major;
sufficient collection of HSCs (CD34+) and transduction
of HSCs at the therapeutic level (72,73). Use
of an insufficient quantity of cells may result in graft rejection,
and in-vivo trials suggested that use of an insufficient
quantity of cells did not provide any notable therapeutic benefits
to patients (74). Lentiviral
vectors are required in high quantities for efficient therapeutic
effects; however, their high cost and sensitive nature of
production limit their use in gene therapy for patients with
β-thalassemia. Additionally, integration of viral vectors at
regions other than the target region may result in the activation
of proto-oncogenes resulting in different types of cancer (75,76).
Gene therapy medicines contain genes with diagnostic
or therapeutic effects. Recombinant genes are inserted by using
these medicines to treat genetic disorders (77). ZYNTEGLO® is a gene therapy
medicine containing HSCs transduced with vectors encoding the
β-globin gene, which has conditionally been authorized. However,
follow-up for 15 years after ZYNTEGLO® therapy is
required to determine its long-term efficacy and safety (78). ZYNTEGLO® works in patients
who are able to produce limited quantities of β-globin and are
suited for gene therapy. In this procedure patients' stem cells are
modified using a vector containing a normal functional gene, and
these cells are infused into the patient's body. One side effect
which has been reported is thrombocytopenia. Additionally,
production of ZYNTEGLO® is performed on a per patient
basis, thus increasing its costs (79).
An advanced approach for treating genetic disorders
is a method of genome editing which utilizes targeted nucleases to
correct the mutations in specific DNA sequences and restore them to
the wild-type sequence. Such genome editing tools include
transcription activator-like effector nucleases (TALENs), zinc
fingers nucleases (ZFNs) and clustered regularly interspaced short
palindromic repeats (CRISPR)/Cas9. These techniques have
diversified the approach to the use of therapies based on stem
cells to treat various diseases. However, ZFNs and TALENs are
lagging in treating haemoglobinopathies due their very low
efficiency rates towards modifying targeted genes in HSCs (80-82).
To produce significant levels of fully functional
haemoglobin, a large number of corrected HSCs are required. ZFNs
are chimeric nucleases consisting of Zinc Finger Protein motifs
which specifically recognize 3-4 bases of DNA (83). ZFNs coupled with FokI, a non-specific
endonuclease domain, introduce a double-stranded break in a
specific DNA sequence (84). The ZFN
performs its role as a dimer resulting in reduced off-target
effects. Similar to ZFN, TALENs are also chimeric; composed of a
nuclease, FokI and sequence binding domain (85,86). The
DNA binding domains are composed of an array of individual TALE
repeats each with the ability to determine a single nucleotide. To
promote specific binding, TALEN repeats also work in a similar
manner to that of a heterodimer (87). However, both these genome editing
tools are not robust enough and new protein sets are required for
re-engineering or re-designing target specific sequences in the
genome. The principle on which ZFNs and TALENs work is protein-DNA
interaction-based which is associated with high toxicity (88,89).
Furthermore, the design of these techniques is complex, and they
are unable to modulate the molecular expression of multiple target
genes (88).
CRISPR/Cas9 is an efficient, accurate and
revolutionary genome-editing tool; it is a robust, simple and
flexible RNA-DNA interaction-based system which has been used in
numerous genetic engineering programs to edit the genome at the
required sites (85,90-92).
The CRISPR/Cas9 system has two major components, a guide RNA (gRNA)
and an endonuclease, Cas9. The Cas9 endonuclease cuts the genome
whereas the gRNA directs the Cas9 to the specified position and
programs its cutting activity. Nucleases are known to be the most
accurate tools for making alterations in the genome with a high
degree of accuracy (93). This
system can be utilized to knockout different single nucleotide
polymorphisms as well as genes, or delete or insert various bases
in animal models and mammalian cell lines (94).
CRISPRs are DNA sequences that are present in
prokaryotes, archaea, and bacteria. These sequences are acquired by
the DNA remains of viruses that had previously infected the host,
and protect the host from any further attack by the same virus. The
RNA guided nucleases hence give protection to the host organisms
(95), as the host gains an adaptive
immune system which prevents it from any further attack (96). The CRISPR/Cas9 system has an
exceptional array of genome locus establishment, composition of
proteins, adaptation of mechanisms, pre-CRISPR/Cas9 RNA involvement
and processing and effecter complex structures (97-99).
Various transcription factors are important in
switching gene expression from γ-globin to β-globin. Shariati et
al (111) described one such
transcription factor, SOX6. A mutation was introduced in the SOX6
binding gene region with the γ-globin gene promoter using
CRISPR/Cas9 preventing its binding, and this resulted in
reactivation of γ-globin gene expression. Increased levels of
γ-globin mRNA expression was observed in K562 cells transfected
with the CRISPR/Cas9 vectors. Thus, CRISPR/Cas9 can be used as a
therapeutic approach for treating patients with β-thalassemia
(111). It is hypothesized that the
CRISPR/Cas9 system may be used to correct specifically harmful
mutations of the HBB gene, and this could be confirmed by normal
erythrocyte differentiation and their normal expression.
Patient derived induced pluripotent stem cells
(iPSCs) have been corrected using the CRISPR/Cas9 system
ex-vivo. The corrected iPSCs were then differentiated into
fully developed red blood cell precursors which were used for
transplantation (112). However,
iPSC differentiation into normal functional HSCs is not possible at
present. An investigational stem cell therapy, CTX001, using
CRISPR/Cas9 technology is able to target BCL11A in clinical
trials, and has been approved in several countries for the
treatment of β-thalassemia (44).
However, there are certain financial constraints involved when
translating the therapeutics assessed in clinical trials for
wide-scale use as specific mutation causing reagents (110). Additionally, the specificity and
safety of the CRISPR/Cas9 system for gene editing is under
discussion, and additional studies are required to confirm its
safety (113).
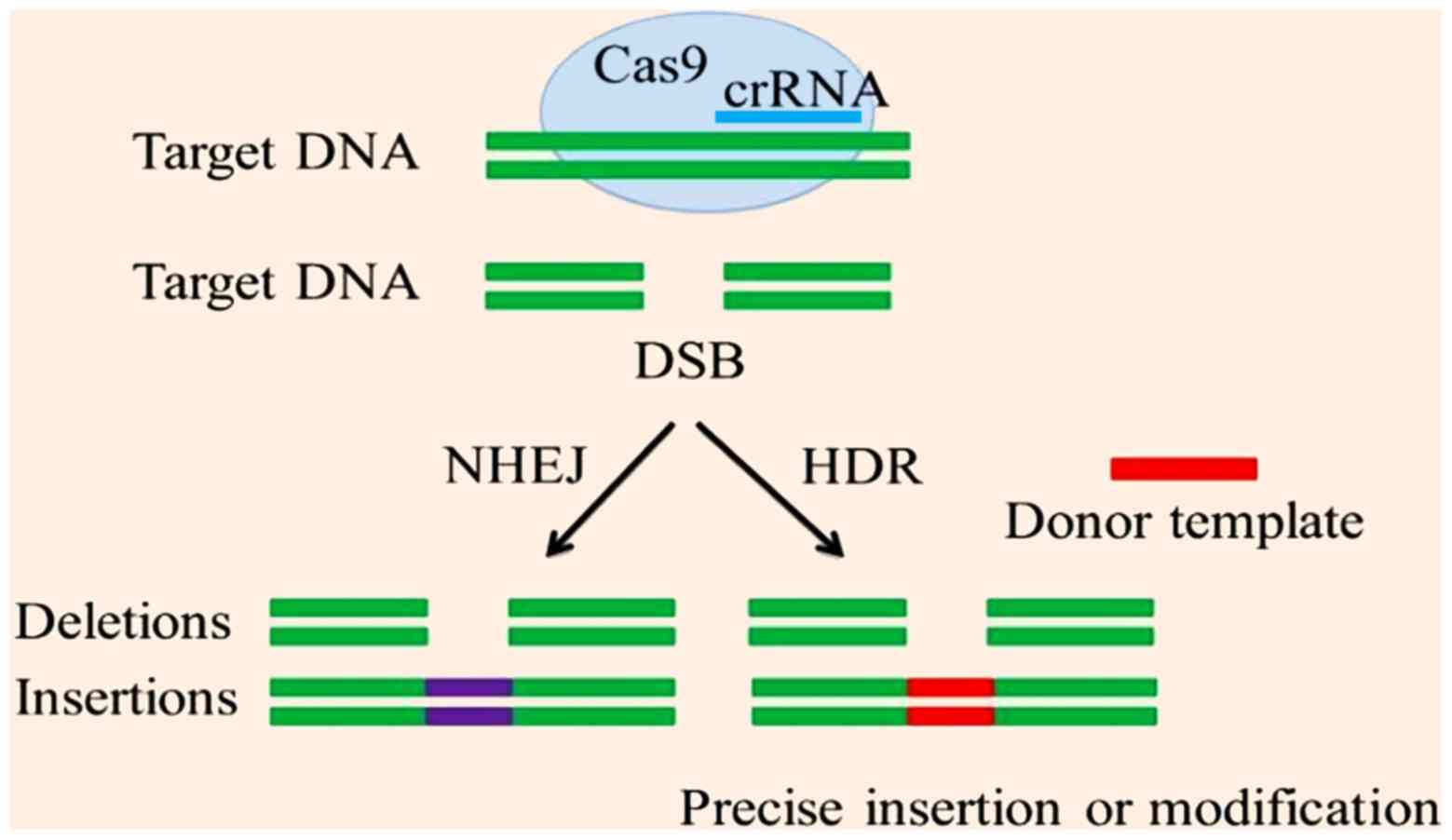
CRISPR/cas9 causes double strand breaks at a
particular sequence. This event activates repair systems; HR and
NHEJ. NHEJ causes repair following insertions or deletions, whereas
HR repair requires a template DNA molecule for the synthesis of the
other strand using complementary base binding (Fig. 3) (114). In gene editing, HR repair is
usually employed to cause changes in the nucleotide sequence when
used for treatment of β-thalassemia major (115).
The potential of RNA guided CRISPR/Cas9 technology
in correcting mutations lies in its ability to generate targeted
double-strand breaks followed by NHEJ-mediated repair. Limitations
of CRISPR/Cas9 are: Off-target activity of endonucleases, and
deletions, insertion or translocations inserted due to NHEJ. The
cellular homology-directed repair (HDR) can be used to address
these limitations of NHEJ. HDR is usually limited to actively
dividing cells. Point mutations account for various genetic
disorders (116), and single base
editors (BEs) can be used to correct these mutations during
replication (117). BEs are
derivatives of CRISPR/Cas9 and deaminases that work without
introducing a double-stranded break. Two classes of BEs have been
described in the literature: i) BEs that convert CG base pairs into
TA, cytosine BEs (CBEs); and ii) BEs that convert AT base pairs
into GC, adenine BEs (ABEs).
CBEs consist of cytidine deaminase, uracil DNA
glycosylase inhibitor and Cas9. Efficient base pair changes have
been achieved in yeast, plants, mouse zygote and human cells (in
vitro) (118-123).
HBB-28 polymorphism is one of the most common mutations. Liang
et al (124). successfully
used CBEs to correct HBB-28 in primary fibroblast cultures obtained
from a patient with β-thalassemia. They also showed that BEs had a
23% efficacy rate in correcting mutations in embryos
in-vitro (125). BEs may
thus be used to correct mutations in embryos and somatic cells.
There are still some limitations in the use of CBEs: Reduced base
editing purity, targeted random mutagenesis, off-target activity
and generation of indels (117).
Conventional therapy for thalassemia consists of
regular blood transfusions, although this is a double-edged sword;
it can ameliorate the clinical severity of the disease temporarily,
but may result in iron accumulation in the body. Pharmaceutical
drugs can be used to augment Hb; however, for long-term curative
effects, there is a need for extended genetic analysis of the
patient. Life-long cures for thalassemia is possible by
transplantation, gene therapy and genome editing. In developing
countries, interest is shifting towards HSCT for permanent cures.
This approach puts both the donor and recipient at risk. Thus, the
scientific community is looking towards to gene therapy as an
alternative, as there is no need for a donor, although this runs
the risk of vector toxicity and tumor formation. CRISPR/Cas9 is
proving to be a suitable for treatment of several human genetic
diseases, and genome editing tools are under clinical trials.
CRISPR/Cas9 can be used for precise transcriptional regulation,
genome modification and epigenetic editing. However, CRISPR/Cas9
may show off-target activity and there are legal and ethical
considerations regarding its use.
Not applicable.
No funding was received.
Not applicable.
FA, TaF, TuF, MAK and MIQ contributed equally to the
drafting and revising of the manuscript. All authors have read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Galanello R and Origa R: Beta-thalassemia.
Orphanet J Rare Dis. 5(11)2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
De Sanctis V, Kattamis C, Canatan D,
Soliman AT, Elsedfy H, Karimi M, Daar S, Wali Y, Yassin M, Soliman
N, et al: β-Thalassemia distribution in the old world: An ancient
disease seen from a historical standpoint. Mediterr J Hematol
Infect Dis. 9(e2017018)2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Haldane JBS: The rate of mutation of human
genes. Hereditas. 35:267–273. 1949.
|
|
4
|
Saeed U and Piracha ZZ: Thalassemia:
Impact of consanguineous marriages on most prevalent monogenic
disorders of humans. Asian Pacific J Tropical Dis. 6:837–840.
2016.
|
|
5
|
Hu L, Shang X, Yi S, Cai R, Li Z, Liu C,
Liang Y, Cai D, Zhang F and Xu X: Two novel copy number variations
involving the α-globin gene cluster on chromosome 16 cause
thalassemia in two Chinese families. Mol Genet Genomics.
291:1443–1450. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Muncie HL Jr and Campbell JS: Alpha and
beta thalassemia. Am Fam Physician. 80:339–344. 2009.PubMed/NCBI
|
|
7
|
Martin A and Thompson AA: Thalassemias.
Pediatr Clin North Am. 60:1383–1391. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Galanello R and Cao A: Alpha-thalassemia.
Genet Med. 13(83)2011.
|
|
9
|
Borgio JF, AbdulAzeez S, Al-Nafie AN,
Naserullah ZA, Al-Jarrash S, Al-Madan MS, Al-Muhanna F, Steinberg
MH and Al-Ali AK: A novel HBA2 gene conversion in cis or trans:
‘α12 allele’ in a Saudi population. Blood Cells Mol Dis.
53:199–203. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
AbdulAzeez S, Almandil NB, Naserullah ZA,
Al-Jarrash S, Al-Suliman AM, ElFakharay HI and Borgio JF:
Co-inheritance of alpha globin gene deletion lowering serum iron
level in female beta thalassemia patients. Mol Biol Rep.
47:603–606. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Haghpanah S, Vahdati S and Karimi M:
Comparison of quality of life in patients with β-Thalassemia
intermedia and β-thalassemia major in Southern Iran. Hemoglobin.
41:169–174. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Choudhry VP: Thalassemia minor and major:
Current management. Indian J Pediatr. 84:607–611. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Thein SL: The molecular basis of
β-thalassemia. Cold Spring Harb Perspect Med.
3(a011700)2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Musallam K, Cappellini MD and Taher A:
Challenges associated with prolonged survival of patients with
thalassemia: Transitioning from childhood to adulthood. Pediatrics.
121:e1426–e1429. 2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ishfaq K, Naeem SB and Ali J:
Socio-economic factors of thalassemia major on Patients 'families:
A case study of the Children's hospital and the institute of child
health Multan, Pakistan. Int J Med Appl Health. 1:2013.
|
|
16
|
Goodnough LT, Brecher ME, Kanter MH and
AuBuchon JP: Transfusion medicine-blood transfusion. N Engl J Med.
340:438–447. 1999.
|
|
17
|
Brittenham GM: Iron-chelating therapy for
transfusional iron overload. N Engl J Med. 364:146–156.
2011.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Borgna-Pignatti C, Rugolotto S, De Stefano
P, Zhao H, Cappellini MD, Del Vecchio GC, Romeo MA, Forni GL,
Gamberini MR, Ghilardi R, et al: Survival and complications in
patients with thalassemia major treated with transfusion and
deferoxamine. Haematologica. 89:1187–1193. 2004.PubMed/NCBI
|
|
19
|
Low LC: Growth of children with
β-thalassemia major. Indian J Pediatr. 72:159–164. 2005.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wu K, Tsai F and Peng C: Growth hormone
(GH) deficiency in patients with β-thalassemia major and the
efficacy of recombinant GH treatment. Ann Hematol. 82:637–640.
2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
de Dreuzy E, Bhukhai K, Leboulch P and
Payen E: Current and future alternative therapies for
beta-thalassemia major. Biomed J. 39:24–38. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
de Montalembert M, Ribeil JA, Brousse V,
Guerci-Bresler A, Stamatoullas A, Vannier JP, Dumesnil C, Lahary A,
Touati M, Bouabdallah K, et al: Cardiac iron overload in
chronically transfused patients with thalassemia, sickle cell
anemia, or myelodysplastic syndrome. PLoS One.
12(e0172147)2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang M, Liu R, Liang Y, Yang G, Huang Y,
Yu C, Sun K, Lai Y and Xia Y: Iron overload correlates with serum
liver fibrotic markers and liver dysfunction: Potential new methods
to predict iron overload-related liver fibrosis in thalassemia
patients. United European Gastroenterol J. 5:94–103.
2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Iqbal A, Ansari SH, Parveen S, Khan IA,
Siddiqui AJ and Musharraf SG: Hydroxyurea treated β-thalassemia
children demonstrate a shift in metabolism towards healthy pattern.
Sci Rep. 8(15152)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Pilo F and Angelucci E: Iron toxicity and
hemopoietic cell transplantation: Time to change the paradigm.
Mediterr J Hematol Infect Dis. 11(e2019030)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Anurathapan U, Locatelli F, Kwiatkowski
JL, Rasko JEJ, Schiller GJ, Porter J, Sauer MG, Thrasher AJ,
Chabannon C, Elliot H, et al: Lentiglobin gene therapy for
transfusion-dependent β-thalassemia: Outcomes from the phase 1/2
Northstar and phase 3 Northstar-2 studies. Biol Blood Marrow
Transplantation. 25 (Suppl):S66–S67. 2019.
|
|
27
|
Ribeil JA, Arlet JB, Dussiot M, Moura IC,
Courtois G and Hermine O: Ineffective erythropoiesis in
β-thalassemia. ScientificWorldJournal. 2013(394295)2013.
|
|
28
|
Al-Sharifi LM, Murtadha J, Shahad A,
Mohammed Y, Sura J, Waleed Z, Raheeq M, Sura A, Ehab H, Shahad M,
et al: Prevalence of hepatitis B and C in thalassemic patients and
its relation with type of thalassemia, frequency of blood
transfusion, and spleen status. Med J Babylon. 16:108–111.
2019.
|
|
29
|
Mettananda S, Pathiraja H, Peiris R,
Wickramarathne N, Bandara D, de Silva U, Mettananda C and
Premawardhena A: Blood transfusion therapy for β-thalassemia major
and hemoglobin E β-thalassemia: Adequacy, trends, and determinants
in Sri Lanka. Pediatr Blood Cancer. 66(e27643)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sharma S, Sharma P and Tyler LN:
Transfusion of blood and blood products: Indications and
complications. Am Fam Physician. 83:719–724. 2011.PubMed/NCBI
|
|
31
|
Roberts DJ, Field S, Delaney M and Bates
I: Problems and approaches for blood transfusion in the developing
countries. Hematol Oncol Clin North Am. 30:477–495. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Mahmoud RA, El-Mazary AA and Khodeary A:
Seroprevalence of hepatitis C, hepatitis B, cytomegalovirus, and
human immunodeficiency viruses in multitransfused thalassemic
children in upper Egypt. Adv Hematol. 2016(9032627)2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Stainsby D: ABO incompatible
transfusions-experience from the UK Serious Hazards of Transfusion
(SHOT) scheme: Transfusions ABO incompatible. Transfus Clin Biol.
12:385–388. 2005.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bird EM, Parameswaran U, William T, Khoo
TM, Grigg MJ, Aziz A, Marfurt J, Yeo TW, Auburn S, Anstey NM and
Barber BE: Transfusion-transmitted severe Plasmodium knowlesi
malaria in a splenectomized patient with beta-thalassaemia major in
Sabah, Malaysia: A case report. Malar J. 15(357)2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Stainsby D, Jones H, Asher D, Atterbury C,
Boncinelli A, Brant L, Chapman CE, Davison K, Gerrard R, Gray A, et
al: Serious hazards of transfusion: A decade of hemovigilance in
the UK. Transfus Med Rev. 20:273–282. 2006.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Papanikolaou G, Tzilianos M, Christakis
JI, Bogdanos D, Tsimirika K, MacFarlane J, Goldberg YP,
Sakellaropoulos N, Ganz T and Nemeth E: Hepcidin in iron overload
disorders. Blood. 105:4103–4105. 2005.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Origa R: β-thalassemia. Genet Med.
19(609)2017.
|
|
38
|
Williamson L, Lowe S, Love EM, Cohen H,
Soldan K, McClelland DB, Skacel P and Barbara JA: Serious hazards
of transfusion (SHOT) initiative: Analysis of the first two annual
reports. BMJ. 319:16–19. 1999.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ansari SH, Lassi ZS, Khowaja SM, Adil SO
and Shamsi TS: Hydroxyurea (hydroxycarbamide) for
transfusion-dependent β-thalassaemia. Cochrane Database Syst Rev.
3(CD012064)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ansari SH, Lassi ZS, Ali SM, Adil SO and
Shamsi TS: Hydroxyurea for β-thalassaemia major. Cochrane Database
Syst Rev. 3(CD012064)2016.
|
|
41
|
Chandy M: Stem cell transplantation in
India. Bone Marrow Transplant. 42 (Suppl 1):S81–S84.
2008.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Jagannath VA, Fedorowicz Z, Al Hajeri A
and Sharma A: Hematopoietic stem cell transplantation for people
with β-thalassaemia major. Cochrane Database Syst Rev.
11(CD008708)2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Krishnamurti L, Bunn HF, Williams AM and
Tolar J: Hematopoietic cell transplantation for hemoglobinopathies.
Curr Probl Pediatr Adolesc Health Care. 38:6–18. 2008.PubMed/NCBI View Article : Google Scholar
|
|
44
|
El-Beshlawy A and El-Ghamrawy M: Recent
trends in treatment of thalassemia. Blood Cells Mol Dis. 76:53–58.
2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Anasetti C: Use of alternative donors for
allogeneic stem cell transplantation. Hematology Am Soc Hematol
Educ Program. 2015:220–224. 2015.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Angelucci E: Hematopoietic stem cell
transplantation in thalassemia. Hematology Am Soc Hematol Educ
Program. 2010:456–462. 2010.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Angelucci E: Hematopoietic stem cell
transplantation in thalassemia. Hematology. 2010:456–462.
2010.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kyvernitakis A, Mahale P, Popat UR, Jiang
Y, Hosry J, Champlin RE and Torres HA: Hepatitis C virus infection
in patients undergoing hematopoietic cell transplantation in the
era of direct-acting antiviral agents. Biol Blood Marrow
Transplant. 22:717–722. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Hong KT, Kang HJ, Choi JY, Hong CR, Cheon
JE, Park JD, Park KD, Song SH, Yu KS, Jang IJ and Shin HY:
Favorable outcome of post-transplantation cyclophosphamide
haploidentical peripheral blood stem cell transplantation with
targeted Busulfan-based myeloablative conditioning using intensive
pharmacokinetic monitoring in pediatric patients. Biol Blood Marrow
Transplant. 24:2239–2244. 2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Gaziev D, Polchi P, Galimberti M,
Angelucci E, Giardini C, Baronciani D, Erer B and Lucarelli G:
Graft-versus-host disease after bone marrow transplantation for
thalassemia: An analysis of incidence and risk factors.
Transplantation. 63:854–860. 1997.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Mehta PA and Faulkner LB: Hematopoietic
cell transplantation for thalassemia: A global perspective BMT
tandem meeting 2013. Biol Blood Marrow Transplant. 19 (1
Suppl):S70–S73. 2013.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Taher AT, Weatherall DJ and Cappellini MD:
Thalassaemia. Lancet. 391:155–167. 2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Elborai Y, Uwumugambi A and Lehmann L:
Hematopoietic stem cell transplantation for thalassemia.
Immunotherapy. 4:947–956. 2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Bernaudin F, Pondarré C, Galambrun C and
Thuret I: Allogeneic/matched related transplantation for
β-thalassemia and sickle cell Anemia. Adv Exp Med Biol.
1013:89–122. 2017.
|
|
55
|
Pavone ME, Manuel S and Thompson A:
Fertility Preservation in a Female Adolescent with a
Hemoglobinopathy. In: Textbook of Oncofertility Research and
Practice. Woodruff T, Shah D and Vitek W (eds). Springer, Cham,
pp551-557, 2019.
|
|
56
|
Naldini L: Gene therapy returns to centre
stage. Nature. 526:351–360. 2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Kumar SR, Markusic DM, Biswas M, High KA
and Herzog RW: Clinical development of gene therapy: Results and
lessons from recent successes. Mol Ther Methods Clin Dev.
3(16034)2016.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Nienhuis AW: Development of gene therapy
for blood disorders: An update. Blood. 122:1556–1564.
2013.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Goswami R, Subramanian G, Silayeva L,
Newkirk I, Doctor D, Chawla K, Chattopadhyay S, Chandra D,
Chilukuri N and Betapudi V: Gene therapy leaves a vicious cycle.
Front Oncol. 9(297)2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Hacein-Bey-Abina S, Von Kalle C, Schmidt
M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS,
Pawliuk R, Morillon E, et al: LMO2-associated clonal T cell
proliferation in two patients after gene therapy for SCID-X1.
Science. 302:415–419. 2003.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Naldini L: Ex vivo gene transfer and
correction for cell-based therapies. Nat Rev Genet.
12(301)2011.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Cavazzana-Calvo M, Payen E, Negre O, Wang
G, Hehir K, Fusil F, Down J, Denaro M, Brady T, Westerman K, et al:
Transfusion independence and HMGA2 activation after gene therapy of
human β-thalassaemia. Nature. 467:318–322. 2010.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Srivastava A and Shaji RV: Cure for
thalassemia major-from allogeneic hematopoietic stem cell
transplantation to gene therapy. Haematologica. 102:214–223.
2017.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Montini E, Cesana D, Schmidt M, Sanvito F,
Bartholomae CC, Ranzani M, Benedicenti F, Sergi LS, Ambrosi A,
Ponzoni M, et al: The genotoxic potential of retroviral vectors is
strongly modulated by vector design and integration site selection
in a mouse model of HSC gene therapy. J Clin Invest. 119:964–975.
2009.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Miccio A, Cesari R, Lotti F, Rossi C,
Sanvito F, Ponzoni M, Routledge SJ, Chow CM, Antoniou MN and
Ferrari G: In vivo selection of genetically modified erythroblastic
progenitors leads to long-term correction of β-thalassemia. Proc
Natl Acad Sci USA. 105:10547–10552. 2008.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Roselli EA, Mezzadra R, Frittoli MC,
Maruggi G, Biral E, Mavilio F, Mastropietro F, Amato A, Tonon G,
Refaldi C, et al: Correction of beta-thalassemia major by gene
transfer in haematopoietic progenitors of pediatric patients. EMBO
Mol Med. 2:315–328. 2010.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Lidonnici MR, Paleari Y, Tiboni F,
Mandelli G, Rossi C, Vezzoli M, Aprile A, Lederer CW, Ambrosi A,
Chanut F, et al: Multiple integrated non-clinical studies predict
the safety of lentivirus-mediated gene therapy for β-thalassemia.
Mol Ther Methods Clin Dev. 11:9–28. 2018.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Rasko J, Walters M, Kwiatkowski J, Hongeng
S, Porter J, Sauer M, Thrasher A, Thuret I, Schiller G, Elliot H,
et al: Efficacy and safety of LentiGlobin gene therapy in patients
with transfusion-dependent β-thalassemia and
non-β0/β0 genotypes: Updated results from the
completed phase 1/2 Northstar and ongoing phase 3 Northstar-2
studies. Cytotherapy. 21(S14)2019.
|
|
69
|
Morgan RA, Gray D, Lomova A and Kohn DB:
Hematopoietic stem cell gene therapy: Progress and lessons learned.
Cell Stem Cell. 21:574–590. 2017.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Khosravi MA, Abbasalipour M, Concordet JP,
Berg JV, Zeinali S, Arashkia A, Azadmanesh K, Buch T and Karimipoor
M: Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal
hemoglobin reactivation: A promising approach for gene therapy of
beta thalassemia disease. Eur J Pharmacol. 854:398–405.
2019.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Barzel A, Paulk NK, Shi Y, Huang Y, Chu K,
Zhang F, Valdmanis PN, Spector LP, Porteus MH, Gaensler KM, et al:
Promoterless gene targeting without nucleases ameliorates
haemophilia B in mice. Nature. 517:360–364. 2015.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Sadelain M, Rivière I, Wang X, Boulad F,
Prockop S, Giardina P, Maggio A, Galanello R, Locatelli F and
Yannaki E: Strategy for a multicenter phase I clinical trial to
evaluate globin gene transfer in beta-thalassemia. Ann N Y Acad
Sci. 1202:52–58. 2010.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Yannaki E and Stamatoyannopoulos G:
Hematopoietic stem cell mobilization strategies for gene therapy of
beta thalassemia and sickle cell disease. Ann N Y Acad Sci.
1202:59–63. 2010.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Mansilla-Soto J, Riviere I, Boulad F and
Sadelain M: Cell and gene therapy for the beta-thalassemias:
Advances and prospects. Hum Gene Ther. 27:295–304. 2016.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Wu C and Dunbar CE: Stem cell gene
therapy: The risks of insertional mutagenesis and approaches to
minimize genotoxicity. Front Med. 5:356–371. 2011.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Karponi G and Zogas N: Gene therapy for
beta-thalassemia: Updated perspectives. Appl Clin Genet.
12(167)2019.PubMed/NCBI View Article : Google Scholar
|
|
77
|
European Medicines Agency: Advanced
therapy medicinal products: Overview 2018. https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview.
Accessed August 1, 2019.
|
|
78
|
Schuessler-Lenz M, Enzmann H and Vamvakas
S: Regulators' advice can make a difference: European medicines
agency approval of Zynteglo for beta thalassemia. Clin Pharmacol
Ther. 107(492)2020.PubMed/NCBI View Article : Google Scholar
|
|
79
|
European Medicines Agency: Zynteglo.
https://www.ema.europa.eu/en/medicines/human/EPAR/zynteglo#product-information-section.
Accessed June 3, 2019.
|
|
80
|
Hockemeyer D, Soldner F, Beard C, Gao Q,
Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA,
Zeitler B, et al: Efficient targeting of expressed and silent genes
in human ESCs and iPSCs using zinc-finger nucleases. Nat
Biotechnol. 27:851–857. 2009.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Mushtaq M, Bhat JA, Mir ZA, Sakina A, Ali
S, Singh AK, Tyagi A, Salgotra RK, Dar AA and Bhat R: CRISPR/Cas
approach: A new way of looking at plant-abiotic interactions. J
Plant Physiol. 224:156–162. 2018.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Gupta RM and Musunuru K: Expanding the
genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J Clin
Invest. 124:4154–4161. 2014.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Scott CT: The zinc finger nuclease
monopoly. Nat Biotechnol. 23:915–918. 2005.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Perez-Pinera P, Ousterout DG and Gersbach
CA: Advances in targeted genome editing. Curr Opin Chem Biol.
16:268–277. 2012.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Charpentier E and Doudna JA:
Biotechnology: Rewriting a genome. Nature. 495:50–51.
2013.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Christian M, Cermak T, Doyle EL, Schmidt
C, Zhang F, Hummel A, Bogdanove AJ and Voytas DF: Targeting DNA
double-strand breaks with TAL effector nucleases. Genetics.
186:757–761. 2010.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Gaj T, Gersbach CA and Barbas CF III: ZFN,
TALEN, and CRISPR/Cas-based methods for genome engineering. Trends
Biotechnol. 31:397–405. 2013.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Baliou S, Adamaki M, Kyriakopoulos AM,
Spandidos DA, Panayiotidis M, Christodoulou I and Zoumpourlis V:
CRISPR therapeutic tools for complex genetic disorders and cancer
(Review). Int J Oncol. 53:443–468. 2018.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Kim EJ, Kang KH and Ju JH: CRISPR-Cas9: A
promising tool for gene editing on induced pluripotent stem cells.
Korean J Intern Med. 32:42–61. 2017.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Stella S and Montoya G: The genome editing
revolution: A CRISPR-Cas TALE off-target story. Inside Cell.
1:7–16. 2016.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Murugan K, Babu K, Sundaresan R, Rajan R
and Sashital DG: The revolution continues: Newly discovered systems
expand the CRISPR-Cas toolkit. Mol Cell. 68:15–25. 2017.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Bhattacharyya S and Mukherjee A: CRISPR:
The revolutionary gene editing tool with Far-reaching applications.
In: Biotechnology Business-Concept to Delivery, Springer, pp47-56,
2020.
|
|
93
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protocols. 8(2281)2013.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Wang T, Wei JJ, Sabatini DM and Lander ES:
Genetic screens in human cells using the CRISPR-Cas9 system.
Science. 343:80–84. 2014.PubMed/NCBI View Article : Google Scholar
|
|
95
|
van Erp PB, Bloomer G, Wilkinson R and
Wiedenheft B: The history and market impact of CRISPR RNA-guided
nucleases. Curr Opin Virol. 12:85–90. 2015.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Sontheimer EJ and Barrangou R: The
bacterial origins of the CRISPR genome-editing revolution. Hum Gene
Ther. 26:413–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Hsu PD, Lander ES and Zhang F: Development
and applications of CRISPR-Cas9 for genome editing. Call.
157:1262–1278. 2014.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Makarova KS and Koonin EV: Annotation and
classification of CRISPR-Cas systems. In: CRISPR. Springer
Protocols, pp47-75, 2015.
|
|
99
|
Ishino Y, Krupovic M and Forterre P:
History of CRISPR-Cas from encounter with a mysterious repeated
sequence to genome editing technology. J Bacteriol. 200:e00580–17.
2018.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Koonin EV, Makarova KS and Zhang F:
Diversity, classification and evolution of CRISPR-Cas systems. Curr
Opin Microbiol. 37:67–78. 2017.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Shmakov S, Smargon A, Scott D, Cox D,
Pyzocha N, Yan W, Abudayyeh OO, Gootenberg JS, Makarova KS, Wolf
YI, et al: Diversity and evolution of class 2 CRISPR-Cas systems.
Nat Rev Microbiol. 15:169–182. 2017.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Makarova KS, Wolf YI, Alkhnbashi OS, Costa
F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E,
Haft DH, et al: An updated evolutionary classification of
CRISPR-Cas systems. Nat Rev Microbiol. 13:722–736. 2015.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Moon SB, Ko JH and Kim YS: Recent advances
in the CRISPR genome editing tool set. Exp Mol Med. 51:1–11.
2019.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Li Y and Peng N: Endogenous CRISPR-Cas
System-based genome editing and antimicrobials: Review and
prospects. Front Microbiol. 10(2471)2019.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Hidalgo-Cantabrana C and Barrangou R:
Characterization and applications of type I CRISPR-Cas systems.
Biochem Soc Trans. 28:15–23. 2020.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Zhang H and McCarty N: CRISPR-Cas9
technology and its application in haematological disorders. Br J
Haematol. 175:208–225. 2016.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Grevet JD, Lan X, Hamagami N, Edwards CR,
Sankaranarayanan L, Ji X, Bhardwaj SK, Face CJ, Posocco DF,
Abdulmalik O, et al: Domain-focused CRISPR screen identifies HRI as
a fetal hemoglobin regulator in human erythroid cells. Science.
361:285–290. 2018.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Dulmovits BM, Appiah-Kubi AO, Papoin J,
Hale J, He M, Al-Abed Y, Didier S, Gould M, Husain-Krautter S,
Singh SA, et al: Pomalidomide reverses γ-globin silencing through
the transcriptional reprogramming of adult hematopoietic
progenitors. Blood. 127:1481–1492. 2016.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Sankaran VG, Menne TF, Xu J, Akie TE,
Lettre G, Van Handel B, Mikkola HK, Hirschhorn JN, Cantor AB and
Orkin SH: Human fetal hemoglobin expression is regulated by the
developmental stage-specific repressor BCL11A. Science.
322:1839–1842. 2008.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Jensen TI, Axelgaard E and Bak RO:
Therapeutic gene editing in haematological disorders with
CRISPR/Cas9. Br J Haematol. 185:821–835. 2019.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Shariati L, Rohani F, Heidari Hafshejani
N, Kouhpayeh S, Boshtam M, Mirian M, Rahimmanesh I, Hejazi Z,
Modarres M, Pieper IL and Khanahmad H: Disruption of SOX6 gene
using CRISPR/Cas9 technology for gamma-globin reactivation: An
approach towards gene therapy of β-thalassemia. J Cell Biochem.
119:9357–9363. 2018.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Savić N and Schwank G: Advances in
therapeutic CRISPR/Cas9 genome editing. Transl Res. 168:15–21.
2016.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Porter J: Beyond transfusion therapy: New
therapies in thalassemia including drugs, alternate donor
transplant, and gene therapy. Hematology Am Soc Hematol Educ
Program. 2018:361–370. 2018.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Tang XD, Gao F, Liu MJ, Fan QL, Chen DK
and Ma WT: Methods for enhancing clustered regularly interspaced
short palindromic repeats/Cas9-mediated homology-directed repair
efficiency. Front Genet. 10(551)2019.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Dever DP, Bak RO, Reinisch A, Camarena J,
Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB,
Mantri S, et al: CRISPR/Cas9 β-globin gene targeting in human
haematopoietic stem cells. Nature. 539:384–389. 2016.
|
|
116
|
Chapman JR, Taylor MR and Boulton SJ:
Playing the end game: DNA double-strand break repair pathway
choice. Mol Cell. 47:497–510. 2012.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Rees HA and Liu DR: Base editing:
Precision chemistry on the genome and transcriptome of living
cells. Nat Rev Genet. 19:770–788. 2018.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Malzahn AL Lowder L and Yiping Qi: Plant
genome editing with TALEN and CRISPR. Cell Biosci.
7(21)2017.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Bortesi L and Fischer R: The CRISPR/Cas9
system for plant genome editing and beyond. Biotechnol Adv.
33:41–52. 2015.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Kim S, Kim D, Cho SW, Kim J and Kim JS:
Highly efficient RNA-guided genome editing in human cells via
delivery of purified Cas9 ribonucleoproteins. Genome Res.
24:1012–1019. 2014.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Enkler L, Richer D, Marchand AL, Ferrandon
D and Jossinet F: Genome engineering in the yeast pathogen Candida
glabrata using the CRISPR-Cas9 system. Sci Rep.
6(35766)2016.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Mou H, Kennedy Z, Anderson DG, Yin H and
Xue W: Precision cancer mouse models through genome editing with
CRISPR-Cas9. Genome Med. 7(53)2015.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Hendel A, Bak RO, Clark JT, Kennedy AB,
Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB, et
al: Chemically modified guide RNAs enhance CRISPR-Cas genome
editing in human primary cells. Nat Biotechnol. 33:985–989.
2015.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Liang P, Ding C, Sun H, Xie X, Xu Y, Zhang
X, Sun Y, Xiong Y, Ma W, Liu Y, et al: Correction of β-thalassemia
mutant by base editor in human embryos. Protein Cell. 8:811–822.
2017.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Zhang XH, Tee LY, Wang XG, Huang QS and
Yang SH: Off-target effects in CRISPR/Cas9-mediated genome
engineering. Mol Ther Nucleic Acids. 4(e264)2015.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Lai K, Huang G, Su L and He Y: The
prevalence of thalassemia in mainland China: Evidence from
epidemiological surveys. Sci Rep. 7(920)2017.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Mondal SK and Mandal S: Prevalence of
thalassemia and hemoglobinopathy in eastern India: A 10-year
high-performance liquid chromatography study of 119,336 cases.
Asian J Transfus Sci. 10:105–110. 2016.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Ansari SH, Shamsi TS, Ashraf M, Bohray M,
Farzana T, Tahir Khan M, Perveen K, Erum S, Nadeem M, Ahmed M and
Raza F: Molecular epidemiology of β-thalassemia in Pakistan: Far
reaching implications. Int J Mol Epidemiol Genet. 2:403–408.
2011.PubMed/NCBI
|
|
129
|
Hammoud H, Ghanem H, Abdallah R, Semaan P,
Azzi J, Parra Prada E and Haidar Hassan K: Genetic mutations of
beta thalassemia in middle east countries *corresponding
aurthor. World J Pharm Pharmaceutical Sci. 9:134–150. 2020.
|
|
130
|
Şanlidağ B, Çağin B, Özenli Ö, Şahaloğlu
Ö, Dalkan C, Galip N, Babayiğit Hocaoğlu A and Bahçeciler N:
Prevalence of thalassemia trait & Iron deficiency anemia during
infancy in 2011-2013 in a thalassemia prevalent region: North
Cyprus. Iran J Public Health. 45:1038–1043. 2016.PubMed/NCBI
|
|
131
|
Kountouris P, Kousiappa I, Papasavva T,
Christopoulos G, Pavlou E, Petrou M, Feleki X, Karitzie E,
Phylactides M Fanis P, et al: The molecular spectrum and
distribution of haemoglobinopathies in Cyprus: A 20-year
retrospective study. Sci Re. 6(26371)2016.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Angastiniotis M, Vives Corrons JL,
Soteriades ES and Eleftheriou A: The impact of migrations on the
health services for rare diseases in Europe: The example of
haemoglobin disorders. The Scientific World Journal.
2013(727905)2013.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Guler E, Caliskan U, Ucar Albayrak C and
Karacan M: Prevalence of beta-thalassemia and sickle cell anemia
trait in premarital screening in Konya urban area, Turkey. J
Pediatr Hematol. 29:783–785. 2007.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Mir SA, Alshehri BM, Alaidarous M, Banawas
SS, Dukhyil AAAB and Alturki MK: Prevalence of Hemoglobinopathies
(β-Thalassemia and Sickle Cell Trait) in the adult population of Al
Majma'ah, Saudi Arabia. Hemoglobin. 44:47–50. 2020.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Fucharoen S and Weatherall DJ: Progress
toward the control and management of the thalassemias. Hematol
Oncol Clin North Am. 30:359–371. 2016.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Persons DA: Gene therapy: Targeting
β-thalassaemia. Nature. 467:277–278. 2010.
|
|
137
|
Panigrahi I and Marwaha R: Mutational
spectrum of thalassemias in India. Indian J Hum Genet. 13:36–37.
2007.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Ansari SH, Shamsi TS, Ashraf M, Farzana T,
Bohray M, Perveen K, Erum S, Ansari I, Ahmed MN, Ahmed M and Raza
F: Molecular epidemiology of β-thalassemia in Pakistan: Far
reaching implications. Indian J Hum Genet. 18:193–197.
2012.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Al-Sultan A, Phanasgaonkar S, Suliman A,
Al-Baqushi M, Nasrullah Z and Al-Ali A: Spectrum of β-thalassemia
mutations in the eastern province of Saudi Arabia. Hemoglobin.
35:125–134. 2011.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Hamamy HA and Al-Allawi NA:
Epidemiological profile of common haemoglobinopathies in Arab
countries. J Community Genet. 4:147–167. 2013.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Amato A, Cappabianca MP, Colosimo A, Perri
M, Grisanti P, Zaghis I, Ponzini D and Lerone M: Current genetic
epidemiology of β-Thalassemias and structural hemoglobin variants
in the lazio region (Central Italy) following recent migration
movements. Adv Hematol. 2010(317542)2010.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Boussiou M, Karababa P, Sinopoulou K,
Tsaftaridis P, Plata E and Loutradi-Anagnostou A: The molecular
heterogeneity of beta-thalassemia in Greece. Blood Cells Mol Dis.
40:317–319. 2008.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Sultana G, Begum R, Akhter H, Shamim Z,
Rahim MA and Chubey G: The complete spectrum of beta (β)
thalassemia mutations in Bangladeshi population. Austin Biomark
Diagn. 3(1024)2016.
|
|
144
|
Kleinstiver BP, Pattanayak V, Prew MS,
Tsai SQ, Nguyen N, Zheng Z and Joung JK: High-fidelity CRISPR-Cas9
variants with undetectable genome-wide off-targets. Nature.
529:490–495. 2016.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Acharya S, Mishra A, Paul D, Ansari AH,
Azhar M, Kumar M, Rauthan R, Sharma N, Aich M, Sinha D, et al:
Francisella novicida Cas9 interrogates genomic DNA with very
high specificity and can be used for mammalian genome editing. Proc
Natl Acad Sci USA. 116:20959–20968. 2019.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Lee CM, Cradick TJ and Bao G: The
Neisseria meningitidis CRISPR-Cas9 system enables specific
genome editing in mammalian cells. Mol Ther. 24:645–654.
2016.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Müller M, Lee CM, Gasiunas G, Davis TH,
Cradick TJ, Siksnys V, Bao G, Cathomen T and Mussolino C:
Streptococcus thermophilus CRISPR-Cas9 systems enable
specific editing of the human genome. Mol Ther. 24:636–644.
2016.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Dugar G, Leenay RT, Eisenbart SK, Bischler
T, Aul BU, Beisel CL and Sharma CM: CRISPR RNA-dependent binding
and cleavage of endogenous RNAs by the Campylobacter jejuni
Cas9. Mol Cell. 69:893–905.e7. 2018.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Moon SB, Lee JM, Kang JG, Lee NE, Ha DI,
Kim DY, Kim SH, Yoo K, Kim D, Ko JH, et al: Highly efficient genome
editing by CRISPR-Cpf1 using CRISPR RNA with a uridinylate-rich
3'-overhang. Nat Commun. 9(3651)2018.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Yamano T, Zetsche B, Ishitani R, Zhang F,
Nishimasu H and Nureki O: Structural basis for the canonical and
non-canonical PAM recognition by CRISPR-Cpf1. Mol Cell.
67:633–645.e3. 2017.PubMed/NCBI View Article : Google Scholar
|