Introduction
Congenital cataracts are a common heterogeneous
disease that causes variable degrees of visual impairment and
blindness in partially affected children, and the etiology involves
both genetic and environmental components (1). Congenital cataracts typically appear
as opacification in any position of the lens before or after birth.
Any obstruction in the visual axis that is left untreated may lead
to permanent impairment of vision or even blindness, even if it is
removed at a later stage (2).
Congenital cataracts may manifest as isolated cataracts or
syndrome-associated cataracts (3).
Worldwide, the prevalence of congenital cataracts in live births
ranges from 1-15/10,000 children, and it is responsible for 10-30%
of cases of blindness in children (4).
The causes of congenital cataracts include gene
defects, environmental factors, chromosomal abnormalities,
metabolic disorders and perinatal infections (1,2,4).
Inherited cataracts, caused by genetic mutations, contribute to
half of all cases of congenital cataracts (2). Several gene mutations are considered
to participate in the pathology of congenital cataracts, amongst
which, mutations of the crystallin gene are the most widely
recorded, and account for ~1/2 of all reported mutations associated
with congenital cataracts (5).
Mutations in β-crystallin B2 (CRYBB2) that cause congenital
cataracts are commonly reported (6). At present, 22 mutations in the
CRYBB2 gene have been identified (7). There are also several studies on the
underlying pathological mechanisms of the CRYBB2 mutations
that lead to congenital cataracts (8,9).
Although CRYBB2:c.62T>A(p.I21N) is a recurrent variant,
its pathogenic mechanism has not been previously reported to the
best of our knowledge.
Implementation of genetic testing using whole-exome
sequencing (WES) has permitted the streamlining of the process of
identifying pathogenic genetic variants, especially when it is
widely used in the diagnosis of a variety of single-gene diseases
(10). In the present study, WES
was used to examine the genetic cause in a four-generation Chinese
family with congenital nuclear cataracts. In addition, the
pathological mechanism of CRYBB2:c.62T>A(p.I21N) was
evaluated in vitro. This variant has been identified in one
other family (11), but the
phenotype was completely distinct from the family examined in the
present study. Patients in the aforementioned study were born with
regular nuclear cataracts in both eyes (11). It was hypothesized that this site
may be a mutation hotspot, hence it is essential to investigate the
mechanism via which this variant may cause congenital cataracts.
This may provide a reference and possible therapeutic target for
gene therapy of congenital cataracts in the future.
Materials and methods
Subjects
A four-generation Chinese family with congenital
nuclear cataracts and 100 healthy controls (age range, 20-55 years,
50 women, 50 men) were recruited from July 2018 to July 2019 from
the Yin Hai Eye Hospital (Chengdu, China). There were a total of 21
family members (12 males, 9 females; age range, 2-77 years; average
age, 33.4±20.7 years) in this family, of which, 8 were affected and
13 were unaffected. Except for the member I. 1 who passed away,
peripheral venous blood was collected from all remaining family
members. All participants signed informed consent forms or this was
signed by the guardian, and the study was performed in accordance
with the Declaration of Helsinki (12). The pedigree medical history was
recorded and clinical examinations were performed. The
Institutional Review Board of Chengdu University of Traditional
Chinese Medicine approved the present study (approval no.
CMEC2010-21).
DNA collection and whole exome
sequencing analysis
Paired-end sequencing for reads of 150 bp was
performed using the Illumina X-10 platform (Illumina, Inc.).
Peripheral venous blood was extracted from all participants using a
vacuum blood collector (BD Biosciences). DNA was extracted using a
DNA Extraction kit (cat. no. 69504; Qiagen China Co., Ltd.) and
purified using a DNA Purification kit (cat. no. K0512; Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The purity and quantity of DNA samples were subsequently
measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher
Scientific, Inc.) and agarose gel electrophoresis. A high-quality
genomic DNA sample was broken using Covaris Technologies (200 and
300 bp), as previously described (13). The sample was then prepared using a
Truseq DNA Sample Preparation kit (Illumina, Inc.,) and captured
using a SureSelect all exons Enrichment kit (Agilent Technologies,
Inc.). Captured products were exposed to an Agilent 2100
Bioanalyzer (14) to detect the
mass concentration of the library, and the final library
concentration was 5.14 ng/µl. Agarose gel electrophoresis detected
clear and slightly diffused bands at 200-500 bp, indicated that the
library had been successfully constructed.
The raw data were filtered to obtain clean reads
according to the following steps: ⅰ) Removing reads containing
sequencing adapter; ⅱ) removing reads whose low-quality base ratio
(base quality ≤5) is >50%; and ⅲ) removing reads whose unknown
base (‘N’ base) ratio is >10%. Statistical analysis of data and
downstream bioinformatics analysis were performed on this filtered,
high-quality data, referred to as the ‘clean data’. The alignment
software, Burrows-Wheeler Aligner (version, 0.7.17) (15), was used to align the clean data to
the human reference genome (GRCh37/HG19). The Picard tool (version,
1.119) (16) was used to remove
duplicate reads, and GATK (version, 3.8) (17) was used for local realignment and
base quality recalibration.
Processing of sequencing data
In order to ensure high-quality sequencing data, a
strict data quality control system was set up for use throughout
the analytical process. Data were further annotated using ANNOVAR
(version, 2.3) (18) and evaluated
using multiple databases, including The 1000 Genomes Project
(ncbi.nlm.nih.gov/variation/tools/1000genomes),
ESP6500.exon.program (evs.gs.washington.edu/EVS), dbSNP (ftp://ftp-trace.ncbi.nih.gov/snp/organisms), ExAC
(exac.broadinstitute.org), Inhouse
(MyGenostics), HGMD (hgmd.cf.ac.uk/ac/index.php), OMIM (omim.org/), ClinVar (ncbi.nlm.nih.gov/clinvar) and gnomAD (gnomad.broadinstitute.org/about). SIFT
(19), PolyPhen-2(20), MutationTaster (21) and GERP++ (22) were used to predict the effect of
variations on protein function. During data analysis, the
variations were selected according to the following conditions: i)
Variants that are reported in dbSNP v141; ii) the subset of
variants with MAF <1% in The 1000 Genome Project; iii) subset of
coding non-synonymous single nucleotide polymorphisms (SNPs) with
SIFT score <0.05; or iv) intergenic variants with GERP++ score
>2. The identified variations were assessed according to the
variant interpretation guidelines of the American College of
Medical Genetics and Genomics (ACMG) (23,24).
Next, the putative variations in the affected members, the
relatives and the healthy controls were compared with the OMIM
database, as well as with previously published literature (5,9).
Sanger sequencing
A pair of primers were designed by Thermo Fisher
Scientific, Inc.; the sequences were: CRYBB2 forward,
5'-CCTTCAGCATCCTTTGGGTTCTCT-3' and reverse,
5'-GCAGTTCTAAAAGCTTCATCAGTC-3'. Sanger sequencing was performed on
all family members and control participants using an ABI 3730
Genetic analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol.
Bioinformatics analysis
Pymol (version, 2.5; https://pymol.en.softonic.com/; Schrödinger, LLC)
(25) showed the protein structure
of wild-type (Wt)-CRYBB2 and mutant (I21N)-CRYBB2,
and the potential functional influence of this amino acid change
detected from the variant was calculated using PolyPhen-2(20) and SIFT (19). ProtScale (version 3.0; web.expasy.org/protscale) was used to evaluate
protein hydrophobicity.
Cell culture and transfection
HeLa cells were obtained from the American Type
Culture Collection, and cultured in DMEM (standard low-glucose
DMEM, 1 g/l; cat. no. 11054001; Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (cat. no. 10100147; Gibco; Thermo
Fisher Scientific, Inc). Cells were maintained at 37˚C in a
humidified incubator with 5% CO2. Subsequently, cells in
the logarithmic growth phase were seeded into a 6-well plate, and
the seeding density reached 70-90% for transfection at room
temperature. A total of 2 µg Wt- or I21N-CRYBB2 plasmid/well were
transfected into cells using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol, at 37˚C for 48 h. When the concentration
of transfected cells reached 80%, the transfected cells were
analyzed.
Reverse transcription-quantitative
(RT-q)PCR
To determine the relative expression of the empty
vector, Wt- and I21N-CRYBB2 in transfected cells, RT-qPCR was
performed. Total RNA was extracted using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), qPCR SYBR®
Green Master Mix (Vazyme Biotech, Co., Ltd.) and the RNA was
reverse transcribed into cDNA using a QuantiTect Reverse
Transcription kit (cat. no. 205313, Qiagen China Co., Ltd.),
according to the manufacturer's protocol. qPCR was performed using
a PCR amplifier (26). The method
of gene quantification used was 2-ΔΔCq as previously
described (27). PCR primers were
designed by Invitrogen (Thermo Fisher Scientific, Inc.) as follows:
CRYBB2 forward, 5'-GTAGCCAGGATTCTGCCATAGGAA-3' and reverse,
5'-GTGCCCTCTGGAGCATTTCATAGT-3'; GAPDH forward,
5'-TTCCGAGTTCCTGTCCCTAATG-3' and reverse,
5'-GCCTCCTTCACCTTCTGCTTG-3'. qPCR was performed using the FTC2000
(Funglyn Biotech). Samples were set up in 50 µl final volumes
containing 6 µl 5X PCR buffer, 0.6 µl 2X primers (25 pmol/µl), 0.3
µl probe (25 pmol/µl) or 0.3 µl SYBR-Green, 1 µl dNTPs (10 mM), 0.3
µl Taq enzyme (5 U/µl), 3 µl Mg2+ (25 mM), 1 µl template
and 17.2 µl DEPC water (Sigma-Aldrich; Merck KGaA). Thermocycling
conditions were as follows: Initial denaturation at 94˚C, followed
by 40 cycles of 20 sec at 94˚C and 30 sec at 60˚C. The relative
expression was calculated based on the expression of GAPDH.
Western blotting
After cells were lysed in a mix containing SDS Lysis
Buffer (cat. no. p0013; Beyotime Institute of Biotechnology) and
protease inhibitors (Sigma-Aldrich; Merck KGaA), proteins were
extracted. Proteins were quantified using a BCA Protein Assay kit
(cat. nos. CW0014S and CWBIO). A total of 25 µg of each protein
sample was loaded per lane on a 12% SDS-gel, resolved using
SDS-PAGE and transferred to PVDF membranes. The membrane was then
incubated with the following antibodies: anti-green fluorescent
protein (GFP; cat. no. ab290; Abcam; 1:1,000), anti-GAPDH (cat. no.
ab8245; Abcam; 1:1,000) and anti-BiP/HSPA5 (cat. no. ab21685;
Abcam; 1:1,000), at 4˚C overnight. Subsequently, the membranes were
then incubated with the secondary antibody horseradish peroxidase
(HRP) Goat anti-Rabbit IgG antibody (cat. no. ab6721; Abcam,
1:5,000) or (HRP) Goat anti-Mouse IgG antibody (cat. no. ab6721;
Abcam; 1:5,000) diluted with 5% skimmed milk powder blocking
solution for 1 hour at room temperature. Signals were visualized
using an ECL reagent (Thermo Fisher Scientific, Inc.), the
membranes were subsequently scanned and densitometry analysis was
performed. Protein band intensities were quantified using ImageJ
software (version, 1.8.0_172; National Institutes of Health).
Immunofluorescence and apoptosis
analysis
The transfected cells were washed with PBS three
times, fixed with 100% pre-cooled acetone (stored at 4˚C) for 10
min at room temperature, washed again with PBS three times and then
stained with DAPI for 10 min at room temperature. The slides were
sealed and cells were observed using a confocal microscope
(magnification, x200). The number of aggregated cells was counted
using ImageJ software (version, 1.8.0_172; National Institutes of
Health).
Apoptosis was observed using Hoechst 33342 staining.
After 24 h, the cells transfected with Wt- and I21N-CRYBB2
plasmids were placed into a 6-well plate at 2x105/well
and cultured for another 24 h in DMEM supplemented with 10% FBS.
When the cell seeding density reached 70-90%, Hoechst 33342 was
added to the wells at room temperature for 10 min, and the number
of apoptotic nuclei were counted using a confocal microscope
(magnification, x200). Cells transfected with Wt- or
I21N-CRYBB2 plasmid were transferred to 6-well plates, 400
µM H2O2 was used to induce stimulation, and
an apoptosis assay was then performed on a flow cytometer (Attune
NxT; Thermo Fisher Scientific, Inc.) using Annexin V-FITC/PI
Apoptosis Detection kit (cat. no. A211-01/02; Vazyme Biotech Co.,
Ltd.) according to the manufacturer's instructions. The cells
apoptotic rate was calculated by the percentage of early plus late
apoptotic cells.
Molecular analysis of UPR-related
genes induced by mutant crystallin
RT-PCR was performed for X-box-binding protein
(Xbp1) splicing and to detect the expression levels of other
UPR-related genes, as previously described. The following primers
were used: Xbp1 forward, 5'-GAACCAGGAGTTAAGAACACG-3' and
reverse, 5'-AGGCAACAGTGTCAGAGTCC-3' for Xbp1 splicing; heat
shock protein family A (Hsp70) member 5 (HSPA5) forward,
5'-CCAAGAGAGGGTTCTTGAATCTCG-3' and reverse,
5'-AGGCAACAGTGTCAGAGTCC-3'; DNA damage-inducible transcript 3
(DDIT3) forward, 5'-AGCCGTTCATTCTCTTCAG-3' and reverse,
5'-CCTCACTCTCCAGATTCCA-3'; GAPDH forward,
5'-AGGCTGTTGTCATACTTCTC-3' and reverse, 5'-CATCACCATCTTCCAGGAG-3';
inositol-requiring enzyme 1 (Ire1) forward,
5'-CGGTCAGGAGGTCAATAACA-3' and reverse, 5'-GGACAGGCTCAATCAAATGG-3';
and β2-microglobulin, forward, 5'-GGATGGATGAAACCCAGACACATAG-3' and
reverse, 5'-CGGGCATTCCTGAAGCTGA-3'.
Statistical analysis
Statistical analysis was performed using SPSS 23.0
software (IBM Corp.). Each experiment was repeated ≥3 times. The
percentage of cells with aggregates was calculated from 200
positively transfected cells in 10 random viewing fields. The
numbers of apoptotic nuclei were counted in five random viewing
fields. All data are expressed as the mean ± SD. One-way ANOVA and
Bonferroni multiple comparison were used to test the differences
between three groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Clinical information of the
patients
According to the sequencing and genetic
characteristics, the family enrolled in the present study possessed
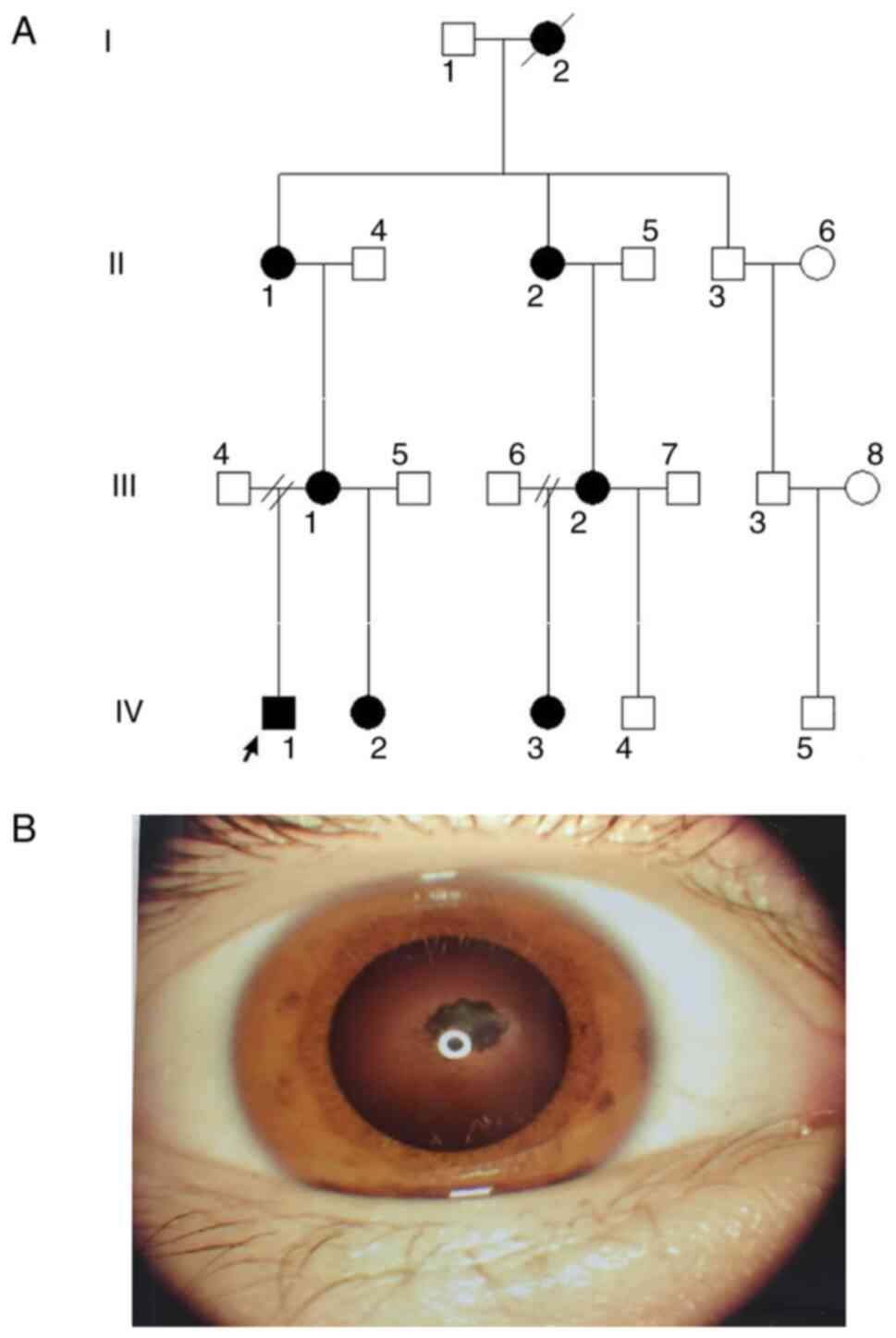
an autosomal dominant inheritance pattern (Fig. 1A), the proband IV.1 was a
4-year-old boy who was found to have congenital binocular nuclear
cataracts at birth and underwent cataract phacoemulsification
without intraocular lens implantation at the age of 2 months
(Fig. 1B). At the age of 2 years,
he underwent binocular sequential intraocular lens implantation.
However, the left eye developed secondary glaucoma and corneal
ulcers within 2 years after cataract surgery. At the age of 4
years, he underwent a left eye prosthesis implantation operation.
At present, his best corrected visual acuity (BCVA) in his right
eye is 0.2, and his intraocular pressure is normal. He has slight
nystagmus and esotropia without fundus abnormalities. Family member
IV.2 was the half-sister of the proband, who also presented with
binocular nuclear cataracts after birth. She underwent binocular
cataracts surgery at the age of 2 months and, at present, has a
bilateral aphakic eye. She also has esotropia and nystagmus. The
mother (III.1) of the proband presented with binocular nuclear
cataracts at birth and underwent binocular cataracts surgery with
intraocular lens implantation at the age of 2 years. Currently, her
BCVA is 0.02 in the right eye and 0.1 in the left eye, and shows
notable nystagmus and esotropia without any fundus abnormalities.
The grandmother (II.1) of the proband also had binocular nuclear
cataracts at birth, and underwent binocular cataracts removal
surgery at the age of 12 years without intraocular lens
implantation; her BCVA is 0.01 in the right eye and 0.06 in the
left eye, and shows obvious nystagmus and esotropia without any
fundus abnormalities. Family member IV.3 is a cousin of the
proband, also had binocular nuclear cataracts at birth, and
underwent binocular cataracts removal at the age of 2 years without
intraocular lens implantation. She is still aphakic. Her BCVA is
0.02 in the right eye and 0.06 in the left eye. However, her
half-brother (IV.4) does not have cataracts. Her mother (III.2)
also presented with binocular nuclear cataracts at birth, and
underwent binocular cataracts removal surgery with implantation of
an intraocular lens at the age of 4 years. Her BCVA was 0.1 in the
right eye and 0.06 in the left eye. She also exhibited esotropia
and nystagmus without fundus abnormalities. Family member II.2 is
the mother of III.2, and also presented with binocular nuclear
cataracts at birth. This individual underwent binocular cataract
extraction without intraocular lens implantation at the age of 11
years. Her BCVA was 0.02 in the right eye and 0.01 in the left eye.
She remained aphakic, showing obvious nystagmus and esotropia
without any fundus abnormalities. Specific clinical information is
presented in Table I.
 | Table IClinical information of patients in
this families. |
Table I
Clinical information of patients in
this families.
| Patient | Age, years | Sex | BCVA (R/L) | Type of
cataracts | Age of removal
cataract | IOL v. aphakia | Refractive error at
age examined (R, L) | Nystagmus | Strabismus
subtype | Fundus
anomalies |
|---|
| IV.1 | 4 | M | 0.2/- | Bilateral nuclear
cataract | 2 month | IOL/Prosthetic |
+4.00DS/-2.00DCx10, | Ophthalmic
nystagmus | Esotropia | NA |
| | | | | | | eye | - | | | |
| IV.2 | 2 | F | NA/NA | Bilateral nuclear
cataract | 2 month | Aphakia | NA/NA | Ophthalmic
nystagmus | Esotropia | NA |
| IV.3 | 8 | F | 0.02/0.06 | Bilateral nuclear
cataract | 2 years | Aphakia |
+19.00DS/-0.50DCx60 | Ophthalmic
nystagmus | Esotropia | Normal |
| | | | | | | |
+22.50DS/-1.50DCx30 | | | |
| III.1 | 30 | F | 0.02/0.1 | Bilateral nuclear
cataract | 2 years | IOL |
-0.75DS/-0.50DCx180 | Ophthalmic
nystagmus | Esotropia | Normal |
| | | | | | | |
-2.00DS/-2.75DCx10 | | | |
| III.2 | 35 | F | 0.1/0.06 | Bilateral nuclear
cataract | 4 years | IOL |
-2.50DS/-1.50DCx170 | Ophthalmic
nystagmus | Esotropia | Normal |
| | | | | | | |
-1.50DS/-1.50DCx110 | | | |
| II.1 | 53 | F | 0.01/0.06 | Bilateral nuclear
cataract | 12 years | Aphakia |
+17.50DS/-1.00DCx5 | Ophthalmic
nystagmus | Esotropia | Normal |
| | | | | | | |
+16.00DS/-2.00DCx160 | | | |
| II.2 | 50 | F | 0.02/0.01 | Bilateral nuclear
cataract | 11 years | Aphakia |
+11.25DS/0.75DCx130 | Ophthalmic
nystagmus | Esotropia | Normal |
| | | | | | | |
+20.00DS/-1.50DCx90 | | | |
Sequencing results and mutation
analysis
An average of 98.9025 Mb raw bases were obtained.
After filtration, an average of 14.4726G clean reads were obtained.
The clean reads had a high-quality value (Q30, 0.93) and the
average G and C basepair content was 48.66%. The results of the
analysis of the clean data suggested that the mapping rate on
genome was 99.61%, the average sequencing depth was 127.97 and the
coverage of the sequencing depth ≥20X was 97.65%. Thus, the target
area had been effectively covered, and the sequencing quality was
high (Table II).
| Table IIQuality control data statistics. |
Table II
Quality control data statistics.
| Sample | Proband |
|---|
| Clean reads
(M) | 98.90 |
| Clean bases
(G) | 14.47 |
| Q20 (%) | 97.03 |
| Q30 (%) | 92.96 |
| GC (%) | 48.66 |
| Read length
(bp) | 150.00 |
| Initial bases on
target | 50,390,601.00 |
| Total effective
reads | 82,447,708.00 |
| Total effective
bases (Mb) | 11,908.75 |
| Effective sequences
on target (Mb) | 6,438.89 |
| Capture specificity
(%) | 54.07 |
| Mapping rate on
genome (%) | 99.61 |
| Duplicate rate on
genome (%) | 16.70 |
| Mismatch rate in
target region (%) | 0.49 |
| Average sequencing
depth on target | 127.97 |
| Fraction of target
covered ≥1x (%) | 99.70 |
| Fraction of target
covered ≥4x (%) | 99.43 |
| Fraction of target
covered ≥10x (%) | 98.88 |
| Fraction of target
covered ≥20x (%) | 97.65 |
After bioinformatics analysis, 112,986 SNPs were
identified in the proband, 99.25% of which were annotated in dbSNP
and 94.06% were annotated in The 1000 Genomes Project database. Of
these, 842 SNPs were novel. Amongst all SNPs, 11,046 were
synonymous, 10,293 were missense, 32 were stop-loss, 83 were
stop-gain, 15 were start-loss and 83 were splice-site mutations. A
total of 18,007 insertion/deletions (InDels) were identified. Of
these, 90.88% were listed in dbSNP and 60.87% in The 1000 Genomes
Project. The number of novel InDels was 1,532. Of the overall
InDels, 281 were frameshift, 1 was stop-loss, 1 was start-loss and
65 were splice-site mutations (Table
III). After comparison with mutation sites in dbSNP, the 1000
Genomes Project, ExAC, OMIM, HGMD, ClinVar and gnomAD, variation
sites with a frequency <0.01 were selected. Genes associated
with hereditary eye diseases were analyzed and according to the
ACMG guidelines (23,24), using software calculation and
prediction, a causative genetic variant was obtained,
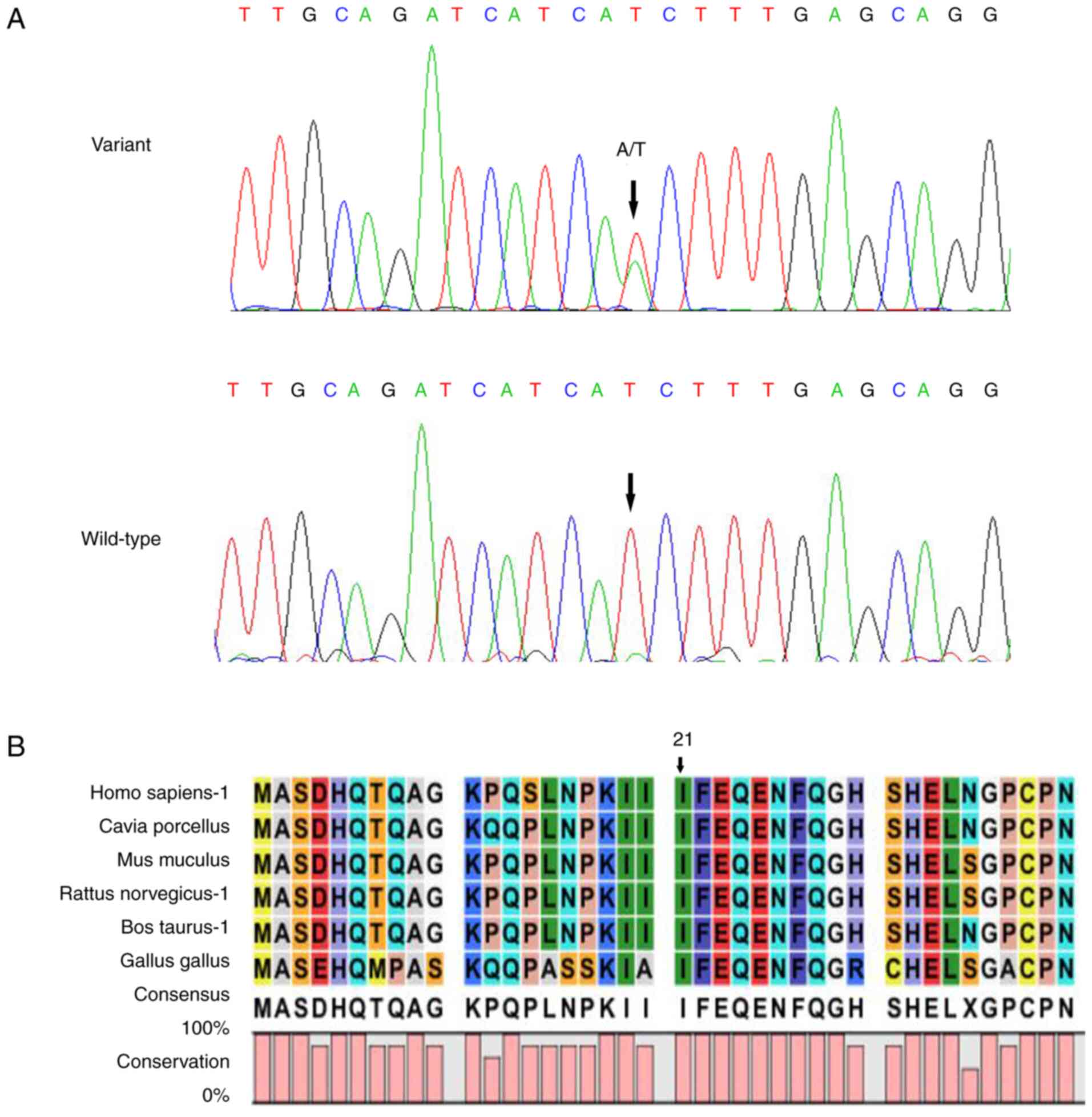
CRYBB2:c.62T>A(p.I21N). Sanger sequencing verified that
CRYBB2:c.62T>A (p.I21N) was found in all patients, but
not in the other relatives and healthy participants (Fig. 2A). The variation and cataracts
phenotype were co-segregated in this family. Multi-species sequence
alignment demonstrated that the locus was highly conserved
(Fig. 2B).
| Table IIISummary statistics for sequencing
data. |
Table III
Summary statistics for sequencing
data.
| Sample | Proband |
|---|
| Total SNPs | 112,986 |
|
Fraction of
SNPs in dbSNP (%) | 99.25 |
|
Fraction of
SNPs in 1000 genomes (%) | 94.06 |
|
Novel | 842.00 |
|
Homozygous | 49,261.00 |
|
Heterozygous | 63,725.00 |
|
Intron | 74,427.00 |
|
5'UTRs | 1,787.00 |
|
3'UTRs | 3,457.00 |
|
Upstream | 3,346.00 |
|
Downstream | 2,736.00 |
|
Intergenic | 4,437.00 |
|
Synonymous | 11,046.00 |
|
Missense | 10,293.00 |
|
Stop-gain | 83.00 |
|
Stop-loss | 32.00 |
|
Start-loss | 15.00 |
|
Splicing | 83.00 |
| Total InDels | 18,007.00 |
|
Fraction of
InDels in dbSNP (%) | 90.88 |
|
Fraction of
InDels in 1000 genomes (%) | 60.87 |
|
Novel | 1,532.00 |
|
Homozygous | 7,595.00 |
|
Heterozygous | 10,412.00 |
|
Intron | 14,422.00 |
|
5'UTRs | 245.00 |
|
3'UTRs | 674.00 |
|
Upstream | 497.00 |
|
Intergenic | 682.00 |
|
Frameshift | 281.00 |
|
Non-frameshift
insertion | 143.00 |
|
Non-frameshift
deletion | 181.00 |
|
Stop-loss | 1.00 |
|
Start-loss | 1.00 |
|
Splicing | 65.00 |
Bioinformatics analysis of the p.I21N
variant at the protein level
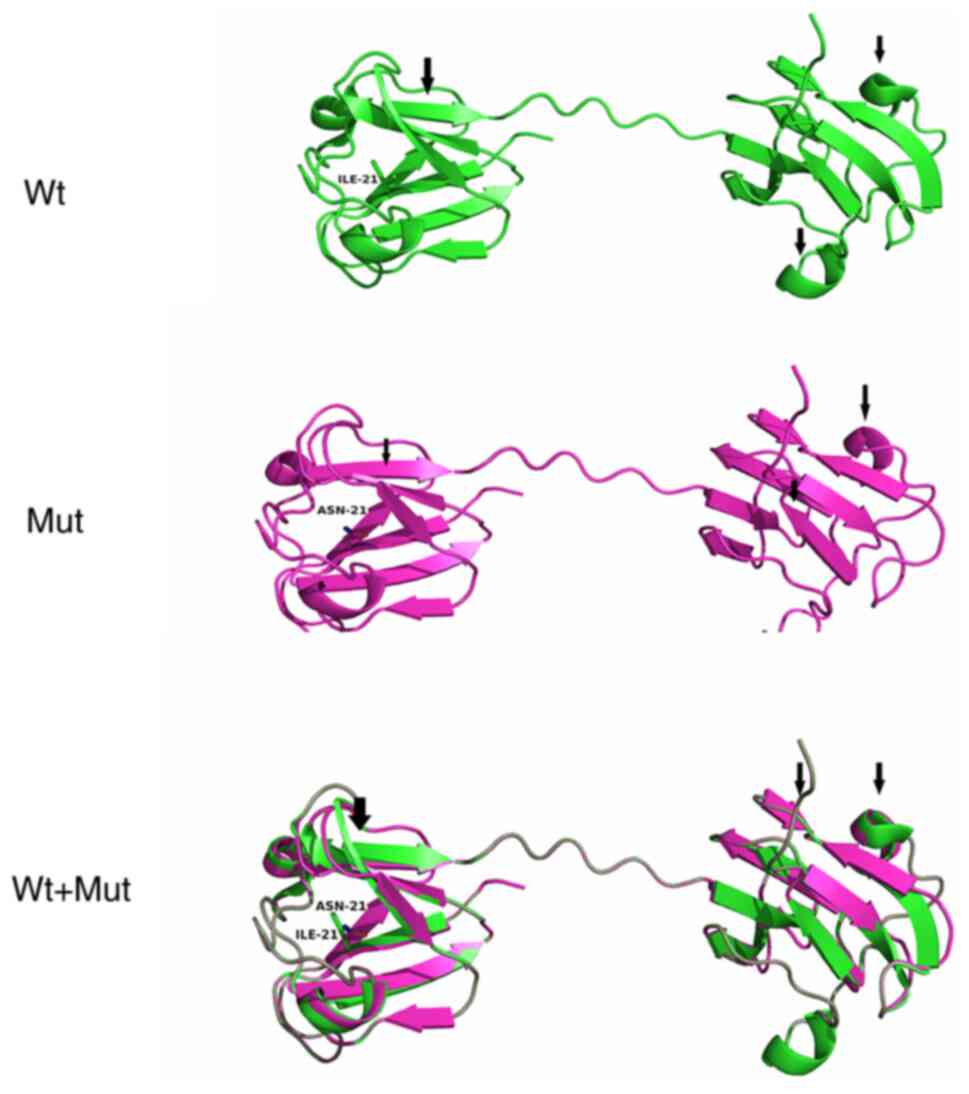
The protein structure of Wt- and Mut-CRYBB2
was predicted using Pymol (28).
It was found that the variant changed the secondary structure of
the protein, specifically altering a portion of the helix and
folding (Fig. 3). In addition, it
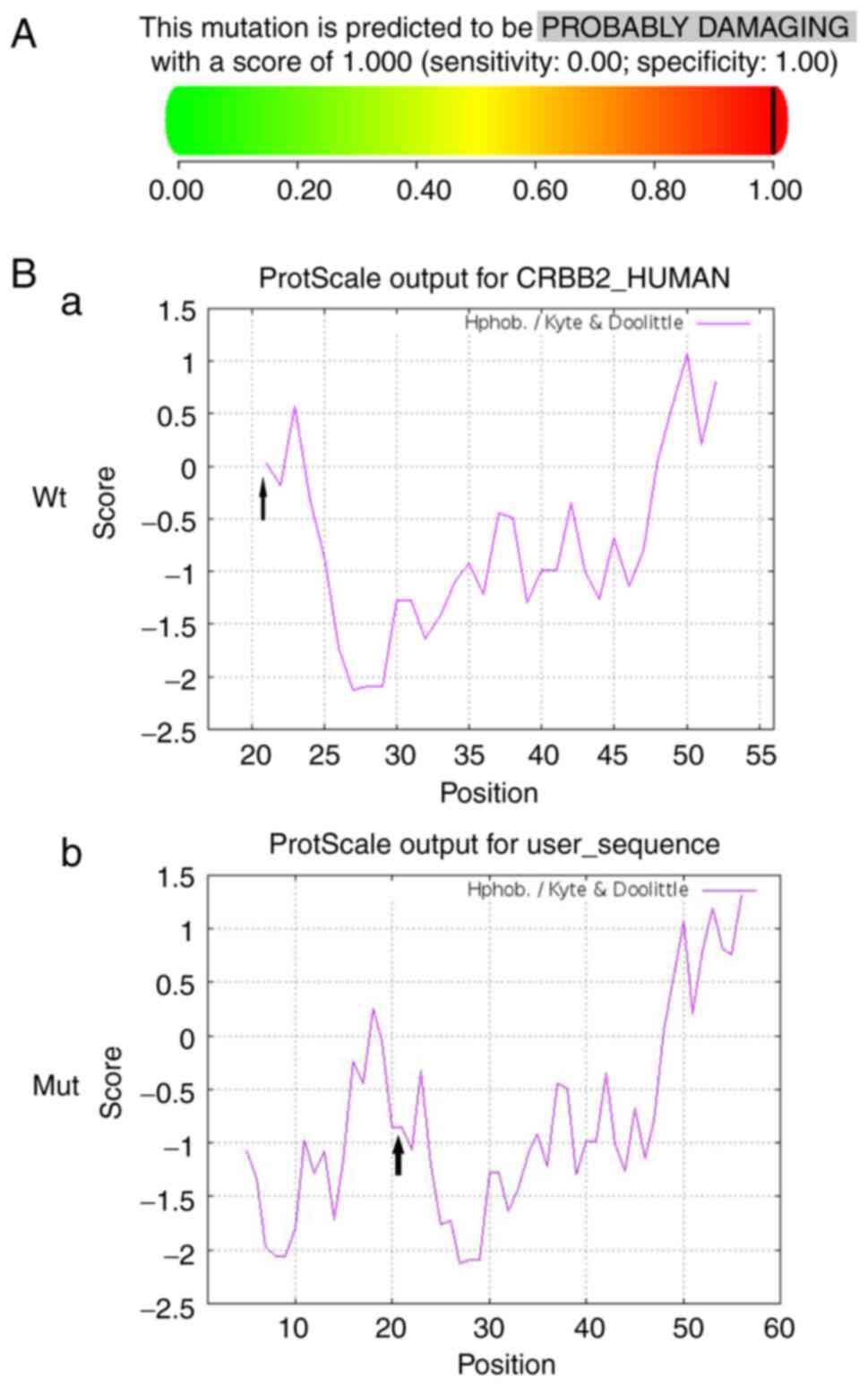
was predicted to cause damage when based on PolyPhen-2, with a
score of 1.0 (Fig. 4A).
Hydrophobicity analysis showed that the hydrophobicity of this site
in the variant protein was lower than that of the Wt protein
(Fig. 4B).
Differences in protein expression and
effect on apoptosis
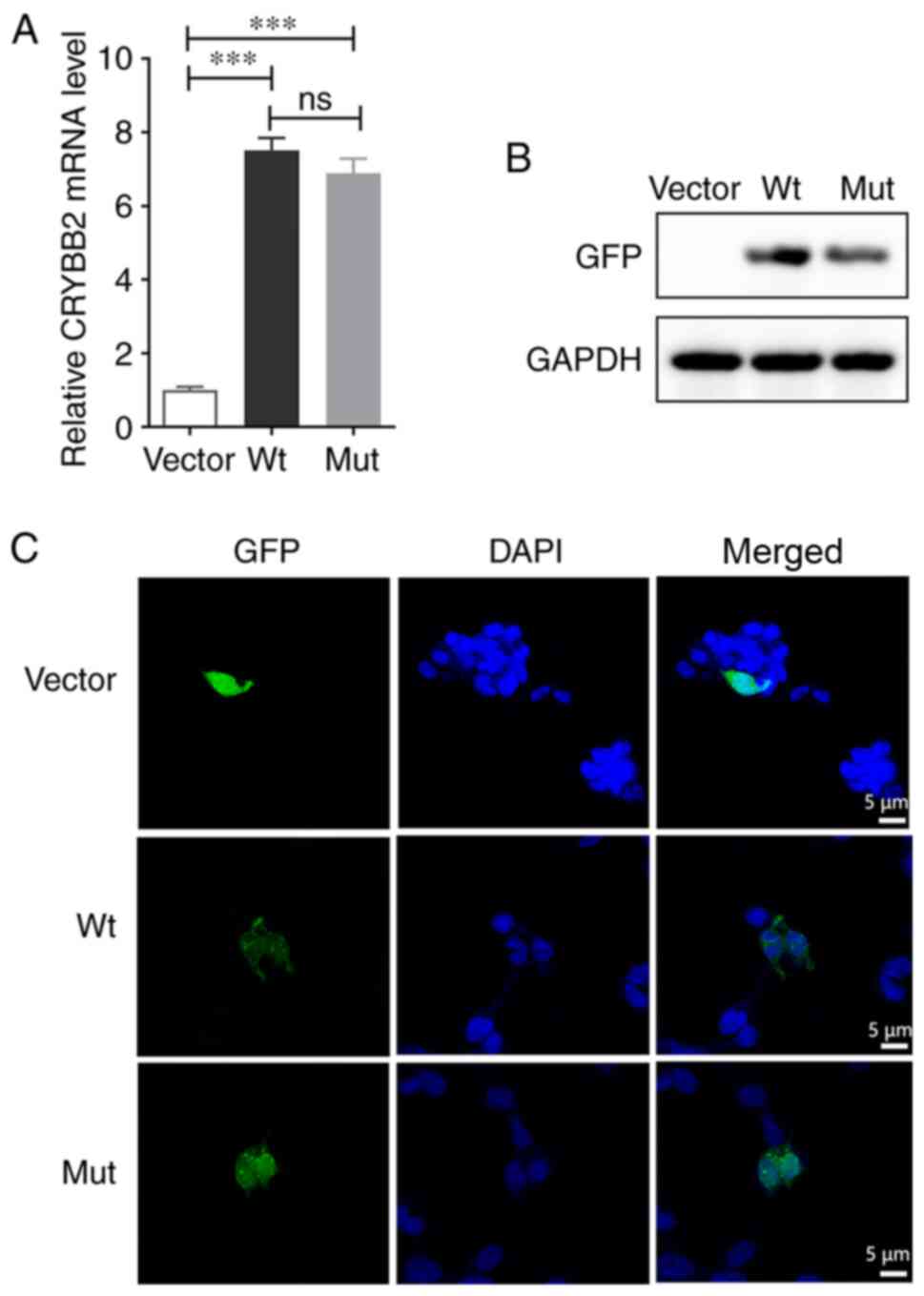
CRYBB2 mRNA was assessed via qPCR analysis,
which revealed a significantly higher transcript abundance than in
empty vector-cells, and there was no difference in mRNA expression
levels between Wt- and Mut-CRYBB2 (Fig. 5A). The expression levels of the
CRYBB2 proteins were further evaluated via western blotting using
an anti-GFP antibody, a specific cross-reactive band was observed
in transfected cells, whereas no such band was detected in empty
vector-cells cell. These results demonstrated this gene, that with
or without mutation, was successfully expressed in transfected
cells; however, the protein expression levels of Mut-CRYBB2
were notably lower than that of Wt-CRYBB2 (Fig. 5B). The expression level of the
fusion protein carrying GFP was observed via confocal microscopy,
and it was found that the Wt-CRYBB2 was uniformly expressed
in the cytoplasm, whereas the Mut-CRYBB2 primarily
surrounded the nucleus, gathering at the nuclear membrane (Fig. 5C).
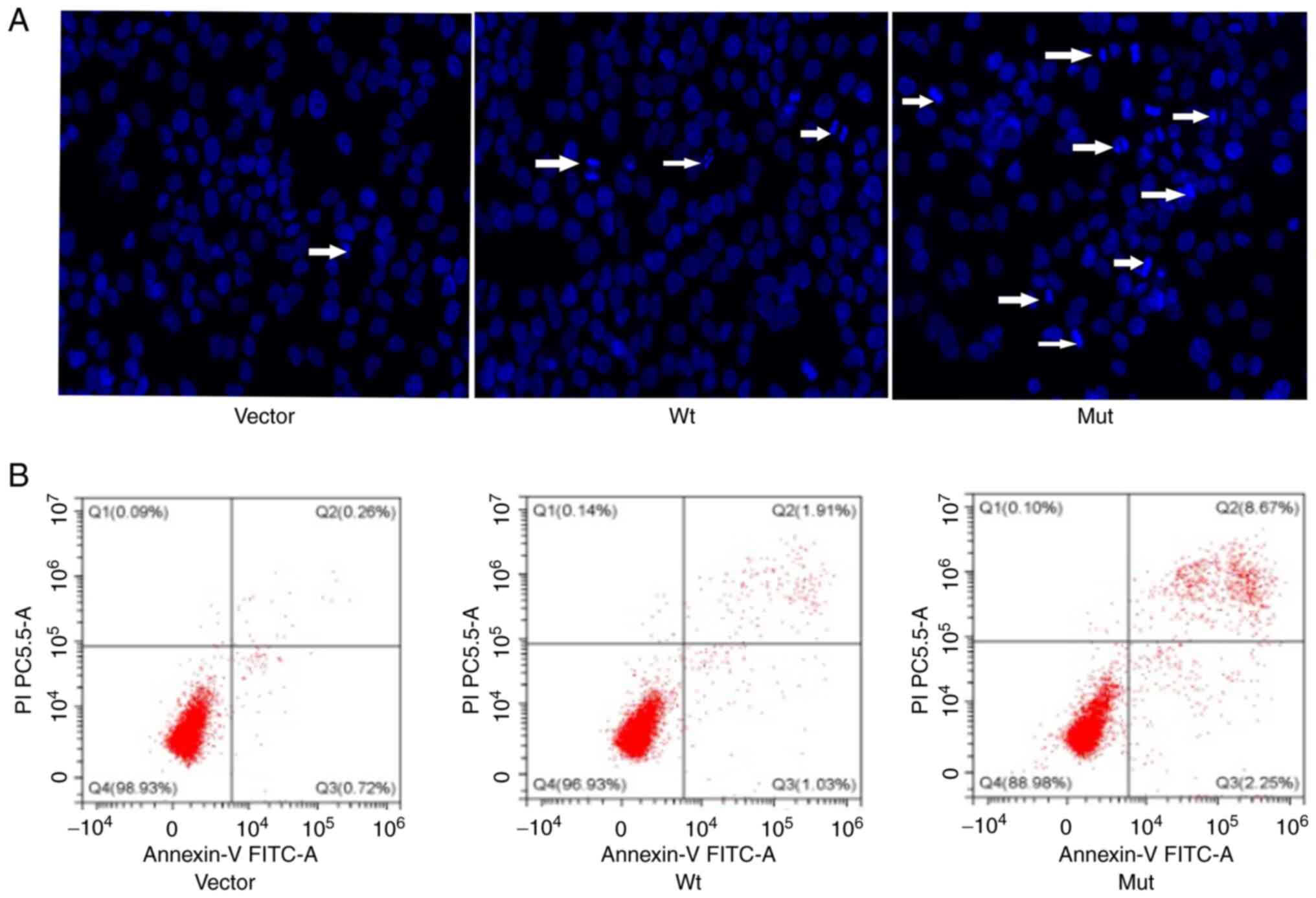
Following nuclear staining with Hoechst 33342, it
was found that the apoptotic rate of cells transfected with
Wt-CRYBB2 plasmid was not affected, but the apoptotic rate
of cells transfected with the Mut-CRYBB2 plasmid was
increased (Fig. 6A). This
conclusion was also verified via flow cytometry analysis; the
apoptosis of cells transfected with the Mut-CRYBB2 plasmid
was increased (Fig. 6B).
UPR-associated gene expression status
in cells
The UPR is a type of adaptive signal induced by
abnormal accumulation of intracellular proteins (29). BiP/HSPA5 is a marker protein of the
endoplasmic reticulum (ER) emergency response, and is a Hsp70
family chaperone localized in the ER lumen (30). It has been confirmed that apoptosis
induced by the UPR is involved in the pathological mechanism of
congenital cataracts (9). In the
present study, following identification of abnormal aggregation of
the variant protein, an UPR was also observed, and this may have
been involved in the formation of congenital nuclear cataracts in
this family. The western blotting results indicated that the
expression of BiP was abnormally increased in the cells transfected
with the Mut-CRYBB2 plasmid (Fig. 7A). In addition, the expression
levels of UPR-related transcription factors were also significantly
increased in the Mut group, including HSPA5, DDIT3
and IRE1 (Fig. 7B-D).
IRE1 is triggered by the splicing of the Xbp1 mRNA (31), and in the present study, increased
expression of XBP1 was also observed in cells transfected
with Mut-CRYBB2 plasmids (Fig.
7E).
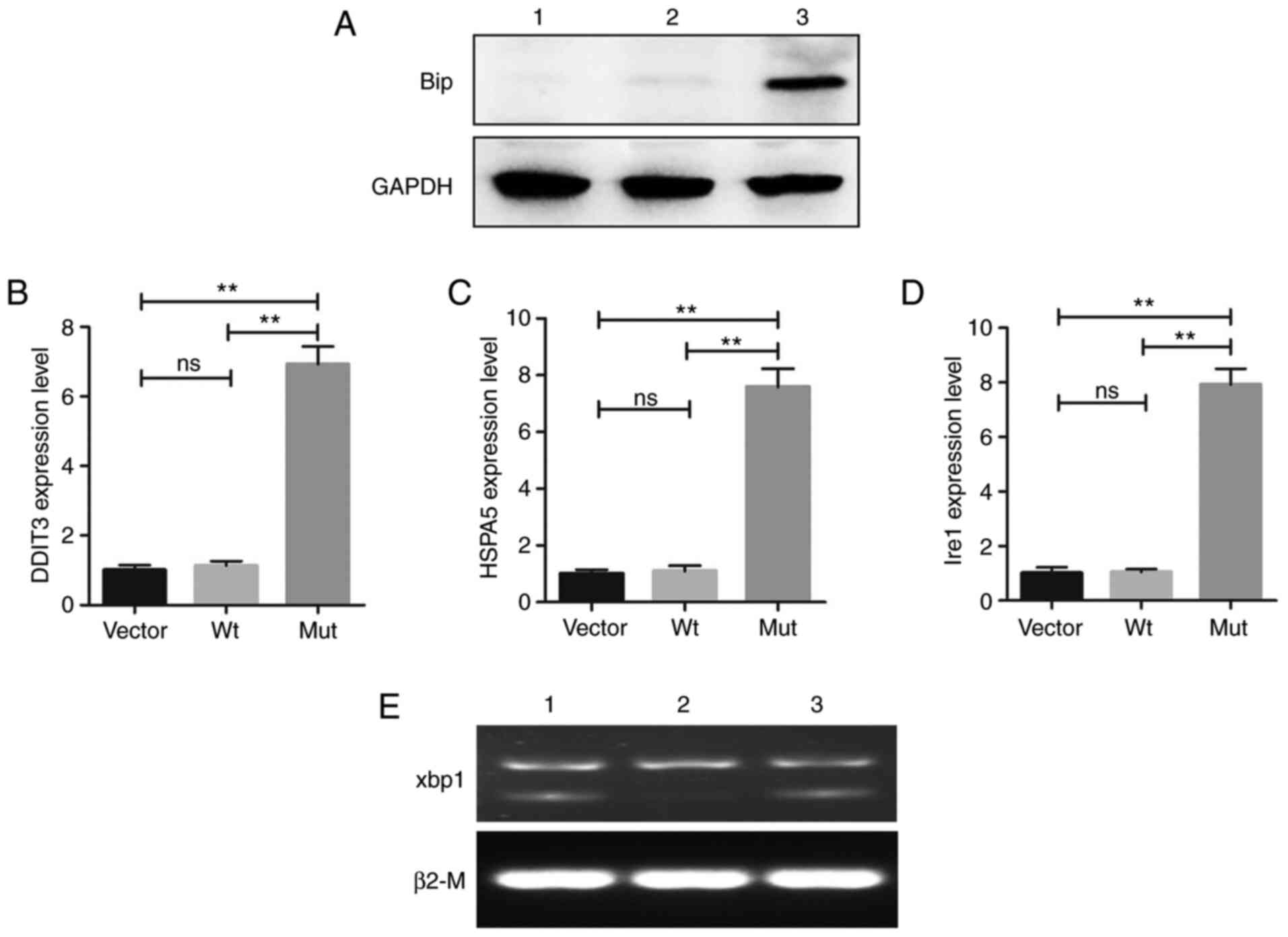 | Figure 7UPR-associated gene expression status
in cells. (A) Western blot analysis confirmed the increase in BiP
expression in cells expressing the Mut-CRYBB2. GAPDH was
used as the loading control. Relative mRNA expression levels of (B)
DDIT3, (C) HSPA5 and (D) IRE1. Values are
presented as the mean ± SD. One way ANOVA and Bonferroni multiple
comparison was used to test the differences between three groups.
**P<0.01. (E) Xbp1 splicing detection (1, Vector; 2,
cells expressing Wt-CRYBB2; 3, cells expressing
Mut-CRYBB2). BiP, binding immunoglobulin protein;
DDIT3, DNA damage-inducible transcript 3; Ire1,
inositol-requiring enzyme 1; Xbp1, X-box-binding protein 1;
HSPA5, heat shock protein family A (Hsp70) member 5; ns, not
significance; Wt, wild-type; Mut, mutant. |
Discussion
Congenital cataracts are a complicated heterogeneous
disease, and it is generally accepted that genetic defects and
environmental factors are the primary causes (32). WES is widely used to identify
genetic defects in complex genetically heterogeneous diseases
(33). In the present study, WES
was used to identify a recurrent variant, CRYBB2:c.62T>A
(p.I21N), in a four-generation Chinese family with congenital
nuclear cataracts. The variant was found to exist in all patients,
but not in other healthy relatives and the controls. There were a
total of 8 affected members and 13 unaffected members in this
family, according to the case records, and all patients presented
with binocular nuclear cataracts at birth, and all had different
degrees of esotropia and nystagmus. Congenital cataracts exhibits
complex genetic heterogeneity. Although it is generally
hypothesized that congenital cataracts are a single-gene disease,
through a large number of clinical observations, no regular
correlation between a specific gene mutation and the cataracts
phenotype has been found (32,34).
Therefore, it is considered that post-transcriptional modifications
or other modified genes and environmental factors may also serve an
important role in the occurrence of congenital cataracts.
Therefore, the present study may only explain the family's
congenital cataracts. Congenital cataracts in other families caused
by the same mutation CRYBB2:c.62T>A (p.I21N) may exhibit
alternative mechanisms. Future investigations will aim to recruit
families with congenital cataracts caused by the same mutation, and
compare the similarities and differences between these
families.
Strabismus of certain patients with congenital
cataracts develops following the occurrence of congenital
cataracts, and nystagmus may be induced by low vision (35,36).
In addition, the operational time of surgery for congenital
cataracts has a certain impact on postoperative vision (37). In childhood, cataracts can prevent
light from entering the eyes and hinder the development of the
retina, resulting in deprivation of amblyopia. All patients in the
present study had poor vision to a certain degree, and this was
accompanied by nystagmus and esotropia. This may also be associated
with the fact that cataract surgery for patients in this family was
performed relatively late compared with the onset of the
disease.
The CRYBB2 gene is located on 22q11.23 and
contains six exons, with the start of translation beginning in the
second exon. Exons 3-6 each encode one Greek key motif (GKM). Its
protein product includes 205 amino acids with a molecular weight of
23, 379 and 20 kDa. At present, congenital cataracts induced by
CRYBB2 variants have been widely reported, most of the
reported CRYBB2 variants are located in the last two exon
regions (7). p.D128V in exon 6
alters the hydrophobicity and charge of the random coil region
between amino acids 126-139 of the CRYBB2 protein (38). p.W151C in exon 6 disrupts the
solubility of CRYBB2 and causes abnormal aggregation of the protein
to form membrane cataracts (39).
In addition, the p.W151C variant identified in an Indian family was
also predicted to disrupt the fourth GKM and increase the protein's
hydrophobicity, thereby leading to the formation of cataracts
(40). p.Q155X in exon 6 results
in a partially unfolded structure and decreased structural order,
inhibiting interactions with other proteins (41). p.A188H in exon 6 is a rare variant
at the carboxyl terminus, which impairs the dimerization of the
CRYBB2 protein by establishing a new hydrogen bond, thereby leading
to increased lens opacity (42).
Xu et al (7) reported that
p.A188H caused congenital cataracts and progressive eye axis
extension in a five-generation Chinese family. p.I21N in exon 3 is
located in the first GKM, on the verge of the N-terminal
translation initiation region, which is highly conserved based on
multi-species sequence analysis. The N-terminal sequence of a
protein has a significant impact on the biological function of the
protein; almost all protein synthesis starts at the N-terminus,
thus the sequence composition of the N-terminus of the protein has
a significant influence on the overall biological function of the
protein (43). For example, the
N-terminal sequence affects the half-life of the protein (44), and regulates the location of the
protein in subcellular organelles (45), which are closely associated with
the function and stability of the protein. Song et al
(46) reported that a heterozygous
variant c.35G>T in exon 1 of α-crystallin A chain (CRYAA),
located at the N-terminus of the CRYAA protein, induced congenital
cataracts and microphthalmia in a four-generation Chinese family,
which also illustrated the influence of the stability of the
N-terminus on protein function. The N- and C-terminal regions of
CRYBB2 have been hypothesized to be essential for the maintenance
of lens transparency (47).
CRYBB2:c.62T>A (p.I21N) was also
determined to be potentially detrimental by PolyPhen-2 and SIFT.
p.I21N was considered as ‘probably damaging’ and ‘deleterious’ by
PolyPhen-2 and SIFT, respectively. In addition, ProtScale indicated
that the hydrophobicity of the mutation site was significantly
reduced. Pymol predicted that this variant caused the α-helix
located in the first GKM to be replaced by a β-strand, and this
change also affected the correct folding of the protein. This
change may also explain why this variant protein lost its function,
consistent with an earlier report (11). There are a large number of
molecular connections between the amino acids in the protein,
including various forces and hydrogen bonds. Changes in the
hydrophobicity of a certain amino acid site may affect the
secondary and tertiary structure of a protein (8,9). The
change in the structure of this variant of CRYBB2 shown by
Pymol may have been due to the variant amino acid site exerting
effects on the forces of the surrounding amino acids. The resulting
conformation re-formed in order to adapt to protein folding. Any
mutation that affects a molecule's inner connections can alter a
protein's stability and capacity to bind to other proteins. In
addition, it has been reported that the lens thermal balance and
the ability to resist oxidative stress in the CRYBB2 gene
knockout mice are reduced, which leads to the occurrence of
cataracts in mice (48).
Gene therapy for diseases caused by genetic defects
is being developed and constantly improved (11), so it is necessary to investigate
the mechanisms via which certain gene mutations may cause
cataracts. In the present study, the variant discovered was a
recurrent pathological variant. It was suggested that the site
where the variant was located was likely to be a mutation hotspot
or a susceptible site. Hence, gene recombination technology was
used to construct mutant and Wt plasmid-transfected cells to assess
the possible pathological mechanisms of
CRYBB2:c.62T>A(p.I21N) in inducing cataracts.
Post-transfection, the mRNA expression levels of Wt- and
I21N-CRYBB2 were not altered, suggesting that the variant
does not affect transcription, but at the protein levels, the
expression levels of I21N-CRYBB2 were lower than in the
Wt-CRYBB2 transfected cells, suggesting that there may be
other processes involved in the regulation of translation, which
leads to decreased protein expression. Post-translational
modifications of proteins are an important mechanism for
maintaining the homeostasis of protein functions in the body
(49). Although it is generally
hypothesized that congenital cataracts are a single-gene disease
(14), complex clinical and
genetic heterogeneity is always observed. In the process of gene
transcription and translation, a variety of transcription modifiers
and protein translation regulation mechanisms are involved
(50), which complicates the
search for a potential target for clinical therapeutics in diseases
caused by gene defects.
In the present study, the immunofluorescence
analysis demonstrated that Wt-CRYBB2 protein predominantly
accumulated in the cytoplasm. However, the I21N-CRYBB2
protein was notably more likely to accumulate around the nucleus.
It is hypothesized that the accumulation of these abnormal proteins
around the nucleus may prevent the denucleation process of lens
fiber cells, thereby destroying the normal lens fiber cells, which
are required to maintain lens transparency, resulting in turbidity
in the lens (51). Additionally,
flow cytometry analysis indicated that the variant may cause cells
to be more prone to apoptosis. This may also be an element in the
formation of cataracts.
The UPR has been shown to be involved in the
pathological process of cataracts caused by certain gene mutations
(52). The UPR is considered to be
a stress response of cells to stimuli (53). When incorrectly folded proteins are
synthesized, they can be terminated in advance or degraded
(30), with the aim of preventing
abnormal protein accumulation, which can cause damage to cells.
There are reports demonstrating the important purpose of the
UPR-related genes for a response to a misfolding protein (54-57).
For instance, CRYBB2: p.V146L was reported to induce cell
apoptosis by activating the UPR, resulting in cataracts (9). It has been shown that the abnormal
expression of the protein leads to the activation of transcription
factors associated with the UPR, which initiates the UPR in an
all-round way, and this leads to the apoptosis of fibroblasts
(58). In the present study, it
was also found that the CRYBB2:p.I21N variant induced
activation of the UPR, and this led to increased apoptosis of the
transfected cells; the present study findings consolidates these
previous studies (9), and supports
the hypothesis that the activation of the UPR induces partial
cataracts.
The formation of cataracts in this family may be
caused by abnormal an abnormal N-terminal structure and in the
first GKM of the protein in I21N-CRYBB2, which does not
hinder transcription and translation. However, abnormal N-terminal
products may affect the half-life of the protein and also affect
the normal anchoring of proteins (59,60)
The aggregation of abnormal proteins activates the UPR, which leads
to a decrease in the aggregation of abnormal proteins (30). The original purpose may be a
protective mechanism, but the activation of UPR also increases the
risk of the cell apoptosis. The accumulation of several factors may
have thus finally lead to the formation of nuclear cataracts in
this four-generation Chinese family.
In conclusion, in the present study, a recurrent
variant, CRYBB2:c.62T>A(p.I21N), in a four-generation
Chinese family with congenital nuclear cataracts was identified
using WES, and the effects of this mutation were assessed in
vitro. These results provide novel evidence via which
CRYBB2:p.I21N may cause congenital nuclear cataracts.
Specifically, the variant exhibited an abnormal N-terminal
structure and in the first GKM in the protein, which likely caused
the abnormal protein aggregation, that triggered the UPR, induced
excessive cell apoptosis, and thus gave rise to the occurrence of
congenital nuclear cataracts in this four-generation Chinese
family.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 51573101), Beijing Natural Science
Foundation (grant no. 7172056) and the Xinglin Scholar Scientific
Research Promotion Program of Chengdu University of Traditional
Chinese Medicine (grant no. BSH2019025).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are not publicly available due to the patient privacy
protection and local policy (Regulations of the People's Republic
of China on the Management of Human Genetic Resources, State Order
no. 717) prohibiting data sharing but are available from the
corresponding author on reasonable request.
Authors' contributions
DDC performed the molecular genetic study,
participated in the sequence alignment and drafted the manuscript.
DDC also prepared the figures, edited the manuscript and drafted
the molecular results. SQZ conceived the study, participated in its
design and coordination and drafted the manuscript. Both authors
read and approved the final manuscript. SQZ and DDC checked the
data and approved the authenticity of the raw data.
Ethics approval and consent to
participate
The study protocol was approved by the Yin Hai Eye
Hospital affiliated with Chengdu University of Traditional Chinese
Medicine Ethics Committee (Chengdu, China; approval no.
CMEC2010-21). Written informed consent was obtained from the
participants or their guardians for genetic testing.
Patient consent for publication
Written informed consent was obtained from the
participants or their guardians for publication of clinical and
genetic data and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Naz S, Sharif S, Badar H, Rashid F, Kaleem
A and Iqtedar M: Incidence of environmental and genetic factors
causing congenital cataract in children of Lahore. J Pak Med Assoc.
66:819–822. 2016.PubMed/NCBI
|
|
2
|
Li J, Chen X, Yan Y and Yao K: Molecular
genetics of congenital cataracts. Exp Eye Res.
191(107872)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lhussiez V, Dubus E, Cesar Q, Acar N,
Nandrot EF, Simonutti M, Audo I, Lizé E, Nguyen S, Geissler A, et
al: Cohen syndrome-associated cataract is explained by VPS13B
functions in lens homeostasis and is modified by additional genetic
factors. Invest Ophthalmol Vis Sci. 61(18)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sheeladevi S, Lawrenson JG, Fielder AR and
Suttle CM: Global prevalence of childhood cataract: A systematic
review. Eye (Lond). 30:1160–1169. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhuang J, Cao Z, Zhu Y, Liu L, Tong Y,
Chen X, Wang Y, Lu C, Ma X and Yang J: Mutation screening of
crystallin genes in Chinese families with congenital cataracts. Mol
Vis. 25:427–437. 2019.PubMed/NCBI
|
|
6
|
Gerasimovich ES, Strelkov SV and Gusev NB:
Some properties of three αB-crystallin mutants carrying point
substitutions in the C-terminal domain and associated with
congenital diseases. Biochimie. 142:168–178. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Xu LJ, Lv ZG, Liu Y, Zhang XX, Cui YX, Li
XC, Zhu YJ and He J: A novel CRYBB2 mutation causes autosomal
dominant cataract: A report from a Chinese family. Eur J
Ophthalmol. 4(1120672120926450)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chen W, Chen X, Hu Z, Lin H, Zhou F, Luo
L, Zhang X, Zhong X, Yang Y, Wu C, et al: A missense mutation in
CRYBB2 leads to progressive congenital membranous cataract by
impacting the solubility and function of βB2-crystallin. PLoS One.
8(e81290)2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Li L, Fan DB, Zhao YT, Li Y, Kong DQ, Cai
FF and Zheng GY: Two novel mutations identified in ADCC families
impair crystallin protein distribution and induce apoptosis in
human lens epithelial cells. Sci Rep. 7(17848)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Guo W, Lai Y, Yan Z, Wang Y, Nie Y, Guan
S, Kuo Y, Zhang W, Zhu X, Peng M, et al: Trio-whole-exome
sequencing and preimplantation genetic diagnosis for unexplained
recurrent fetal malformations. Hum Mutat. 41:432–448.
2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang KJ, Wang BB, Zhang F, Zhao Y, Ma X
and Zhu SQ: Novel beta-crystallin gene mutations in Chinese
families with nuclear cataracts. Arch Ophthalmol. 129:337–343.
2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
World Medical Association. World medical
association declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194.
2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chun SM, Sung CO, Jeon H, Kim TI, Lee JY,
Park H, Kim Y, Kim D and Jang SJ: Next-generation sequencing using
S1 nuclease for poor-quality formalin-fixed, paraffin-embedded
tumor specimens. J Mol Diagn. 20:802–811. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhao S, Zhang C, Mu J, Zhang H, Yao W,
Ding X, Ding J and Chang Y: All-in-one sequencing: An improved
library preparation method for cost-effective and high-throughput
next-generation sequencing. Plant Methods. 16(74)2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Li H and Durbin R: Fast and accurate short
read alignment with burrows-wheeler transform. Bioinformatics.
25:1754–1760. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Broad Institute: Picard Tools. http://broadinstitute.github.io/picard/. Accessed
September 10, 2019.
|
|
17
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The genome analysis toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–11.10.33. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38(e164)2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Cooper GM, Stone EA and Asimenos G: NISC
Comparative Sequencing Program. Green ED, Batzoglou S and Sidow A:
Distribution and intensity of constraint in mammalian genomic
sequence. Genome Res. 15:901–913. 2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
ACMG Board Of Directors: ACMG policy
statement. Updated recommendations regarding analysis and reporting
of secondary findings in clinical genome-scale sequencing. Genet
Med. 17:68–69. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Seeliger D and de Groot BL: Ligand docking
and binding site analysis with PyMOL and autodock/vina. J Comput
Aided Mol Des. 24:417–422. 2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Balaji S and Vanniarajan A: Implication of
pseudo reference genes in normalization of data from reverse
transcription-quantitative PCR. Gene. 757(144948)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mooers B: Shortcuts for faster image
creation in PyMOL. Protein Sci. 29:268–276. 2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Hetz C and Papa FR: The unfolded protein
response and cell fate control. Mol Cell. 69:169–181.
2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bashir S, Banday M, Qadri O, Bashir A,
Hilal N, Nida-I-Fatima Rader S and Fazili KM: The molecular
mechanism and functional diversity of UPR signaling sensor IRE1.
Life Sci. 265(118740)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Sharma RB, Darko C and Alonso LC:
Intersection of the ATF6 and XBP1 ER stress pathways in mouse islet
cells. J Biol Chem. 295:14164–14177. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Berry V, Georgiou M, Fujinami K, Quinlan
R, Moore A and Michaelides M: Inherited cataracts: Molecular
genetics, clinical features, disease mechanisms and novel
therapeutic approaches. Br J Ophthalmol. 104:1331–1337.
2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Liu H, Wei X, Sha Y, Liu W, Gao H, Lin J,
Li Y, Tang Y, Wang Y, Wang Y and Su Z: Whole-exome sequencing in
patients with premature ovarian insufficiency: Early detection and
early intervention. J Ovarian Res. 13(114)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Astiazarán MC, García-Montaño LA,
Sánchez-Moreno F, Matiz-Moreno H and Zenteno JC: Next generation
sequencing-based molecular diagnosis in familial congenital
cataract expands the mutational spectrum in known congenital
cataract genes. Am J Med Genet A. 176:2637–2645. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Hwang SS, Kim WS and Lee SJ: Clinical
features of strabismus and nystagmus in bilateral congenital
cataracts. Int J Ophthalmol. 11:813–817. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Magli A, Carelli R, Forte R, Vecchio EC,
Esposito F and Torre A: Congenital and developmental cataracts:
Focus on strabismus outcomes at long-term follow-up. Semin
Ophthalmol. 32:358–362. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kuhli-Hattenbach C, Fronius M and Kohnen
T: Timing of congenital cataract surgery: Amblyopia versus aphakic
glaucoma. Ophthalmologe. 117:190–198. 2020.PubMed/NCBI View Article : Google Scholar : (In German).
|
|
38
|
Pauli S, Söker T, Klopp N, Illig T, Engel
W and Graw J: Mutation analysis in a German family identified a new
cataract-causing allele in the CRYBB2 gene. Mol Vis. 13:962–967.
2007.PubMed/NCBI
|
|
39
|
Zhao WJ, Xu J, Chen XJ, Liu HH, Yao K and
Yan YB: Effects of cataract-causing mutations W59C and W151C on
βB2-crystallin structure, stability and folding. Int J Biol
Macromol. 103:764–770. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Santhiya ST, Manisastry SM, Rawlley D,
Malathi R, Anishetty S, Gopinath PM, Vijayalakshmi P,
Namperumalsamy P, Adamski J and Graw J: Mutation analysis of
congenital cataracts in Indian families: Identification of SNPS and
a new causative allele in CRYBB2 gene. Invest Ophthalmol Vis Sci.
45:3599–3607. 2004.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Li FF, Zhu SQ, Wang SZ, Gao C, Huang SZ,
Zhang M and Ma X: Nonsense mutation in the CRYBB2 gene causing
autosomal dominant progressive polymorphic congenital coronary
cataracts. Mol Vis. 14:750–755. 2008.PubMed/NCBI
|
|
42
|
Weisschuh N, Aisenbrey S, Wissinger B and
Riess A: Identification of a novel CRYBB2 missense mutation causing
congenital autosomal dominant cataract. Mol Vis. 18:174–180.
2012.PubMed/NCBI
|
|
43
|
Kim L, Kwon DH, Heo J, Park MR and Song
HK: Use of the LC3B-fusion technique for biochemical and structural
studies of proteins involved in the N-degron pathway. J Biol Chem.
295:2590–2600. 2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Friemel M, Marelja Z, Li K and Leimkühler
S: The N-terminus of iron-sulfur cluster assembly factor ISD11 is
crucial for subcellular targeting and interaction with l-cysteine
desulfurase NFS1. Biochemistry. 56:1797–1808. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Skach WR, Shi LB, Calayag MC, Frigeri A,
Lingappa VR and Verkman AS: Biogenesis and transmembrane topology
of the CHIP28 water channel at the endoplasmic reticulum. J Cell
Biol. 125:803–815. 1994.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Song Z, Si N and Xiao W: A novel mutation
in the CRYAA gene associated with congenital cataract and
microphthalmia in a Chinese family. BMC Med Genet.
19(190)2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Carver JA, Grosas AB, Ecroyd H and Quinlan
RA: The functional roles of the unstructured N- and C-terminal
regions in αB-crystallin and other mammalian small heat-shock
proteins. Cell Stress Chaperones. 22:627–638. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zhang J, Li J, Huang C, Xue L, Peng Y, Fu
Q, Gao L, Zhang J and Li W: Targeted knockout of the mouse
betaB2-crystallin gene (Crybb2) induces age-related cataract.
Invest Ophthalmol Vis Sci. 49:5476–5483. 2008.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Song L and Luo ZQ: Post-translational
regulation of ubiquitin signaling. J Cell Biol. 218:1776–1786.
2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Frye M, Harada BT, Behm M and He C: RNA
modifications modulate gene expression during development. Science.
361:1346–1349. 2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Zhang J, Cui WW, Du C, Huang Y, Pi X, Guo
W, Wang J, Huang W, Chen D, Li J, et al: Knockout of DNase1l1l
abrogates lens denucleation process and causes cataract in
zebrafish. Biochim Biophys Acta Mol Basis Dis.
1866(165724)2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Shiels A and Hejtmancik JF: Inherited
cataracts: Genetic mechanisms and pathways new and old. Exp Eye
Res. 209(108662)2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Chan WC, Tsang KY, Cheng YW, Ng VC, Chik
H, Tan ZJ, Boot-Handford R, Boyde A, Cheung KN, Cheah KS and Chan
D: Activating the unfolded protein response in osteocytes causes
hyperostosis consistent with craniodiaphyseal dysplasia. Hum Mol
Genet. 26:4572–4587. 2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Berry V, Pontikos N, Moore A, Ionides A,
Plagnol V, Cheetham ME and Michaelides M: A novel missense mutation
in HSF4 causes autosomal-dominant congenital lamellar cataract in a
British family. Eye (Lond). 32:806–812. 2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Ghavami S, Yeganeh B, Zeki AA, Shojaei S,
Kenyon NJ, Ott S, Samali A, Patterson J, Alizadeh J, Moghadam AR,
et al: Autophagy and the unfolded protein response promote
profibrotic effects of TGF-β(1) in human lung fibroblasts. Am J
Physiol Lung Cell Mol Physiol. 314:L493–L504. 2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Schinzel RT, Higuchi-Sanabria R, Shalem O,
Moehle EA, Webster BM, Joe L, Bar-Ziv R, Frankino PA, Durieux J,
Pender C, et al: The hyaluronidase, TMEM2, promotes ER homeostasis
and longevity independent of the UPR ER. Cell. 179:1306–1318.
2019.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Muñoz JP, Ivanova S, Sánchez-Wandelmer J,
Martínez-Cristóbal P, Noguera E, Sancho A, Díaz-Ramos A,
Hernández-Alvarez MI, Sebastián D, Mauvezin C, et al: Mfn2
modulates the UPR and mitochondrial function via repression of
PERK. EMBO J. 32:2348–2361. 2013.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Andley UP and Goldman JW: Autophagy and
UPR in alpha-crystallin mutant knock-in mouse models of hereditary
cataracts. Biochim Biophys Acta. 1860:234–239. 2016.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Nguyen KT, Lee CS, Mun SH, Truong NT, Park
SK and Hwang CS: N-terminal acetylation and the N-end rule pathway
control degradation of the lipid droplet protein PLIN2. J Biol
Chem. 294:379–388. 2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Nguyen KT, Mun SH, Lee CS and Hwang CS:
Control of protein degradation by N-terminal acetylation and the
N-end rule pathway. Exp Mol Med. 50:1–8. 2018.PubMed/NCBI View Article : Google Scholar
|