Introduction
Although rapid progress has been made in the
development of treatment strategies for acute lymphoblastic
leukemia (ALL) over the past decades, prognosis for patients with
relapsed or treatment-refractory ALL remains poor (1,2). The
bone marrow microenvironment serves a critical role in protecting
leukemic cells against chemotherapy (3-8),
with cell adhesion-mediated drug resistance (CAM-DR) being one of
the underlying mechanisms (9-12).
Integrins belong to a family of glycoprotein cell surface receptors
that are comprised of two subunits, α and β (13), which primarily mediates cell
adhesion in the extracellular matrix (ECM). The integrin α4 chain,
also known as CD49d, non-covalently associates with the β1 integrin
chain, also known as CD29, to form very-late-antigen-4 (VLA-4)
(14). VLA-4 in turn binds to
counter receptors, including vascular cell adhesion molecule-1
(VCAM-1), fibronectin and osteopontin (14). During normal development and
hematopoiesis, α4 is crucial for homeostasis, regeneration and the
homing of hematopoietic stem and progenitor cells in the bone
marrow (15,16). In particular, integrin α4 is
expressed in B-ALL cells (17) and
has been previously implicated in CAM-DR which serves a key role in
tumor microenvironment (7), since
the binding of α4 to its ligand VCAM-1 mediates signaling to
maintain the survival of leukemic cells in the presence of
chemotherapy (8,18,19).
In previous study, its was shown that natalizumab
(NZM), a humanized anti-integrin α4 monoclonal antibody, can cause
the de-adhesion of B-ALL cells from VCAM-1, thus sensitizing ALL
cells to chemotherapy (20).
However, to date there have been no clinically approved agents that
target α4 for the treatment of B-ALL. Previously, a non-peptidic
small molecule α4 inhibitor, TBC3486, which is a small molecule
mimetic of VCAM-1, was tested in mice with primary ALL, which
prolonged the survival of the mice (21). However, TBC3486 remain unavailable
in an oral formulation and no in-depth preclinical studies have
been previously conducted. AVA4746 is an analog of TBC3486 that is
also a small molecule mimetic of VCAM-1 (22,23).
Notably, AVA4746 was found to be well tolerated as an oral
formulation in previous human clinical trials conducted (22,23).
AVA4746 is ~five times more potent in dislodging VLA-4 from VCAM-1
compared with TBC3486, according to in vitro cell assays
(unpublished data from Aviara Pharmaceuticals, Inc.). In the
present study, the in vitro competitive ligand-binding and
anti-adhesive properties of AVA4746 were evaluated. In addition,
its chemotherapy sensitization effects in vivo by reversing
CAM-DR in B-ALL cells were also assessed.
Materials and methods
Patient samples and cell lines
As described previously (20,24-26)
bone marrow cells were obtained from patients with B-ALL after
informed consent was obtained under Institutional Review Board of
Children's Hospital Los Angeles approved protocols. Primary B-ALL
blasts were obtained from the bone marrow aspirates by separation
using Ficoll (Cytiva) gradient centrifugation of 450 x g at 20˚C
for 30 min then pellets were harvested. Harvested mononuclear cells
(0.01-1x106/200 µl PBS/mouse) were intravenously
injected into
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG)
female mice (Jackson Laboratory; total of 129 mice; age, 5-7 weeks;
weight, 17-24 g), which were conditioned with a single sub-lethal
dose of 250 cGy of whole body irradiation at 21-23˚C -room
temperature, and expanded. Mice had free access to food and water
and were housed at 21-23˚C room temperature, at 30-70% humidity and
with a 6:00 am-7:00 pm light cycle. The mice were sacrificed using
CO2 at 30% air displacement rate. Engrafted human
leukemia cells were harvested from the bone marrow and spleen 2-26
weeks after injection, before they were cultured at 37˚C with 5%
CO2 further in the presence of murine stromal OP-9 cells
(American Type Culture Collection, ATCC) in vitro. Primary
B-ALL cells (LAX7, LAX7R, LAX53, LAX56, TXL3 and ICN24 from the Lab
of Dr. Yong-Mi Kim) were co-cultured with OP-9 stromal cells in
MEM-α (Invitrogen; Thermo Fisher Scientific, Inc.) supplemented
with 20% fetal bovine serum (FBS; Invitrogen; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin. The B cell lineage-ALL (B-ALL) cell lines were all
incubated at 37˚C with 5% CO2. TOM-1 (DSMZ) and BV173
(DSMZ) were cultured in 80% RPMI-1640 (Invitrogen; Thermo Fisher
Scientific, Inc.) + 20% FBS. SupB15 (ATCC) was cultured in 80% IMDM
(Invitrogen; Thermo Fisher Scientific, Inc.) + 20% FBS. REH (ATCC),
RS4;11 (ATCC), Kasumi-2 (DSMZ), 697 (DSMZ), BEL1 (DSMZ), RCH (DSMZ)
were in cultured in 90% RPMI 1640 + 10% FBS. Cytogenetic
information and α-integrin expression percentages of all B-ALL cell
lines used for the present study are shown in Table SI (25,26).
Human umbilical vein endothelial cells (HUVECs; Thermo Fisher
Scientific, Inc.) were cultured in Medium 200 supplemented with
0.2% LSGS (Invitrogen; Thermo Fisher Scientific, Inc.) at 37˚C with
5% CO2.
Compounds
AVA4746 and TBC3486 were obtained from Aviara
Pharmaceuticals, Inc. for research use. MG132 was obtained from
Selleck Chemicals. Vincristine, Dexamethasone and L-asparaginase
were obtained from Children Hospital of Los Angeles.
Flow cytometry
After harvesting, B-ALL cells or HUVECs
(0.1-1x106) were centrifuged at 500 g for 5 min at room
temperature and resuspended in 100 µl PBS containing
anti-human CD19 (#302206, 1:20 dilution, Biolegend, Inc.),
anti-human CD49d (#304304, 1:20 dilution, Biolegend, Inc.) and
anti-human CD29 (#303008, 1:20 dilution, Biolegend, Inc.)
antibodies, alongside the isotype control mouse IgG1κ (Biolegend,
Inc.). Following incubation at 4˚C for 30 min, cells were washed
with 1 ml PBS and resuspended in 100 µl DAPI/PBS (0.1µg/ml)
for 15 min at 4˚C for assessment using a flow cytometer (BD
FACSCanto II, BD Biosciences). Flow cytometry data were analyzed
from the viable cells in the DAPI-negative population, based on an
isotype gating strategy using Flow Jo (version 10, BD
Biosciences).
For apoptosis assay, B-ALL cells were treated for 48
h with either DMSO or AVA4746 25 µM, with or without VDL
chemotherapy (vincristine 5 nM, dexamethasone 0.05 nM,
L-asparaginase 2.5x10-3 IU/ml) in the presence or
absence of OP9. Annexin V (#640947, 1:20; Biolegend, Inc.) and DAPI
were used to stain the 1x106 cells for 15 min at room
temperature (25˚C), in the dark.
VLA-4 conformation activation
assay
As previously described (27,28),
LAX7R or TXL3 cells were suspended in the HEPES buffer (110 mM
NaCl, 10 mM KCl, 10 mM glucose, 1 mM MgCl2, 1.5 mM CaCl2 and 30 mM
HEPES, pH 7.4 containing 0.1% HSA Sigma-Aldrich; Merck KGaA) at a
density of 1x106 cells/ml. B-ALL cells were then
incubated with either AVA4746 of 0.00, 0.01 µM, 0.1
µM, 1 µM, 10 µM, or 100 µM or human
VCAM-1 (PeproTech, Inc.) of 0 µM, 1.35 µM, 13.5
µM, 135 µM, or 1350 µM in the presence of 10%
phycoerythrin (PE)-conjugated HUTS-21 antibodies (#556049, BD
Biosciences) with or without 5mM Mn2+ for 30 min at
37˚C. Subsequently, 0.5 µl DAPI (200 µg/ml) was added to
each reaction 5 min before the end of the incubation time. The
PE-HUTS-21 mean fluorescence intensity (MFI) was analyzed by flow
cytometry (BD FACSCanto II, Flow Jo version 10, BD Biosciences).
Receptor occupancy (RO) assays are designed to quantify the binding
of therapeutics to their targets on a cell's surface (29). The protocol used for calculating
the normalized VLA-4 RO was as follows: The maximal MFI value from
highest concentration of AVA4746 or VCAM-1 was normalized as 100%,
whilst the minimum MFI value from lowest concentration of AVA4746
or VCAM-1was normalized as 0%. EC50 of RO (half maximal occupancy
of VLA-4 receptor concentration) was calculated using GraphPad
Prism 5 software. To determine a level of non-specific binding,
cells were stained in parallel with the 10% isotype control
antibody (PE Mouse IgG2a, κ Isotype Control, #555574, BD
Biosciences).
Cell adhesion and de-adhesion assay. For the cell
adhesion assay, non-tissue culture 96-well plates were coated with
10 µg/ml human VCAM-1 (PeproTech, Inc.) overnight at 4˚C,
washed and blocked with 2% BSA (Sigma-Aldrich; Merck KGaA) for 30
min at room temperature. LAX7R or TXL3 cells
(1x106/well) were incubated with 0.00, 0.01 µM,
0.1 µM, 1 µM, 10.00 or 100 µM AVA4746 at room
temperature for 1 h, added into the plates pre-coated with VCAM-1
and were allowed to adhere for 1 h at 37˚C. Non-adherent cells were
removed before being gently washed once by PBS, whilst adherent
cells were counted using a hemocytometer following Trypan Blue
(Invitrogen; Thermo Fisher Scientific, Inc.) exclusion.
Cell de-adhesion assays were performed
as previously described (20,21)
Similarly, B-ALL cells (1x106/well) were
seeded into the plates pre-coated with 10 µg/ml human VCAM-1
or OP9 cells for 4 h at 37˚C in 5% CO2. Next, either
AVA4746 (25 µM) or DMSO was added as a vehicle control and
the cells were incubated at 37˚C with 5% CO2 overnight.
The non-adherent cells in the supernatant were then removed,
whereas alive adherent cells were detached by pipetting with PBS
prior to counting by using Trypan Blue exclusion of dead cells with
a hemocytometer. Finally, the percentage of adhesion was calculated
by dividing the number of live adherent cells by the total number
of cells.
Western blot analysis
B-ALL cells were treated with AVA4746 (0,1 µM, 5
µM, or 25 µM) for 24 h or 96 h for on-target effect
experiments. Furthermore, following 2 h pre-treated with MG132 0 µM
or 1 µM B-ALL cells were treated with AVA4746 either 0 µM or
25 µM for 96 h as separate experments. B-ALL cells were harvested
and lysed in M-PER™ Mammalian Protein Extraction Reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) containing a 1%
protease inhibitor cocktail (VWR International, LLC). Protein
concentration was measured using Bradford protein assay. Protein
lysates (20 µg/lane) were separated in 4-12% Bis-Tris protein gels
by SDS-PAGE and transferred onto PVDF membranes. Then the membranes
were blocked by 5% dry fat milk for 1 h at room temperature. The
following primary antibodies were incubated overnight at 4˚C:
Anti-integrin α4 (#4600, 1:1,000; Cell Signaling Technology, Inc.),
anti- phosphorylated (p-)-AKTSer473 (#9271, 1:1000; Cell Signaling
Technology, Inc.), anti-AKT (#9272, 1:1,000; Cell Signaling
Technology, Inc.), anti-phosphotyrosine (#05-321MG, 1:1000
dilution, EMD Millipore) and anti-β-actin (#sc-47778, 1:2000
dilution, Santa Cruz Biotechnology, Inc.). Secondary antibody
solution Alk-Phos. conjugated (anti-rabbit #WP20007 or anti-mouse
#WP20006; Thermo Fisher Scientific, Inc.) was applied at room
temperature for 1 h. To visualize bands, chemiluminescent substrate
(#WP20002, Thermo Fisher Scientific, Inc.) was used. Western blots
quantify was analyzed via Image J (version 1.53e, National
Institutions of Health).
Starvation and activation assay for
the detection of phosphorylated proteins
B-ALL cells were first serum-starved by being washed
twice with Dulbecco's PBS (DPBS) and cultured in MEM-α media at
37˚C and 5% CO2 overnight. Following another wash with
DPBS, B-ALL cells were treated with either the vehicle control 0.1%
DMSO or AVA4746 (5 or 25 µM) for 30 min at room temperature.
Subsequently, FBS was added to a final concentration of 20% to all
cells except for those in the no-activation control groups. In
either the human VCAM-1 or OP9 group, 1x106 cells were
seeded into plates pre-coated with either human VCAM-1 (10
µg/ml) or OP9 (0.2x106 cells/well on 12-well
plate were pre-coated at 37˚C for 1 day before experiment). Whole
cell lysates were isolated after incubation for either 1 h or 24 h
at 37˚C for use in western blot analysis for either p-AKT or
phosphotyrosine detection.
In vivo leukemia cell mobilization
study
Mobilization of leukemia cells was tested according
to a previously described protocol (20). Primary B-ALL LAX7R cells
(0.05x106 in 200 µl PBS per mouse) were intravenously
injected into in total of 14 NSG mice (7 mice in PBS group while 7
mice in AVA4746 treatment group). In total, 80 µl peripheral
blood was collected each mouse by retroorbital exsanguination
following 2% isoflurane anesthesia once a week from weeks 2 to 4
after the injection of LAX7R cells. The total duration of the blood
sampling protocol was typically 3-6 min. When ~0.5 or ~2.5%
hCD45+ hCD19+ LAX7R cells were detected by
flow cytometry in the peripheral blood (PB) of NSG mice, the mice
were treated with either PBS or AVA4746 (60 mg/kg) by
intraperitoneal (i.p.) injection. Following 100 µg/ml
purified human IgG (#I2511, Sigma-Aldrich, Inc.) as blocking
reagent for elimination of non-specific binding, 0.1x106
cells were stained with APC-hCD45 (#304012, 1:20 dilution,
BioLegend, Inc.), PE-hCD19(#302254, 1:20 dilution, BioLegend,
Inc.), FITC-mCD45 (#103108, 1:20 dilution, BioLegend, Inc.), and
APC-Cy7 (#304328, 1:20 dilution, BioLegend, Inc.) at 4˚C in dark
for 30 min. Subsequently, 8 or 36 h after treatment, the mice were
sacrificed using CO2 at 30% displacement rate. Following
animal sacrifice whole mononuclear cells were isolated from the PB,
bone marrow (BM) and spleen (SPC) using the ACK lysing buffer
(Invitrogen; Thermo Fisher Scientific, Inc.) at either 8 or 36 h
after treatment. Alive mononuclear cell (MNC) numbers were counted
with a hemocytometer using trypan blue exclusion, whilst the
percentages of either mCD45+ or
hCD45+hCD19+ cells were determined by flow
cytometry (BD FACSCanto II, Flow Jo version 10; BD Biosciences).
Therefore, number of mouse cells or leukemia cells was calculated
as MNC number x percentage of mCD45+ or
hCD45+hCD19+ for mouse and human leukemia
cells, respectively. This animal study was performed in compliance
with a research protocol approved by the Institutional Animal Care
and Use Committee at the Saban Research Institute of Children's
Hospital Los Angeles.
Endothelial tube formation assay
A Matrigel-coated 48-well plate (#354508,
Biocoat™; BD Biosciences) was first pre-warmed at 37˚C
for 30 min. HUVECs (4-6x104/well) that were treated with
AVA4746 (0, 5 or 25 µM) or TBC3486 (0, or 25 µM) at
37˚C for 30 min were seeded into each well and incubated for 4-6 h
at 37˚C in 5% CO2. After incubation, images of three
representative fields of view per well were obtained using phase
contrast light microscopy (x100 magnification), before being
analyzed using the ‘angiogenesis analyzer’ tool (Gilles Carpentier)
in ImageJ (30) (version 1.53e,
National Insititutes of Health). Tube formation was assessed by
analyzing the number of nodes (pixels with ≥ 3 neighboring elements
corresponding to a bifurcation), segments (elements delimited by
two junctions), meshes (areas enclosed by segments or master
segments and made by tube-like structures) and total area, before
being quantified.
In vivo survival study
Primary B-ALL cells (0.05x106) were
intravenously injected into each NSG mouse. In total of 91 mice
were divided into four treatment groups: PBS (5 mice in LAX7R
exp#1, 4 mice in LAX7R exp#2, 7 mice in RS4;11, 5 mice in TXL3),
AVA4746 group (5 mice in LAX7R exp#1, 5 mice in LAX7R exp#2, 7 mice
in RS4;11, 6 mice in TXL3), VDL group (6 mice in LAX7R exp#1, 5
mice in LAX7R exp#2, 6 mice in RS4;11, 6 mice in TXL3), and
VDL+AVA4746 group (6 mice in LAX7R exp#1, 5 mice in LAX7R exp#2, 8
mice in RS4;11, 5 mice in TXL3). AVA4746 (15 or 30 mg/kg dissolved
in PBS) was then administered by oral gavage twice a day
continuously for 14, 21 or 28 days, with treatment starting 2 days
after B-ALL cell injection. Subsequently, starting at 3 days after
B-ALL cell injection, vincristine (0.5 mg/kg) was administered
(i.p.) once a week for 2 or 4 weeks, whilst dexamethasone (10.5
mg/kg) and L-asparaginase (1500 IU/kg) were administered (i.p.)
once a day (but not at weekends) for 2 or 4 weeks. Animals were
monitored daily and the survival time of the mice was then
recorded. Animals were sacrificed by CO2 euthanasia at
an air displacement rate of 30% if >15% weight loss was observed
or physical changes, including loss of ambulation, were
observed.
Statistical analysis
Statistical significance was assessed using a
two-tailed Student's t-test and the data are presented as the mean
± SD. One-way analysis of variance (ANOVA) with Tukey's post hoc
test was used for comparisons of > two groups. Survival data
were analyzed by Kaplan-Meier survival curves and comparisons were
performed using the log-rank test for two groups without
adjustment. P<0.05 was considered to indicate a statistically
significant difference. All statistical analysis was performed
using GraphPad Prism 5 software (GraphPad Software, Inc.). At least
two of experimental repeats for in vitro assays.
Results
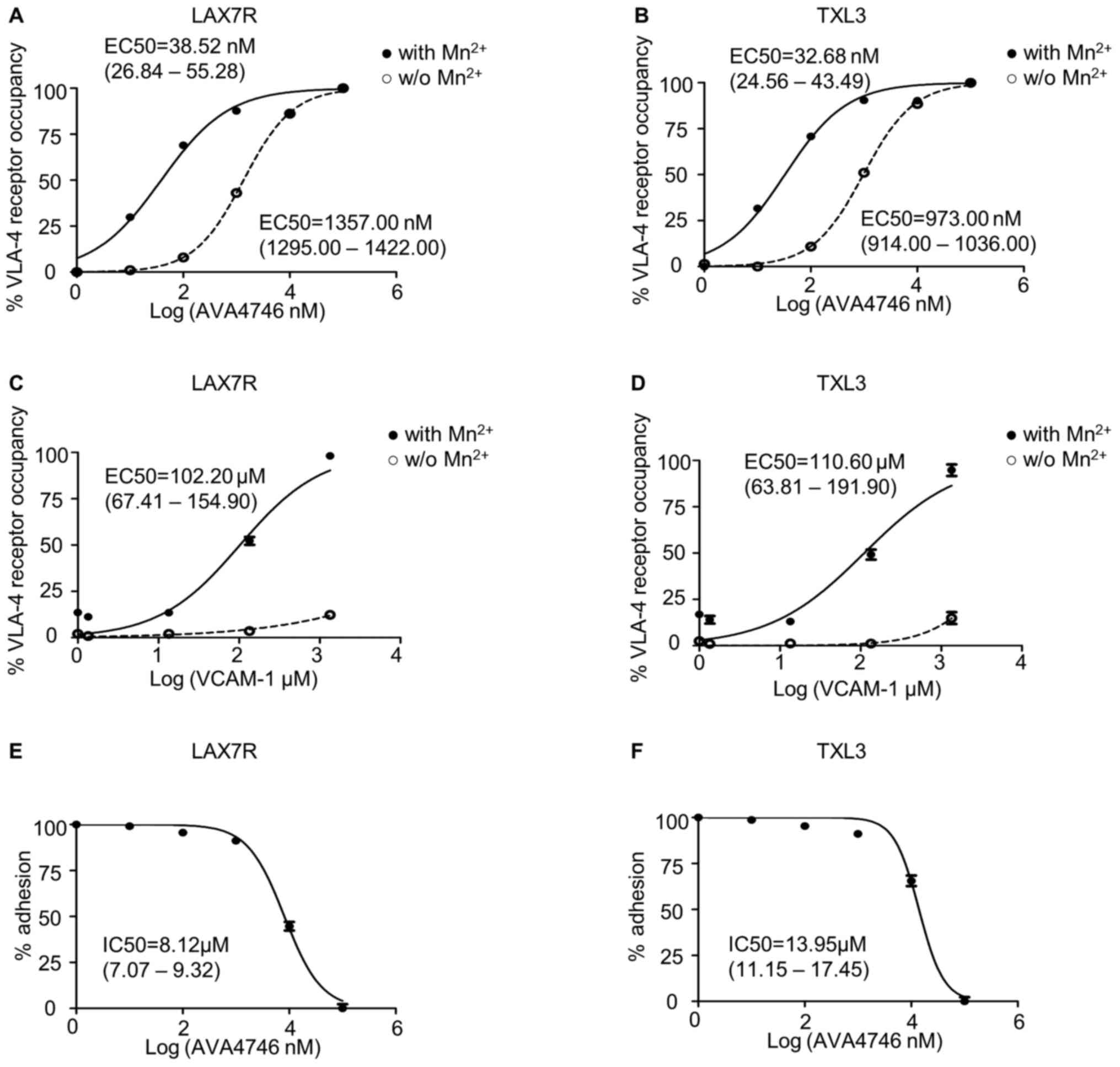
AVA4746 binds VLA-4 on B-ALL cells
with high affinity and efficiently blocks ligand-binding with
VCAM-1
Since AVA4746 is a small molecule mimetic compound
of VCAM-1(23), conformation
activation experiments were performed to determine the half maximal
effective concentration (EC50) of AVA4746. HUTS-21 is a specific
monoclonal antibody that targets an epitope corresponding to the
active conformation of integrin β1(31). Anti-HUTS-21 antibody binding would
occur when VLA-4 binds to certain ligands, such as VCAM-1(31). The HUTS-21 antibody binding curve
represents the VLA-4 RO, which is used as an indicator of binding
affinity (31). In LAX7R cells,
the EC50 of AVA4746 was 38.52 nM (26.84-55.28 nM) and
1357.00 nM (1295.00-1422.00 nM) in the presence (Fig. S1C) or absence of 5 mM
Mn2+ (Fig. S1A),
respectively (Fig. 1A). In TXL3
cells, the EC50 of AVA4746 was 32.68 nM (24.56-43.49 nM)
and 973.00 nM (914.00-1036.00 nM) in the presence (Fig. S1C) or absence of 5 mM
Mn2+ (Fig. S1A),
respectively (Fig. 1B). By
contrast, the EC50 of human VCAM-1 was 102.20 µM
(67.41-154.90 µM) and 110.60 µM (63.81-191.90
µM) in the presence of 5 mM Mn2+ in LAX7R and
TXL3 cells, respectively (Figs.
1C, D and S1D). There was little conformation
changes corresponding to VLA-4 activation by human VCAM-1 without 5
mM Mn2+ (Figs. 1C,
D and S1B). The MFI of PE-HUTS-21 shown in
Table SII are from two
independent experiments performed in triplicate. Taken together,
AVA4746 exhibits nanomolar levels of affinity for the active
conformation of VLA-4, which was ~3,500-fold higher compared with
that of VCAM-1 in the presence of Mn2+.
Other soluble integrin ligands present in the serum
may interfere with VLA-4 ligand binding (31), since the primary B-ALL cells were
cultured in MEM-α in the presence of 20% FBS. Therefore, VCAM-1
cell adhesion assays were used to determine the half maximal
inhibitory concentration (IC50) of AVA4746. The IC50 of
AVA4746 was 8.12 (7.07-9.32 µM) and 13.95 µM
(11.15-17.45 µM) in LAX7R and TXL3 cells, respectively, in
one out of the two independent experimental repeats (Fig. 1E and F). The percentages of adhesion from the
two independent experiments are shown in Table SIII. To achieve a saturating
competitive effect, 5 and 25 µM AVA4746 were used for
subsequent experiments.
AVA4746 decreases cell surface and
total intracellular expression of integrin α4
Primary B-ALL cells derived from four human patients
(LAX7R, LAX7, LAX53 and LAX56) and the B-ALL cell line REH were
used to evaluate the on-target effect of AVA4746 on B-ALL in
vitro. Cells were treated with either AVA4746 (1, 5 and 25
µM) or 0.1% DMSO as a control for either 24 or 96 h, before
they were analyzed by flow cytometry to detect human integrin α4
expression. The MFI of integrin α4 on the B-ALL cells was decreased
by AVA4746 in a dose-dependent manner, whilst the MFIs of integrins
α5 and α6 were not affected (Figs.
2A-E and S2). AVA4746 also
caused a reduction in the protein expression levels of α4 as
determined by western blot analysis, after 24 and 96 h of treatment
(Fig. 2F). Subsequently, the
effects of proteasomal degradation on this decrease in integrin α4
protein expression were subsequently tested. B-ALL cells were
treated for 96 h with either AVA4746 (25 µM), MG132 (1
µM) or DMSO as a vehicle control, alone or in combination.
α4 was partially restored by MG132 in all cases apart from LAX56
(Fig. S3.). These results suggest
that this decrease in α4 expression induced by AVA4746 may also be
independent of the ubiquitination-proteasome pathway. In addition,
AVA4746 slightly downregulated the phosphorylation levels of AKT
following stimulation with human VCAM-1 in serum-starved LAX7R
cells, but no obvious changes were observed following AVA4746
treatment after stimulation with serum (Fig. S4A). Similar tendency was found in
LAX56 (Fig. S4A). These results
suggest that other cell signaling pathways may be involved
(Fig. S4A), since AVA4746 may
also affect phosphotyrosine cell signaling in LAX53 cells (Fig. S4B). Clear decreased band densities
were found in both 5 µM and 25 µM AVA4746 compared
with 0 µM in 1 h treatment group around 50-60 kDa.
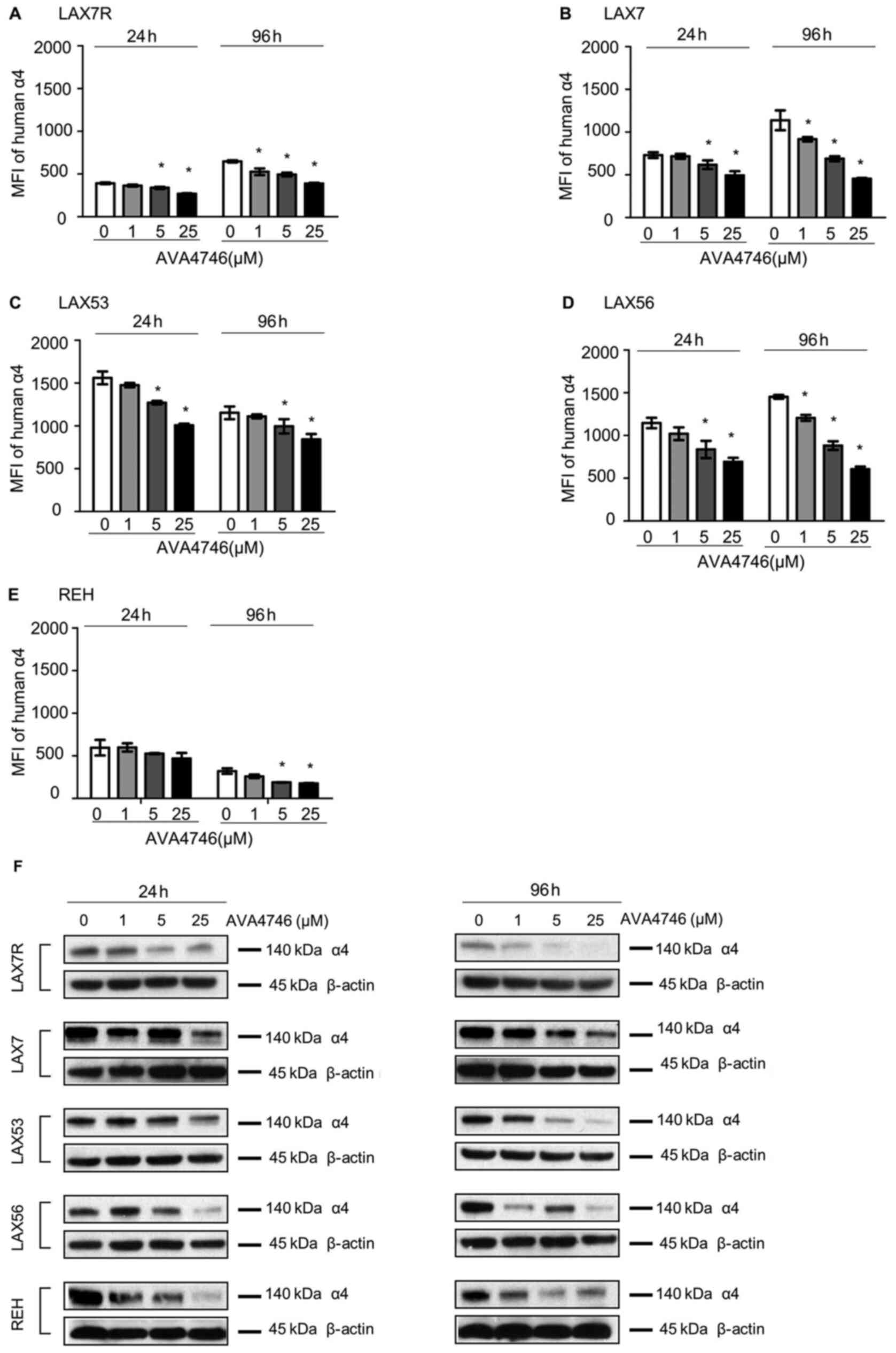 | Figure 2AVA4746 decreases cell surface and
total integrin α4 expression. MFI representing the expression
levels of integrin α4 on the (A) LAX7R, (B) LAX7, (C) LAX53, (D)
LAX56 and (E) REH cell surfaces 24 and 96 h after treatment with 1,
5 and 25 µM AVA4746, with 0 representing the DMSO control.
*P<0.05 vs. 0. Experiments were performed in
triplicates. (F) B-ALL cell lines LAX7R, LAX7, LAX53, LAX56 and REH
were treated for 24 and 96 h with 1, 5 and 25 µM AVA4746, with 0
representing the DMSO control. Following 24 and 96 h treatment with
AVA4746, the integrin α4 protein levels at 140 kDa in LAX7R, LAX7,
LAX53, LAX56 and REH cells were analyzed by western blotting, with
β-actin being used as lthe oading control. MFI, mean fluorescence
intensity; α4, α4 integrin. |
AVA4746 detaches B-ALL cells from
VCAM-1
To investigate the competing ligand effect, the
de-adhesion assays were performed as previously described (21). Five types of primary B-ALL cells,
namely LAX7R, LAX53, LAX56, TXL3 and ICN24 cells (Fig. 3A-E), along with seven B-ALL cell
lines, namely RS4;11, SupB15, Kasumi-2, 697, BEL-1, BV173 and RCH
cells (Fig. 3F-L; Table SI), were plated onto either human
VCAM-1- (10 µg/ml) or 2% bovine serum albumin (BSA)-coated
plates, before being treated with either DMSO or 25 µM
AVA4746 overnight. All 12 types of B-ALL cells showed significant
de-adhesion from the VLA-4 ligand, human VCAM-1, after AVA4746
treatment compared with that in the DMSO control (Fig. 3). In addition, AVA4746 was found to
induce the detachment of eight B-ALL cell lines from the OP9
stromal cell line (Fig. S5A-H).
Since the murine calvaria-derived stromal cell line OP9 has been
found to facilitate survival and drug resistance in B-ALL cells
(20), Annexin V assays were
subsequently performed to determine the apoptotic effects of
AVA4746 in combination with the chemotherapeutic regimen of 0.5
mg/kg vincristine, 10.5 mg/kg dexamethasone and 1500 IU/kg
L-asparaginase (VDL). The small but significant induced more
apoptosis were found in VDL+AVA4746 compared with VDL alone in TXL3
(without OP9, Fig. S6A), LAX56
(without OP9, Fig. S6B) and LAX7R
(with OP9, Fig. S6C). However,
neither AVA4746 alone nor AVA4746 in combination with VDL induced
noTable apoptotic effects in B-ALL cells compared with that in the
corresponding groups that were not treated with AVA4746, regardless
of the presence of OP9 cells (Fig.
S6A-C).
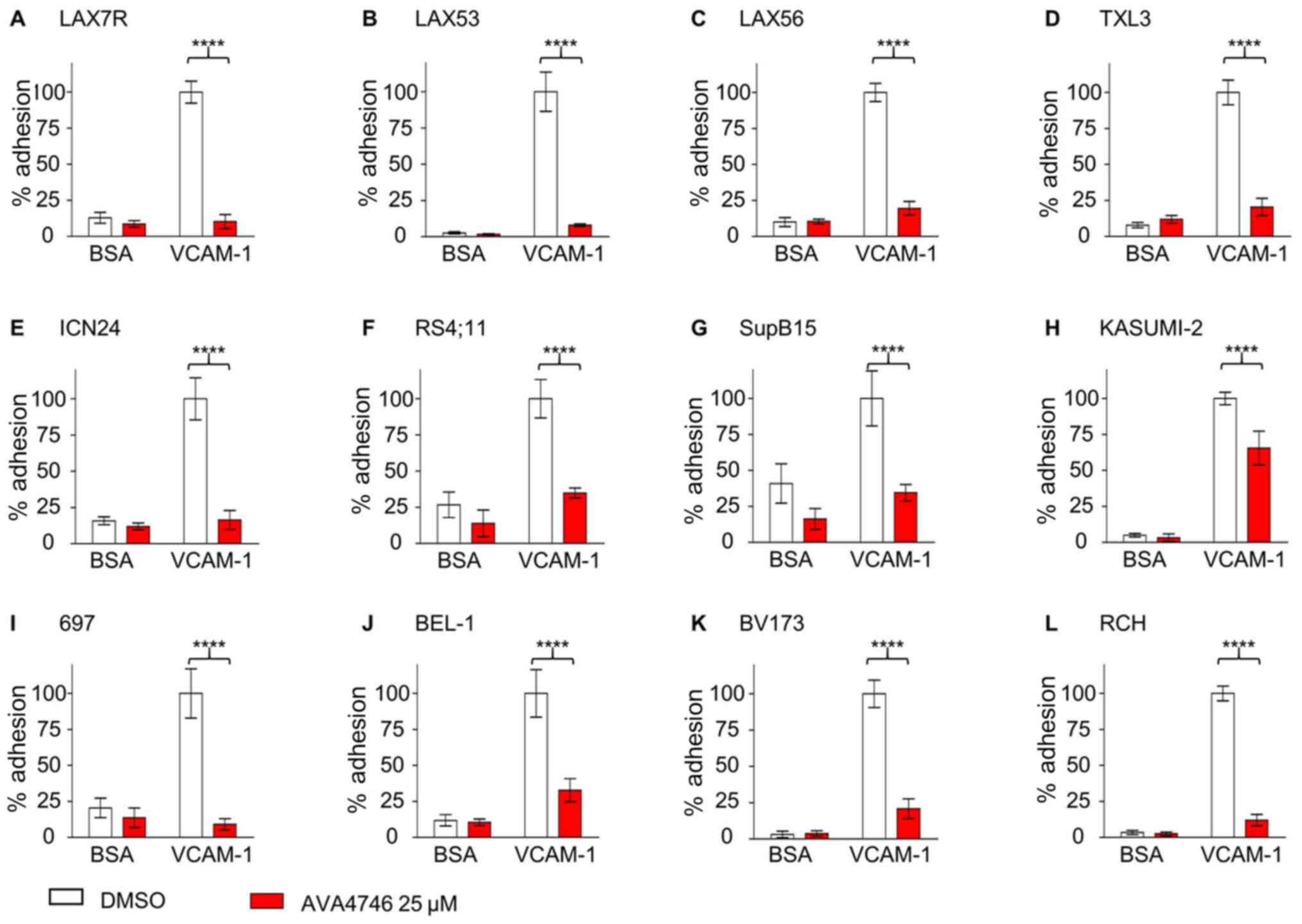 | Figure 3AVA4746 causes the detachment of
B-ALL cells from human VCAM-1. Primary B-ALL cell lines (A) LAX7R,
(B) LAX53, (C) LAX56, (D) TXL3 and (E) ICN24, in addition to B-ALL
cell lines (F) RS4;11, (G) SupB15, (H) KASUMI-2, (I) 697, (J)
BEL-1, (K) BV173 and (L) RCH were seeded into 96-well plates coated
with either human VCAM-1 (10 µg/ml) or 2% BSA as a control for 4 h
prior to treatment with the DMSO control or AVA4746 (25 µM)
overnight. The percentage (%) of viable adherent cells was
calculated based on the cell counts using trypan blue exclusion
which was then normalized to those in the VCAM-1-DMSO group.
****P<0.0001. Experiments were performed in
triplicates. VCAM-1, vascular cell adhesion molecule-1; B-ALL,
B-lineage acute lymphoblastic leukemia. |
AVA4746 moderately inhibits the
migration of LAX7R cells from BM to the peripheral blood
To determine the role of integrin α4 in B-ALL cell
metastasis, mobilization assays were performed. In brief, AVA4746
treatment was initiated when detecTable B-ALL cells could be
observed in PB. Subsequently, mice were sacrificed before the PB,
BM and SPC were harvested for analysis. When ~2.5% LAX7R cells was
detected in the PB (Fig. S7A),
mice were treated with either AVA4746 (60 mg/kg; n=6), PBS as a
control (n=6), VDL (n=6) or VDL + AVA4746 (n=6).
After 8 h of treatment, PB, BM and SPCs were
harvested for a human leukemic population analysis using flow
cytometry. There was an insignificant but decreasing tendency of
leukemia (% of hCD45+hCD19+) in the PB
collected from the AVA4746 group compared with that in the PBS
group (Fig. S7B), whilst there
was no significant difference between BM and SPCs (Fig. S7C and D). The percentage of leukemic cells
(hCD45+hCD19+) was significantly decreased in
both VDL and VDL + AVA4746 groups compared with that in the PBS
groups, but there was no statistical difference between that in the
VDL and VDL + AVA4746 groups (Fig.
S7B). Additionally, a significant decrease in the human α4 MFI
under the leukemic population (hCD45+hCD19+)
was exclusively found in AVA4746 groups compared with that in the
PBS group in the BM samples (Fig.
S7C).
The timeframe of the initial treatment was
subsequently optimized, so that when the leukemia cell
(hCD45+hCD19+) percentage in the PB was 0.5%,
the duration of AVA4746 treatment was prolonged to 36 h (Fig. 4A). Consequently, AVA4746
significantly decreased the percentage of B-ALL cells
(hCD45+hCD19+)in the PB compared with that in
the PBS control (Figs. 4B and
S8A) whilst significantly
increasing that of B-ALL cells
(hCD45+hCD19+)in the BM (Figs. 4C and S8B). No changes were found in the SPC
samples (Figs. 4D and S8C).
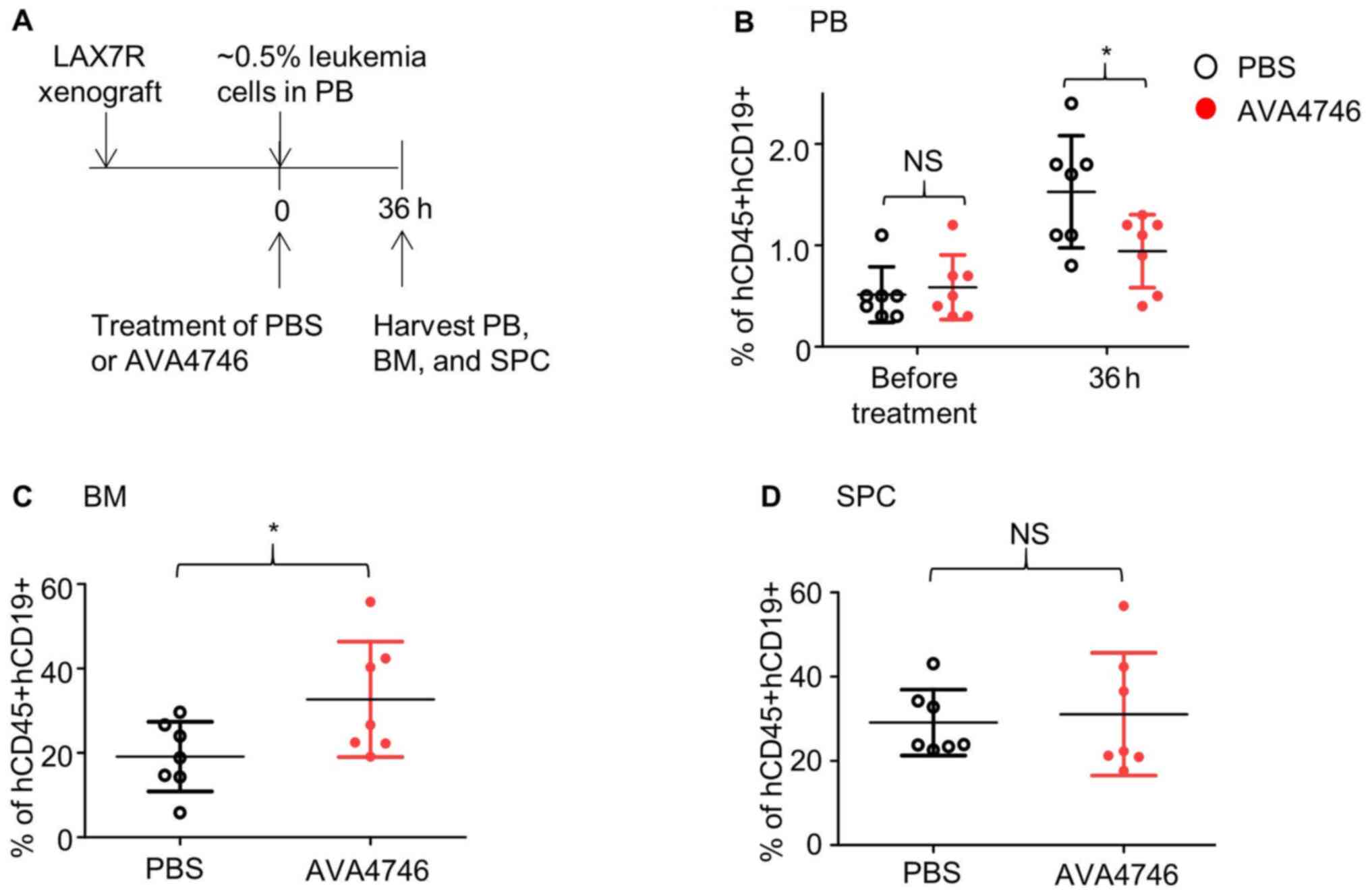 | Figure 4AVA4746 decreases the number of human
B-ALL cells in the mouse peripheral blood samples whilst increasing
the number of ALL cells in the bone marrow. (A) Experimental design
of the in vivo leukemic cell mobilization assay. When 0.5%
of human CD45/CD19 double-positive LAX7R cells were detected by
flow cytometry in the PB of the NSG mice, mice were treated with
either PBS (n=7) or 60 mg/kg AVA4746 (n=7). Mononuclear cells were
isolated from the PB, BM and SPC 36 h after treatment. Percentages
of hCD45+hCD19+cells in the (B) PB, (C) BM
and (D) SPC are shown. *P<0.05. PB, peripheral blood,
BM, bone marrow; SPC, spleen; NSG,
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; NS,
no significant; B-ALL, B-lineage acute lymphoblastic leukemia. |
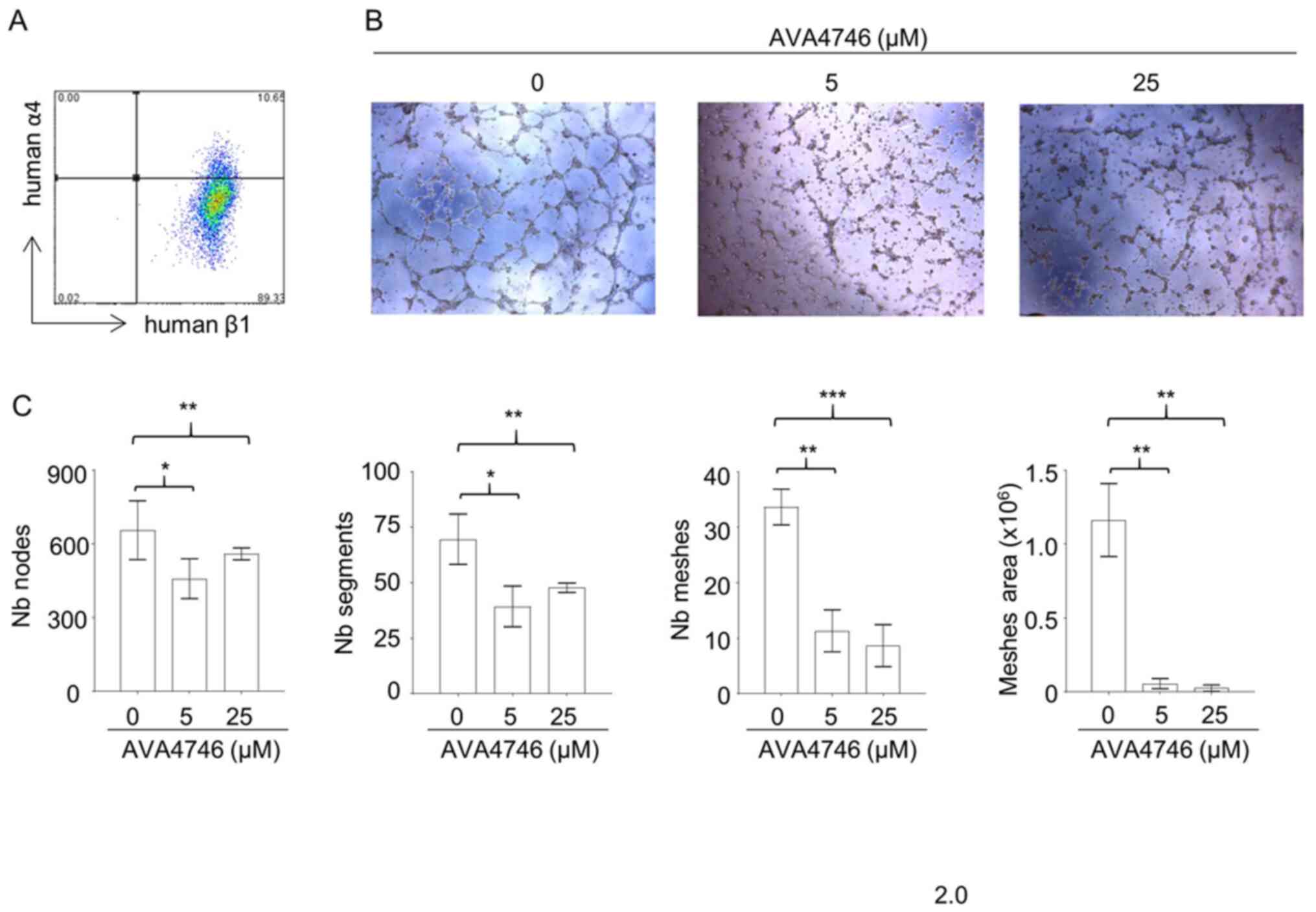
AVA4746 inhibits in vitro
angiogenesis
Results from the in vivo mobilization assays
suggest that AVA4746 can cause the retention of B-ALL cells in the
BM. Since integrin α4 and β1 were found to be expressed on
endothelial cells (Fig. 5A), it
was hypothesized that the underlying mechanism may be due to the
AVA4746-mediated inhibition of α4 on endothelial cells. This may in
turn reduce vessel formation and reduce the release of B-ALL cells
into the peripheral circulation. To test this hypothesis, the
potential effects of AVA4746 on HUVECs were evaluated in
vitro. AVA4746 signficantly attenuated the in vitro
angiogenic potential of HUVECs. Representative images of HUVECs in
Fig. 5B demonstrate that the
number of nodes, segments, meshes and total meshes was all
significantly decreased by both 5 and 25 µM AVA4746
(Fig. 5C). Similar results were
found using TBC3486, an analog of AVA4746 (Fig. S9). However, there were no
statistical differences in angiogenesis inhibition using NZM (data
not shown).
Combination of AVA4746 and
chemotherapy treatment prolongs the survival of mice engrafted with
primary relapsed B-ALL
To further determine the effects of AVA4746 on the
survival of human B-ALL cell mouse xenograft models, LAX7R cells
(0.05x106 cells per mouse) were injected into the NSG
mice intravenously. Subsequently, 2 days post-injection, either
AVA4746 or PBS was administered by oral gavage at 15 mg/kg, twice a
day (30 mg/kg/day) for 14 days. VDL was given 3 days post-injection
for 4 weeks by i.p. (Fig. 6A). A
significant prolongation of overall survival in the group treated
with a combination of AVA4746 and VDL (n=6 MST; 78.5 days) was
found compared with that in the VDL-only group (n=6; MST, 68 days;
Fig. 6B). By contrast, there was
no difference between the PBS (n=5; MST, 38 days) and the AVA4746
groups (n=5; MST, 38 days; Fig.
6B). An increase in the AVA4746 dosage to 60 mg/kg/day and an
increase in the treatment time to 28 days did not result in an
improvement in survival time compared with AVA4746 30 mg/kg/day
(Fig. 6C). However, significantly
prolonged survival in the AVA4746 + VDL group was again observed
(n=5; MST, 87 days) compared with that in the VDL-only group (n=5;
MST, 77 days; Fig. 6D).
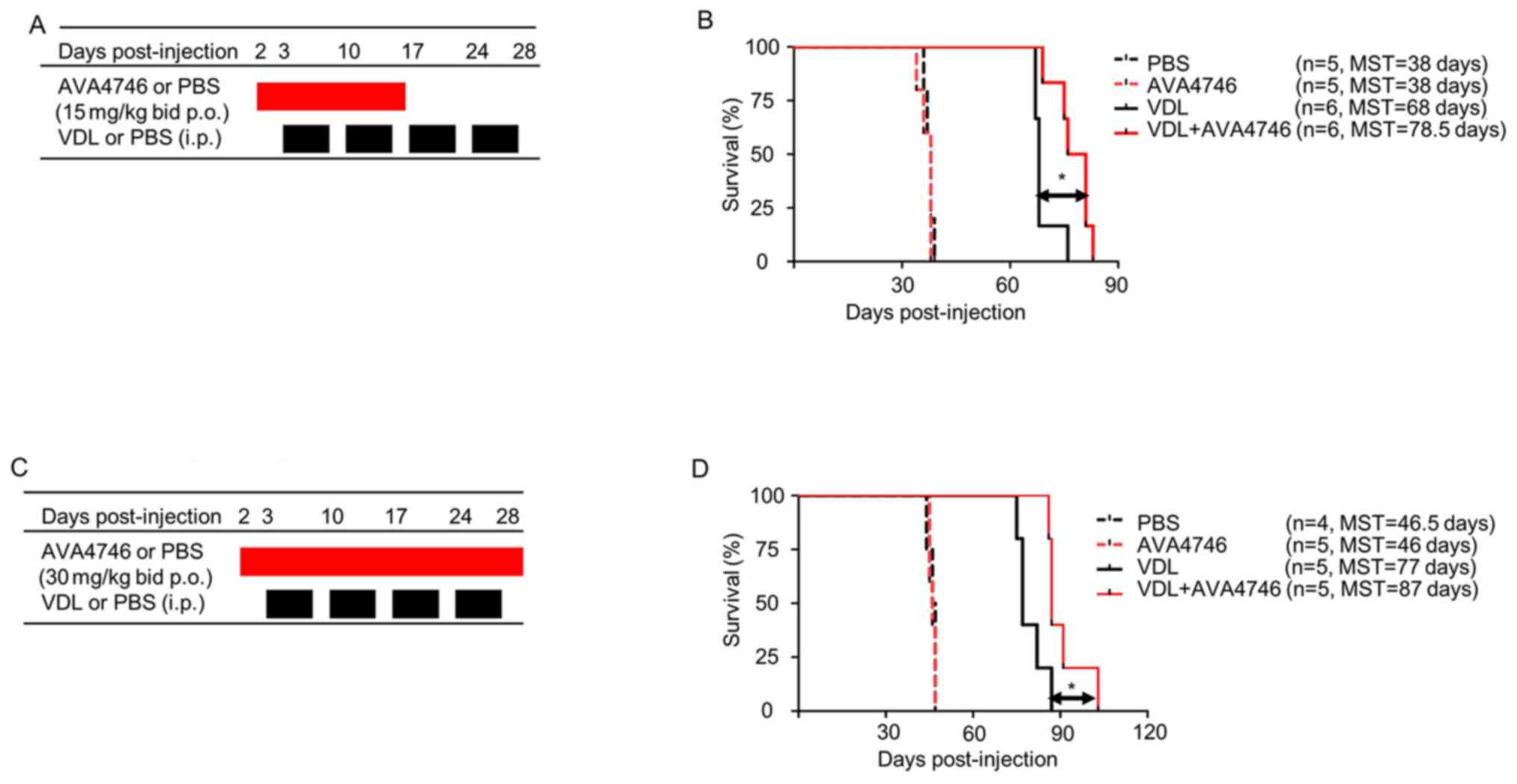 | Figure 6Combined AVA4746 and chemotherapy
treatment prolongs the survival of mice with primary relapsed
B-lineage acute lymphoblastic leukemia. (A) NSG mice were
intravenously injected with 50,000 LAX7R cells per mouse and then
treated with either AVA4746 or the PBS vehicle control (15 mg/kg by
oral gavage twice a day continuously for 14 days) 2 days after
injection with LAX7R cells. In addition, 3 days after LAX7R cell
injection, mice were treated with VDL alone (0.5 mg/kg vincristine
once a week, 10.5 mg/kg dexamethasone five times a week and 1500
IU/kg L-asparaginase five times a week for 4 weeks)
intraperitoneally or in combination with AVA4746 by oral gavage.
(B) Kaplan-Meier survival analysis for the experimental design
shown in (A). The MST was 38 days for both the PBS group (n=5) and
the AVA4746 group (n=5). The 68-day MST of the VDL-treated group
(n=6) was significantly different from the 78.5-day MST of the
AVA4746- and VDL-treated group (n=6). *P=0.01. (C) The
same protocol as that performed in (A) was conducted, except for
that the dose of AVA4746 was higher (30 mg/kg) and the treatment
time was longer (twice a day for 28 days). (D) Kaplan-Meier
survival analysis for the experimental design shown in (C). The MST
was 46.5 days for the PBS group (n=4), whilst the MST was 46 days
for the AVA4746 group (n=5). The 77-day MST of the VDL-treated
group (n=5) was significantly different compared with the 87-day
MST in the group treated with the combination of both AVA4746 and
VDL (n=5). *P=0.023. NSG,
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; MST,
mean survival time; VDL, vincristine, dexamethasone and L-
asparaginase; p.o., per os; i.p. intraperitoneal. |
In addition, mouse xenograft models were established
using cell lines derived from two other cases B-ALL (RS4;11 and
TXL3). AVA4746 in combination with VDL (n=8; MST, 105 days) did not
lead to a statistically significant improvement in the survival of
mice injected with RS4;11 cells in the VDL-alone group (n=6; MST,
95 days; Fig. S10A and B). By contrast, AVA4746 alone (n=6;
MST, 66 days) prolonged the survival of the TXL3 xenograft mice
compared with that in the PBS control (n=5, MST, 59 days; Fig. S10C and D). However, there were no changes in
the survival of mice in the group treated with AVA4746 + VDL
compared with VDL alone in the TXL3 model.
Discussion
Integrin α4 has long been of interest in the
hematology research field because it has been frequently reported
to be a key mediator of adhesion by leukemia cells to the
chemoprotective niche (18,20,21,32-34).
NZM is a humanized mouse monoclonal IgG4κ antibody that recognizes
the human α4 chain of both α4β1 and α4β7 integrin (35,36).
Although the Food and Drug Administration has approved NZM for the
treatment multiple sclerosis (MS), progressive multifocal
leukoencephalopathy can arise in patients treated with NZM
(37). In clinical trials,
antibodies against natalizumab have been reported in 11% patients
with MS and 7% patients with Crohn's disease (38,39).
This issue can be averted using small molecules. Previously, the
beneficial effects of a non-clinical grade small molecule
inhibitor, TBC3486(21), was
reported in a xenograft mouse model of primary B-ALL. Therefore, in
the present study, the effects of AVA4746, a small molecule
antagonist of VLA-4, was evaluated in primary B-ALL.
The epitopes that can be targeted by the HUTS-21
antibody, termed ligand-induced binding site epitopes, becomes
exposed as a result of the conformational change that occur
following activation, which can be exploited to discover novel
allosteric VLA-4 antagonists (27). Therefore, the
conformation-sensitive anti-CD29 HUTS-21 antibody was used to
measure the VLA-4 RO, which indicates ligand-binding affinity
measured in a scale ranging from 0 to 100%, as previously described
(31). VLA-4 RO is upregulated by
the divalent cation Mn2+ (31). AVA4746 was found to have a low
EC50 for VLA-4 RO compared with that of VCAM-1 without
Mn2+, suggesting that AVA4746 binds to integrin α4 with
higher affinity compared with VCAM-1. In addition, the prominent
detachment effect on B-ALL from VCAM-1 mediated by AVA4746 was
consistent with the effects of TBC3486 previously observed
(21). Therefore, AVA4746 may be a
useful tool for the studying VLA-4/VCAM-1 interactions, which are
important for cell-cell adhesion between B-ALL and stromal cells
(20).
Subsequently, it was demonstrated that AVA4746 can
specifically target human integrin α4 on the cell surface whilst
also downregulating the protein expression level of α4 in general.
Previous studies have demonstrated that integrins are predominantly
degraded through the endosome-lysosomal pathway (40,41).
However, data from the present study showed that there was little
or no degradative effects from the proteasome pathway, since
inhibiting the proteasome using MG132 only partially reversed the
downregulation of α4. In addition, there appeared to be no effect
mediated by AVA4746 on AKT signaling, which is one of the pathway
previously reported to be required stromal cell-mediated
chemotherapy protection (42).
However, changes in the phosphotyrosine levels were observed
following AVA4746 treatment, which warrants further study.
According to previous studies, VCAM-1 alone could not confer
resistance to chemotherapy, suggesting the requirement for stromal
cells capable of responding to activated leukemic cells (18,34).
Similar results were found in the present study, where AVA4746
treatment did not result in any additional effects on B-ALL
apoptosis induced by with VDL in vitro.
In addition, it was observed that AVA4746 treatment
increased the percentage of CD45+/CD19+
leukemia cells in the BM, whilst the percentage of
CD45+/CD19+ leukemia cells was decreased in
the PB. In endothelial cells, VCAM-1 clustering, either by antibody
crosslinking or integrin binding, triggers the activation of Rac1,
a Rho-like GTPase (43). The
activation of Rac1 results in the rearrangement of the cytoskeletal
network, which then remodels the tight junctions among the vascular
endothelial cells and mediate transendothelial migration (TEM)
(44). Previously, it was
demonstrated that overexpression of macrophage inflammatory
protein-1α upregulated the expression of the endothelial adhesion
molecule VCAM-1 to enhance TEM by ALL cells (45). Therefore, in the present study the
effects of AVA4746 on endothelial cell physiology and TEM were
investigated. Kim et al (46) previously showed that a specific
monoclonal antibody against VCAM-1 was able to block
VCAM-1-mediated cell-cell contacts and suppress angiogenesis
(46). Similarly, the present
study demonstrated that both AVA4746 and TBC3486 attenuated the
angiogenesis of HUVECs in vitro. Therefore, one possible
hypothesis from this observation is that the B-ALL cells remained
in the BM despite their detachment from the stromal cells due to
the anti-angiogensis effects of AVA4746.
According to a previously established in
vivo B-ALL xenograft model and chemotherapy regimen (20), AVA4746 in combination with VDL
markedly prolonged the survival of mice engrafted with LAX7R cells,
which were derived from a patient with primary relapsed B-ALL, but
not in mice engrafted with RS4;11 and TXL3 cells. Unlike NZM, which
is a pan-α4-integrin antibody that can not only inhibit α4/VCAM-1
but can also inhibit the α4/fibronectin (47) and α4/osteopontin (14) interactions, the VCAM-1 small
molecule mimetic AVA4746 did not prolong the survival of mice for
as long as NZM (20). However, it
was found in the present study that interfering with the
VLA-4/VCAM-1 interaction can enhance the sensitivity of B-ALL to
chemotherapy. One explanation for the contradictory observations of
the effects of AVA4746 in vitro and in vivo is the
possibility that oral bioavailability is not as robust in the NSG
mice used in the present study. High receptor occupancy by integrin
antagonists is required for the sufficient inhibition of
adhesion(31). Therefore, it
remains possible that sufficiently high levels of AVA4746 were not
maintained throughout the course of the in vivo study in NSG
mice. A follow-up study is required to confirm this possibility,
which should include additional, in-depth cell adhesion studies
involving different extracellular matrix components for α4
integrin, including fibronectin and osteopontin (14).
Taken together, AVA4746 is a potent antagonist of
VLA-4 that can efficiently induce the detachment of B-ALL cells
from VCAM-1. It is possible that AVA4746 can exert effects on
normal hematopoietic stem and progenitor cells in the bone marrow,
since these progenitor cells also express integrin α4(48). However, this possibility is beyond
the scope of the present study. The present study showed that the
interruption of VLA-4/VCAM-1 by AVA4746, an orally applicable small
molecule inhibitor, can sensitize LAX7R cells derived from a
patient with relapsed B-ALL to chemotherapy. Therefore, AVA4746
hold promise a favorable tool for the investigation of VLA-4/VCAM-1
interactions in the CAM-DR mechanism of B-ALL.
Supplementary Material
AVA4746 increases the mean
fluorescence intensity of PE-HUTS-21 antibodies in a dose-dependent
manner. Corresponding representative flow cytometry histograms of
the PE-HUTS-21 antibody MFI for Fig.
1. LAX7R or TXL3 cells were treated with progressively
increasing dosages of (A) AVA4746 or (B) VCAM-1 without
Mn2+. LAX7R or TXL3 cells were treated with
progressively increasing dosages of (C) AVA4746 or (D) VCAM-1 with
Mn2+. MFI, mean fluorescence intensity; VCAM-1, vascular
cell adhesion molecule 1.
AVA4746 decreases integrin α4
expression but does not affect α5 and α6 in B-lineage acute
lymphoblastic leukemia cells. (A) LAX7R, (B) LAX7, (C) LAX53, (D)
LAX56 and (E) REH cells were treated with AVA4746 at 1 (red curve),
5 (blue curve) and 25 μM (orange curve) for either 24 or 96 h,
where 0 was the DMSO control (green curve). Histograms representing
the cell surface expression of integrins α4, α5 and α6 using flow
cytometry are shown. ISO, isotype control.
Reductions in the expression of
integrin α4 induced by AVA4746 is partially reversed by the
proteasome inhibitor MG132. (A) Schematic of the experimental
design. (B) LAX7R, LAX53, LAX56 and REH B-lineage acute
lymphoblastic leukemia cells were pre-treated with the proteasome
inhibitor MG132 (1 μM) for 2 h, followed by treatment with either
DMSO (0.1%) or AVA4746 25 μM for 96 h. Cell lysates were analyzed
for integrin α4 protein expression by western blotting. β-actin was
used as loading control. α4, α4-integrin.
AVA4746 does not affect AKT
phosphorylation but may regulate other cell signaling molecules in
B-lineage acute lymphoblastic leukemia cells. (A) LAX56 and LAX7R
cells were serum starved overnight and stimulated with either 20%
FBS or 10 μg/ml human VCAM-1, with or without AVA4746 treatment (25
μM), for 1 h. Western blot analysis of p-AKT, total AKT protein
levels with β-actin as the loading control are shown. (B) Following
overnight serum starvation, LAX53 cells were stimulated with stroma
OP9 cells for 1 or 24 h with AVA4746 at doses of 0, 5 and 25 μM.
Western blot analysis of phosphotyrosine with β-actin as the
loading control is shown. Red box shows potential cell signaling
changes by AVA4746. p-, phosphorylated; hVCAM1, human vascular cell
adhesion molecule 1.
AVA4746 detaches pre-B-ALL cells from
the stromal cell line OP9. (A) RS4;11, (B) SupB15, (C) KASUMI-2,
(D) 697, (E) BEL-1, (F) BV173, (G) RCH and (H) TOM-1 B-ALL cells
were seeded into plates with or without OP9 cells for 4 h and then
treated with either DMSO control or the AVA4746 (25 μM) overnight.
Percentage of adherent cells are shown, which was determined by
cell counting using trypan blue exclusion. Each experiment was
performed in triplicate. *P<0.05 and
**P<0.01, B-ALL, B-lineage acute lymphoblastic
leukemia.
Stromal cells support the viability in
B-ALL cells. (A) TXL3, (B) LAX56 and (C) LAX7R B-ALL cells were
seeded into uncoated wells or wells pre-coated the stromal cell
line OP9 and with treated for 48 h with either DMSO or AVA4746 25
μM, with or without VDL chemotherapy (vincristine 5 nM,
dexamethasone 0.05 nM, L-asparaginase 2.5x10-3 IU/ml). Percentage
of cell apoptosis (% Annexin-V-positive population) was determined
using flow cytometry. Experiments were performed in triplicate.
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. NS, not
significant; B-ALL, B-lineage acute lymphoblastic leukemia; VDL,
vincristine, dexamethasone and L-asparaginase.
AVA4746 does not change leukemia
distribution at 8 h post-treatment. (A) A schematic of the leukemic
cell distribution experimental design. Following 8 h of treatment
with AVA4746 (60 mg/kg) or PBS control, with or without VDL
(vincristine 0.5 mg/kg, dexamethasone 10.5 mg/kg, and
L-asparaginase 1500 IU/kg; n=6 per group), mice were sacrificed
before (B) PB, (C) BM and (D) SPC were collected and analyzed using
flow cytometry. The total MNC number, percentage (%) of human
CD45+CD19+ population, leukemic cell number,
MFI of human α4, % of mouse CD45+ and mouse cell number
are also shown. NS, not significant. **P<0.01 and
***P<0.001. MNC, mononuclear cell; PB, peripheral
blood; BM, bone marrow; SPC, spleen; VDL, vincristine,
dexamethasone and L-asparaginase; Nb, number.
Corresponding representative
hCD45+hCD19+ flow cytometry dot plots for
Fig. 4. Representative
hCD45+ (y axis) and hCD19+ (x axis) dot plots
of whole mononuclear cells isolated from (A) PB, (B) BM or (C) SPC
were shown. PB, peripheral blood; BM, bone marrow; SPC,
spleen.
TBC3486 inhibits tube formation by
HUVEC in vitro. HUVECs treated with either 0 or 25 μM
TBC3486 for 30 min were seeded onto Matrigel-coated plates for 4-6
h. Tube formation was assessed by analyzing the number of (A)
nodes, (B) segments, (C) meshes and (D) the total area. Images were
analyzed with the ImageJ software coupled with the angiogenesis
analyzer plug-in. **P<0.01 and
***P<0.001. Nb, number.
AVA4746 in combination of chemotherapy
does not prolong the survival of NSG mice injected with RS4;11 and
TXL3 cells. (A) Treatment regimen of 21 days with AVA4746 and 4
weeks with VDL after RS4;11 cell injection. (B) Kaplan-Meier
survival curve of RS4;11-engrafted mice. P-values for PBS vs.
AVA4746 and PBS + VDL vs. AVA4746 +VDL were 0.1842 and 0.0515,
respectively. (C) Treatment regimen of 14 days with AVA4746 and 14
days with VD after TXL3 cell injection. (D) P-value of PBS vs.
AVA4746 and PBS + VD vs. AVA4746 + VD were 0.003 and 0.080,
respectively. *P<0.05. VDL, vincristine 0.5 mg/kg
once a week, dexamethasone 10.5 mg/kg five times a week and
L-asparaginase 1500 IU/kg five times a week; i.p., intraperitoneal;
Bid, twice a day. P.o., oral gavage.
Cytogenetic information and integrin
expression of B-ALL cells
MFI of HUST-21 of AVA4746 or VCAM-1
treatment with or without Mn2+ in LAX7R and TXL3.
Combined values indicate average of experiments #1 and #2.
Percentages of adhered cells following
AVA4746 treatment in LAX7R and TXL3. Combined values indicate
average of experiments #1 and #2.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported received funding from
National Institute of Health (R01 CA172896) and the Leukemia and
Lymphoma Society, Translational Research Program Award (Grant no.
#6505).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
DB, ASW and YMK were responsible for the
conceptualization of the present study. YR, EJG, MY and YMK
designed the present study. YR, EJG, HNK, SL and HAO performed the
research, analyzed and interpreted the data. HCL and MY analyzed
the data. All the authors reviewed the draft manuscript, edited the
final draft and approved the final version for submission. YMK, YR,
EJG, HNK, SL and HAO confirm the authenticity of all the raw data.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Bone marrow and peripheral blood samples from
patients with B lineage acute lymphoblastic leukemia were acquired
in compliance with the Institutional Review Board of Children's
Hospital Los Angeles regulations. The human studies were conducted
in accordance with the Declaration of Helsinki. Informed consent
was obtained from the patients under the approved IRB protocol
(approval no. CCI-08-00102). All studies were approved by the
Institutional Animal Care and Use Committee of Children's Hospital
Los Angeles (Los Angeles, USA).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gregory S: Adult Acute Lymphoblastic
Leukemia: Treatment and Management Updates. Semin Oncol Nurs.
35(150951)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gökbuget N, Dombret H, Ribera JM, Fielding
AK, Advani A, Bassan R, Chia V, Doubek M, Giebel S, Hoelzer D, et
al: International reference analysis of outcomes in adults with
B-precursor Ph-negative relapsed/refractory acute lymphoblastic
leukemia. Haematologica. 101:1524–1533. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Konopleva MY and Jordan CT: Leukemia stem
cells and microenvironment: Biology and therapeutic targeting. J
Clin Oncol. 29:591–599. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Witkowski MT, Kousteni S and Aifantis I:
Mapping and targeting of the leukemic microenvironment. J Exp Med.
217(217)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Shafat MS, Gnaneswaran B, Bowles KM and
Rushworth SA: The bone marrow microenvironment - Home of the
leukemic blasts. Blood Rev. 31:277–286. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kumagai M, Manabe A, Pui CH, Behm FG,
Raimondi SC, Hancock ML, Mahmoud H, Crist WM and Campana D:
Stroma-supported culture in childhood B-lineage acute lymphoblastic
leukemia cells predicts treatment outcome. J Clin Invest.
97:755–760. 1996.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Manabe A, Coustan-Smith E, Behm FG,
Raimondi SC and Campana D: Bone marrow-derived stromal cells
prevent apoptotic cell death in B-lineage acute lymphoblastic
leukemia. Blood. 79:2370–2377. 1992.PubMed/NCBI
|
|
8
|
Mudry RE, Fortney JE, York T, Hall BM and
Gibson LF: Stromal cells regulate survival of B-lineage leukemic
cells during chemotherapy. Blood. 96:1926–1932. 2000.PubMed/NCBI
|
|
9
|
Shishido S, Bönig H and Kim YM: Role of
integrin alpha4 in drug resistance of leukemia. Front Oncol.
4(99)2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Damiano JS, Hazlehurst LA and Dalton WS:
Cell adhesion-mediated drug resistance (CAM-DR) protects the K562
chronic myelogenous leukemia cell line from apoptosis induced by
BCR/ABL inhibition, cytotoxic drugs, and gamma-irradiation.
Leukemia. 15:1232–1239. 2001.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Konopleva M, Tabe Y, Zeng Z and Andreeff
M: Therapeutic targeting of microenvironmental interactions in
leukemia: mechanisms and approaches. Drug Resist Updat. 12:103–113.
2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sison EAR, Kurre P and Kim YM:
Understanding the bone marrow microenvironment in hematologic
malignancies: A focus on chemokine, integrin, and extracellular
vesicle signaling. Pediatr Hematol Oncol. 34:365–378.
2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Guo W and Giancotti FG: Integrin
signalling during tumour progression. Nat Rev Mol Cell Biol.
5:816–826. 2004.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hynes RO: Integrins: Bidirectional,
allosteric signaling machines. Cell. 110:673–687. 2002.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Qian H, Georges-Labouesse E, Nyström A,
Domogatskaya A, Tryggvason K, Jacobsen SE and Ekblom M: Distinct
roles of integrins alpha6 and alpha4 in homing of fetal liver
hematopoietic stem and progenitor cells. Blood. 110:2399–2407.
2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Scott LM, Priestley GV and Papayannopoulou
T: Deletion of alpha4 integrins from adult hematopoietic cells
reveals roles in homeostasis, regeneration, and homing. Mol Cell
Biol. 23:9349–9360. 2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Härzschel A, Zucchetto A, Gattei V and
Hartmann TN: VLA-4 Expression and Activation in B Cell
Malignancies: Functional and Clinical Aspects. Int J Mol Sci.
21(21)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Jacamo R, Chen Y, Wang Z, Ma W, Zhang M,
Spaeth EL, Wang Y, Battula VL, Mak PY, Schallmoser K, et al:
Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of
NF-κB mediates chemoresistance. Blood. 123:2691–2702.
2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Matsunaga T, Fukai F, Miura S, Nakane Y,
Owaki T, Kodama H, Tanaka M, Nagaya T, Takimoto R, Takayama T, et
al: Combination therapy of an anticancer drug with the FNIII14
peptide of fibronectin effectively overcomes cell adhesion-mediated
drug resistance of acute myelogenous leukemia. Leukemia.
22:353–360. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hsieh YT, Gang EJ, Geng H, Park E, Huantes
S, Chudziak D, Dauber K, Schaefer P, Scharman C, Shimada H, et al:
Integrin alpha4 blockade sensitizes drug resistant pre-B acute
lymphoblastic leukemia to chemotherapy. Blood. 121:1814–1818.
2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hsieh YT, Gang EJ, Shishido SN, Kim HN,
Pham J, Khazal S, Osborne A, Esguerra ZA, Kwok E, Jang J, et al:
Effects of the small-molecule inhibitor of integrin α4, TBC3486, on
pre-B-ALL cells. Leukemia. 28:2101–2104. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Vanderslice P, Biediger RJ, Woodside DG,
Berens KL, Holland GW and Dixon RA: Development of cell adhesion
molecule antagonists as therapeutics for asthma and COPD. Pulm
Pharmacol Ther. 17:1–10. 2004.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Vanderslice P and Woodside DG: Integrin
antagonists as therapeutics for inflammatory diseases. Expert Opin
Investig Drugs. 15:1235–1255. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen
C, Huantes S, Park E, Naing K, Klemm L, Swaminathan S, et al:
Small-molecule inhibition of CBP/catenin interactions eliminates
drug-resistant clones in acute lymphoblastic leukemia. Oncogene.
33:2169–2178. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Adam E, Kim HN, Gang EJ, Schnair C, Lee S,
Lee S, Khazal S, Kosoyan O, Konopleva M, Parekh C, et al: The PI3Kδ
Inhibitor Idelalisib Inhibits Homing in an in Vitro and
in Vivo Model of B ALL. Cancers (Basel). 9(9)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Gang EJ, Kim HN, Hsieh YT, Ruan Y, Ogana
HA, Lee S, Pham J, Geng H, Park E, Klemm L, et al: Integrin α6
mediates the drug resistance of acute lymphoblastic B-cell
leukemia. Blood. 136:210–223. 2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chigaev A, Wu Y, Williams DB, Smagley Y
and Sklar LA: Discovery of very late antigen-4 (VLA-4, alpha4beta1
integrin) allosteric antagonists. J Biol Chem. 286:5455–5463.
2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tissino E, Benedetti D, Herman SEM, Ten
Hacken E, Ahn IE, Chaffee KG, Rossi FM, Dal Bo M, Bulian P, Bomben
R, et al: Functional and clinical relevance of VLA-4 (CD49d/CD29)
in ibrutinib-treated chronic lymphocytic leukemia. J Exp Med.
215:681–697. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Liang M, Schwickart M, Schneider AK,
Vainshtein I, Del Nagro C, Standifer N and Roskos LK: Receptor
occupancy assessment by flow cytometry as a pharmacodynamic
biomarker in biopharmaceutical development. Cytometry B Clin Cytom.
90:117–127. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Botta C, Cucè M, Pitari MR, Caracciolo D,
Gullà A, Morelli E, Riillo C, Biamonte L, Gallo Cantafio ME,
Prabhala R, et al: MiR-29b antagonizes the pro-inflammatory
tumor-promoting activity of multiple myeloma-educated dendritic
cells. Leukemia. 32:1003–1015. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chigaev A, Waller A, Amit O, Halip L,
Bologa CG and Sklar LA: Real-time analysis of
conformation-sensitive antibody binding provides new insights into
integrin conformational regulation. J Biol Chem. 284:14337–14346.
2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Alachkar H, Santhanam R, Maharry K,
Metzeler KH, Huang X, Kohlschmidt J, Mendler JH, Benito JM, Hickey
C, Neviani P, et al: SPARC promotes leukemic cell growth and
predicts acute myeloid leukemia outcome. J Clin Invest.
124:1512–1524. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Matsunaga T, Takemoto N, Sato T, Takimoto
R, Tanaka I, Fujimi A, Akiyama T, Kuroda H, Kawano Y, Kobune M, et
al: Interaction between leukemic-cell VLA-4 and stromal fibronectin
is a decisive factor for minimal residual disease of acute
myelogenous leukemia. Nat Med. 9:1158–1165. 2003.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tabe Y, Jin L, Tsutsumi-Ishii Y, Xu Y,
McQueen T, Priebe W, Mills GB, Ohsaka A, Nagaoka I, Andreeff M, et
al: Activation of integrin-linked kinase is a critical prosurvival
pathway induced in leukemic cells by bone marrow-derived stromal
cells. Cancer Res. 67:684–694. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Léger OJ, Yednock TA, Tanner L, Horner HC,
Hines DK, Keen S, Saldanha J, Jones ST, Fritz LC and Bendig MM:
Humanization of a mouse antibody against human alpha-4 integrin: A
potential therapeutic for the treatment of multiple sclerosis. Hum
Antibodies. 8:3–16. 1997.PubMed/NCBI
|
|
36
|
Kent SJ, Karlik SJ, Cannon C, Hines DK,
Yednock TA, Fritz LC and Horner HC: A monoclonal antibody to alpha
4 integrin suppresses and reverses active experimental allergic
encephalomyelitis. J Neuroimmunol. 58:1–10. 1995.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ransohoff RM: Natalizumab and PML. Nat
Neurosci. 8(1275)2005.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Miller DH, Khan OA, Sheremata WA,
Blumhardt LD, Rice GP, Libonati MA, Willmer-Hulme AJ, Dalton CM,
Miszkiel KA and O'Connor PW: International Natalizumab Multiple
Sclerosis Trial Group. A controlled trial of natalizumab for
relapsing multiple sclerosis. N Engl J Med. 348:15–23.
2003.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ghosh S, Goldin E, Gordon FH, Malchow HA,
Rask-Madsen J, Rutgeerts P, Vyhnálek P, Zádorová Z, Palmer T and
Donoghue S: Natalizumab Pan-European Study Group. Natalizumab for
active Crohn's disease. N Engl J Med. 348:24–32. 2003.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Tuloup-Minguez V, Hamaï A, Greffard A,
Nicolas V, Codogno P and Botti J: Autophagy modulates cell
migration and β1 integrin membrane recycling. Cell Cycle.
12:3317–3328. 2013.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Lobert VH, Brech A, Pedersen NM, Wesche J,
Oppelt A, Malerød L and Stenmark H: Ubiquitination of alpha 5 beta
1 integrin controls fibroblast migration through lysosomal
degradation of fibronectin-integrin complexes. Dev Cell.
19:148–159. 2010.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wang L, Fortney JE and Gibson LF: Stromal
cell protection of B-lineage acute lymphoblastic leukemic cells
during chemotherapy requires active Akt. Leuk Res. 28:733–742.
2004.PubMed/NCBI View Article : Google Scholar
|
|
43
|
van Wetering S, van den Berk N, van Buul
JD, Mul FP, Lommerse I, Mous R, ten Klooster JP, Zwaginga JJ and
Hordijk PL: VCAM-1-mediated Rac signaling controls endothelial
cell-cell contacts and leukocyte transmigration. Am J Physiol Cell
Physiol. 285:C343–C352. 2003.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Chen Q and Massagué J: Molecular pathways:
VCAM-1 as a potential therapeutic target in metastasis. Clin Cancer
Res. 18:5520–5525. 2012.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Ma YR and Ma YH: MIP-1α enhances Jurkat
cell transendothelial migration by up-regulating endothelial
adhesion molecules VCAM-1 and ICAM-1. Leuk Res. 38:1327–1331.
2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Kim TK, Park CS, Na HJ, Lee K, Yoon A,
Chung J and Lee S: Ig-like domain 6 of VCAM-1 is a potential
therapeutic target in TNFα-induced angiogenesis. Exp Mol Med.
49(e294)2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Layani-Bazar A, Skornick I, Berrebi A,
Pauker MH, Noy E, Silberman A, Albeck M, Longo DL, Kalechman Y and
Sredni B: Redox modulation of adjacent thiols in VLA-4 by AS101
converts myeloid leukemia cells from a drug-resistant to
drug-sensitive state. Cancer Res. 74:3092–3103. 2014.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Karpova D, Rettig MP, Ritchey J, Cancilla
D, Christ S, Gehrs L, Chendamarai E, Evbuomwan MO, Holt M, Zhang J,
et al: Targeting VLA4 integrin and CXCR2 mobilizes serially
repopulating hematopoietic stem cells. J Clin Invest.
129:2745–2759. 2019.PubMed/NCBI View Article : Google Scholar
|