Introduction
Glutamate is a major mediator of excitatory
neurotransmission in the mammalian central nervous system (CNS),
and plays crucial roles in the physiology of numerous CNS functions
ranging from learning to memory and cognition (1). The tonic concentrations of glutamate
are tightly regulated in the synapse by high-affinity glutamate
reuptake systems (2). However, high
concentrations of glutamate in the CNS are potentially neurotoxic.
The neurotoxic effects are generally characterized by neuronal
hyperexcitability and excitotoxicity. Glutamate-induced
excitotoxicity has been associated with inflammation and oxidative
stress, and has emerged as an important pathological mechanism in a
wide range of acute and chronic neurological disorders, including
cerebral ischemia, epilepsy, traumatic brain injury and
neurodegenerative diseases such as Alzheimer's disease, Parkinson's
disease and Huntington's disease (3-6).
The physiological basis for glutamate-induced neurotoxicity is
beginning to emerge, but there is considerable uncertainty
surrounding the underlying mechanisms. Several lines of evidence
point to mitochondrial dysfunction as a defining event in
glutamate-induced neurotoxicity. Mitochondria-dependent calcium
influx and the depletion of the intracellular antioxidant
glutathione promote acute oxidative stress by elevating the levels
of reactive oxygen species (ROS), which in turn leads to an
imbalance in mitochondrial dynamics and, ultimately, cell death
(7).
Mitochondria are bioenergetic and biosynthetic
organelles with a well-recognized role in the production of ATP and
other macromolecules. It has become apparent that mitochondria
perform various signaling functions, as evidence has emerged on
their crucial role in the maintenance of intracellular calcium
[Ca2+]i homeostasis, regulation of the
intracellular level of ROS and, most importantly, programmed cell
death (8,9). Mitochondria are highly dynamic
intracellular entities that undergo biogenesis, clearance and
fission/fusion. In these mitochondrial dynamics, the continuous
cycles of fission and fusion are maintained in a delicate balance
and are essential for mitochondrial homeostasis (10,11).
Fission and fusion events are tightly and coordinately regulated by
several nuclear-encoded proteins with GTPase hydrolysis activities.
Among them, dynamin-related protein 1 (Drp1) and mitochondrial
fission 1 protein (Fis1) mediate mitochondrial fission, while
mitofusin 1 and 2 (Mfn1/2) and optic atrophy 1 (Opa1) proteins
mediate mitochondrial fusion (12).
Drp1, the master regulator of mitochondrial fission, exists
predominantly in the cytoplasm where, when being activated, it
interacts with GTP and lipids to form oligomers that are
subsequently recruited to the mitochondrial outer membrane to form
an integral part of the fission machinery (13). Fis1 is a component protein of the
fission complex that includes mitochondrial fission factor (Mff),
mitochondrial division protein 1, and mitochondrial dynamics
proteins of 49 kDa (MiD49) and 51 kDa (MiD51). Fis1 is anchored in
the outer membrane of the mitochondria where it mediates the
recruitment of oligomerized Drp1 to fission sites on the membrane.
The translocation of Drp1 to the mitochondrial membrane initiates a
series of events that culminates in the division of the
mitochondria or the activation of apoptosis, depending on the
triggering stimulus (10,11,13).
In contrast to mitochondrial fission, the process of mitochondrial
fusion is positively regulated by Mfn1/2 and Opa1, and occurs in
two phases. In the first phase, the dimerization and binding of
mitofusins facilitates the tethering of the outer membranes of
neighboring mitochondria via a process that involves lipid
hydrolysis. In the second phase, Opa1 coordinates the formation of
cristae and fusion of the mitochondrial inner membranes, thus
completing the fusion process (12). The view that mitochondrial fusion is
a requirement for the transport of mitochondria within the cell and
essential for cell survival is commonly accepted.
The small molecule
4-chloro-N-(naphthalen-1-ylmethyl)-5-[3-(piperazin-1-yl)phenoxy]thiophene-2-sulfonamide
(B355252) is an pheneoxythiophene sulfonamide with intrinsic nerve
growth factor (NGF) potentiating activity. B35525 has been shown to
enhance the ability of NGF-primed NS-1 pheocromocytoma cells to
differentiate into a neuron-like phenotype with extensive networks
of branched neurites (14,15). B35525 does not induce neurite
outgrowth alone, but enhances the effect of sub-physiological
concentrations of NGF on neurons in vitro. In addition,
B355252 has exhibited unique activity in several in vitro
models of chronic neurological and neurodegenerative disorders
(16,17). In our previous study, B355252 was
shown to protect HT22 neuronal cell against glutamate-induced
excitotoxicity via the potent suppression of the oxidative injury
caused by ROS production and [Ca2+]i overload
(16). Given that mitochondria are
the major site of [Ca2+]i and ROS production
during glutamate excitotoxicity, the present study examined the
effect of B355252 on HT22 cell viability and markers of
mitochondrial structural dynamics and apoptosis following glutamate
exposure.
Materials and methods
Cell culture and experimental
treatment
HT22 murine hippocampal neuronal cells (donated by
Dr June Panee; University of Hawaii, Honolulu, Hawaii) were
cultured in Dulbecco's Modified Eagle's Medium (DMEM; Invitrogen;
Thermo Fisher Scientific, Inc.) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; cat. no. MT35011CV;
Thermo Fisher Scientific, Inc.), 2 mM L-glutamine and 200 mM
streptomycin/penicillin (Invitrogen; Thermo Fisher Scientific,
Inc.) and maintained at 90-95% relative humidity in 5%
CO2 at 37˚C. Glutamate (Sigma-Aldrich; Merck KGaA) was
dissolved in water while B355252 (donated by Dr Alfred Williams;
North Carolina Central University, Durham, North Carolina) was
dissolved in dimethyl sulfoxide at a concentration of 10 mM. Stock
solutions were diluted with cell culture medium for each
experiment. Cells were subjected to glutamate stress for 18 h alone
or after pretreatment for 2 h with B355252 in full culture medium.
Various concentrations of glutamate (0.25-16 mM) and B355252
(0.16-10 µM) were evaluated to determine their optimal working
concentrations prior to their combined use.
Cell viability assay
The viability of the HT22 cells was quantified in
96-well plates using 7-hydroxy-3H-phenoxazin-3-one 10-oxide
(resazurin). A stock solution of resazurin was prepared in
deionized water at a concentration of 1 mg/ml. Following the
aforementioned treatment of cells with glutamate and/or B355252, a
10 µl aliquot of resazurin was dispensed into each well containing
100 µl DMEM to achieve a final concentration of 0.1 mg/ml. After 3
h of incubation in 5% CO2 at 37˚C, the cells were
equilibrated to room temperature for 15 min and the fluorescence
was measured with a PHERAstar Microplate Reader (BMG Labtech GmbH)
with a 540-20/590-20 nm filter. The relative fluorescence of the
untreated, control cells represented 100% cell viability and the
cell viability of each experimental group was converted to a
percentage relative to the control.
Western blotting
For immunoblotting, treated cells were lysed in RIPA
buffer (cat. no. R0278; Sigma-Aldrich; Merck KGaA) with complete
protease (cat. no. P1860; Sigma-Aldrich; Merck KGaA) and
phosphatase (cat. no. 52-462-51SET; MilliporeSigma) inhibitor
cocktails. Following lysis, subcellular fractions of the cytosol,
mitochondria and nucleus were isolated through differential
centrifugation steps as previously described (18). The purity of the fractions was
verified as previously reported (19). Protein concentrations were
determined using the Bradford assay. Equal amounts of protein (20
µg/lane) in the total cell fractions were separated on 4-12% NuPAGE
SDS-PAGE gels (Invitrogen; Thermo Fisher Scientific, Inc.),
transferred to nitrocellulose membranes, and then probed overnight
at 4˚C with the following antibodies: Phospho-(Ser616)Drp1 (p-Drp1;
1:1,000; cat. no. 3455; Cell Signaling Technology, Inc.), Drp1
(1:500; cat. no. PIPA577924; Invitrogen; Thermo Fisher Scientific,
Inc.), Fis1 (1:500; cat. no. sc-98900; Santa Cruz Biotechnology,
Inc.), Opa1 (1:1,000; cat. no. sc-30572; Santa Cruz Biotechnology,
Inc.), Mfn1 (1:1,000; cat. no. sc-50330; Santa Cruz Biotechnology,
Inc.), Mfn2 (1:1,000; cat. no. sc-50331; Santa Cruz Biotechnology,
Inc.), apoptosis-inducing factor (AIF; 1:500; cat. no. sc-55519;
Santa Cruz Biotechnology, Inc.), mitochondrial cytochrome c
oxidase subunit IV (Cox-IV; 1:1,000; cat. no. ab14744; Abcam) or
β-actin (1:1,000; A1978; Sigma-Aldrich; Merck KGaA). The membranes
were incubated with IRDye® 680RD goat anti-rabbit IgG
(H+L) (1:2,000; cat. no. 926-68171; LI-COR Biosciences) or
IRDye® 800 CW goat anti-mouse IgG (H+L) (1:2,000; cat.
no. 926-32210; LI-COR Biosciences) for 1 h at room temperature.
β-actin, Cox-IV and lamin B (1:1,000; cat. no. ab16048; Abcam) were
used as internal loading controls. Membranes were scanned with an
Odyssey LI-COR imaging system (LI-COR Biosciences). The targeting
band densities were quantified using Image Studio (4.x CLx; LI-COR
Biosciences) and ratios of the targeted proteins and loading
control were calculated and presented.
Mitochondrial imaging
H922 cells were grown on a Lab-Tek™ II Chamber
Slide™ (Thermo Fisher Scientific, Inc.), and were treated with
glutamate and/or B3552525 as aforementioned. After the treatment,
the cells were labeled with MitoTracker™ Red CM-XRos (M7512;
Invitrogen; Thermo Fisher Scientific, Inc.) at 37˚C in a humidified
5% CO2 atmosphere for 30 min. The cells were thereafter
fixed with 4% paraformaldehyde in DMEM culture medium for 15 min at
room temperature. The fixed cells were rinsed twice with PBS,
mounted with Vectashield mounting medium containing DAPI (Vector
Laboratories, Inc.) and analyzed using a BD Pathway™ 855
High-Content Bioimager (BD Biosciences) at x100 final
magnification. The experiment was repeated three times and images
of a minimum of three fields per group were captured. The number of
cells containing fragmented mitochondria was manually counted in
each experimental condition and presented as percentage of total
counted cells.
AIF immunostaining
HT22 cells were grown on Lab-Tek II Chamber Slides
(cat. no. 12-565-5; Thermo Fisher Scientific, Inc.). Following 18 h
of glutamate exposure with or without B355252 pretreatment 37˚C,
the cells were fixed for 20 min at room temperature with 4%
paraformaldehyde, washed with PBS and permeabilized in 0.3% Triton
X-100 for 5 min. The cells were blocked with 10% donkey serum
(5664605ML; MilliporeSigma) for 1 h at room temperature and then
incubated overnight at 4˚C with anti-AIF primary antibody (1:200;
cat. no. sc-55519). The cells were washed with PBS and then
incubated for 2 h at room temperature with Alexa Fluor
488-conjugated secondary antibodies (1:500; cat. no. R37114;
Invitrogen; Thermo Fisher Scientific, Inc.). Finally, the slides
were mounted with Vectashield mounting medium containing DAPI. The
slides were scanned with a BD Pathway 855 High-Content Bioimager.
The experiment was repeated three times and images of a minimum of
three fields per group were captured and processed for
analysis.
Statistical analysis
Data are presented as the mean ± SD of at least
three independent experiments and were analyzed using Graph Pad
Prism 7 software (GraphPad Software, Inc.). One-way ANOVA followed
by Tukey's test was used to analyze differences among groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
B355252 protects HT22 cells against
glutamate-induced cell death
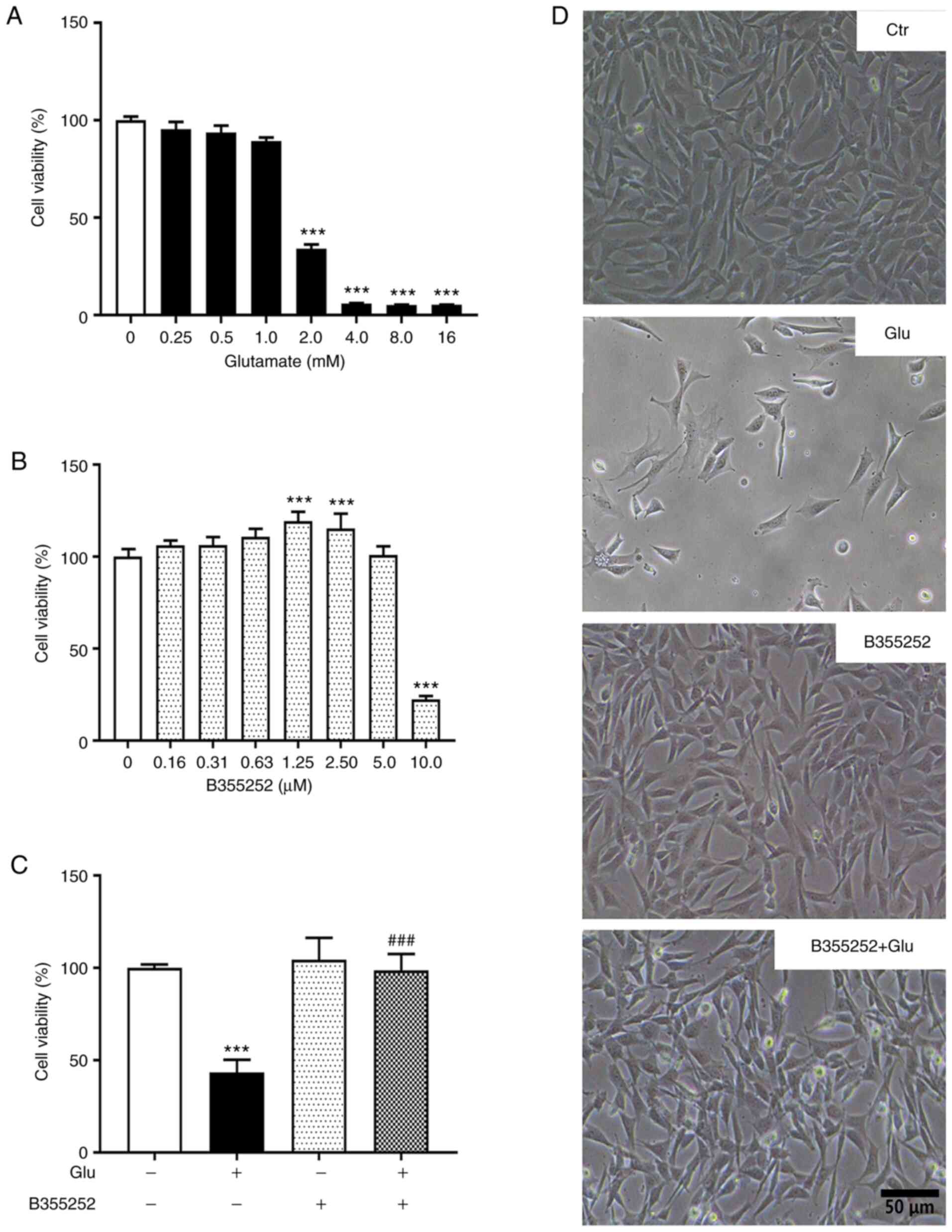
Concentrations of glutamate >1.0 mM caused
significant concentration-dependent reductions in HT22 cell
viability compared with that of the untreated control, with 2.0 mM
decreasing cell viability by ~60% (P<0.001; Fig. 1A and D) and higher concentrations decreasing
cell viability >90%. Based on this result, 2.0 mM glutamate was
selected for subsequent experiments. B355252 alone exhibited no
toxicity on HT22 cell viability at concentrations ≤5 µM relative to
the control (Fig. 1B and D). However, a 75% loss of viability was
observed when the cells were exposed to 10.0 µM B355252. On the
basis of these results, 2.5 µM B355252 was selected for further
experiments.
The pretreatment of HT22 cells with B355252
essentially prevented the toxic effect of 2.0 mM glutamate on
viability (P<0.001; Fig. 1C and
D). Notably, glutamate-induced cell
death was also prevented by the simultaneous application of 5 µM
B355252, although the level of protection was less robust compared
with that of pretreatment with the compound prior to glutamate
exposure (data not shown). These results demonstrate that B355252
pretreatment significantly reduced cell death and improved the
survival of HT22 cells under glutamate exposure.
B355252 suppresses the
glutamate-induced increase of mitochondrial fission proteins Drp1
and Fis1
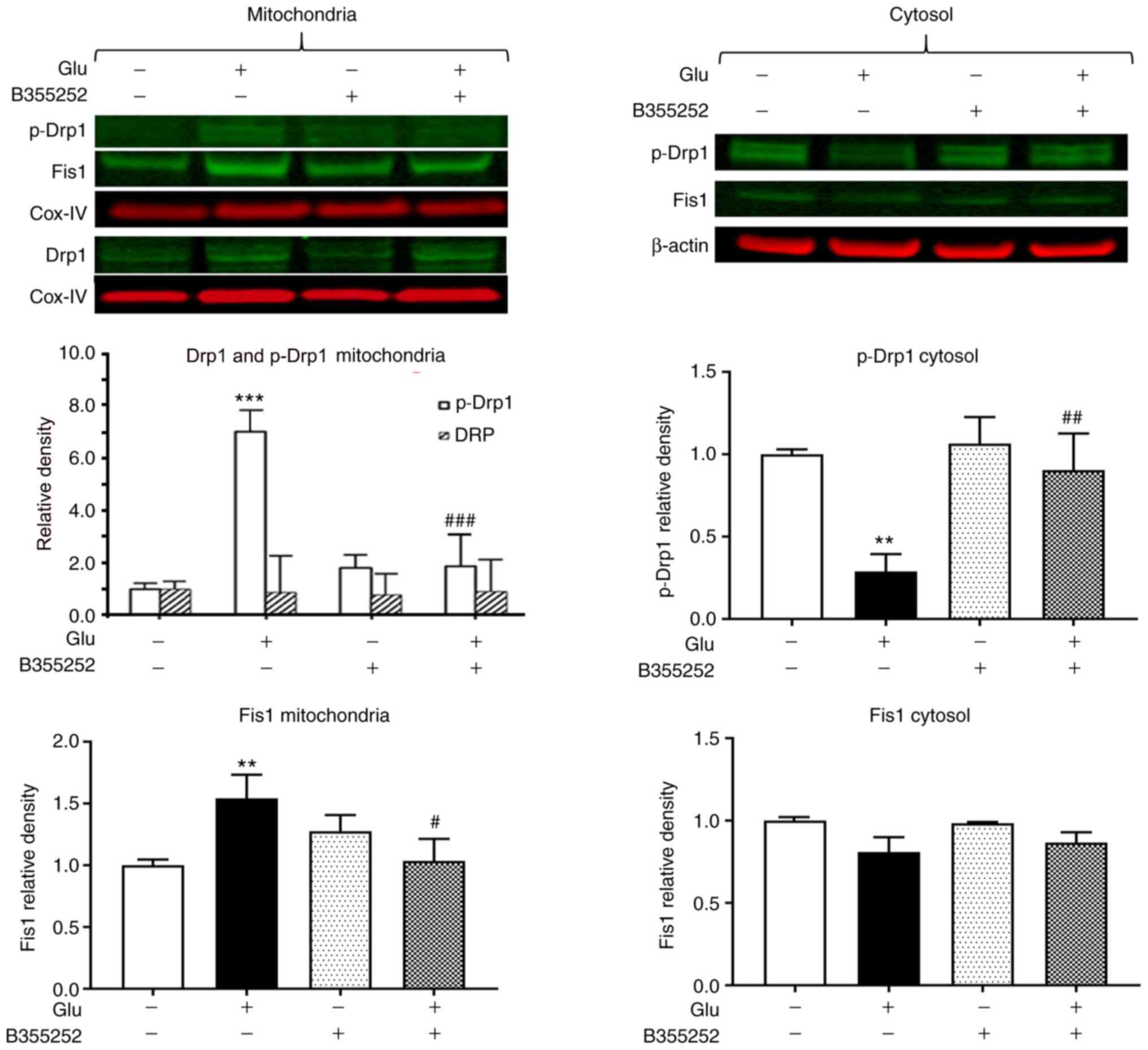
The exposure of HT22 cells to glutamate led to a
significant 7-fold increase in the level of p-Drp1 in the
mitochondrial protein fraction (P<0.001; Fig. 2) and a 4-fold reduction in the
cytosolic level of this protein compared with the respective levels
in unexposed control cells (P<0.01; Fig. 2), which suggests that p-Drp1
translocated from the cytosol to the mitochondria. Pretreatment
with B355252 prior to glutamate exposure blocked the activation and
translocation of Drp1 and thereby restored the balance between
cytosolic and mitochondrial p-Drp1 to levels similar to those in
control cells. In the mitochondrial fraction, the band densities of
total Drp1 relative to those of the internal loading control COX-IV
were not significantly altered by the treatments. Analysis of Fis1
showed a slight but significant 1.5-fold (P<0.01) upregulation
of the protein in the mitochondria following glutamate exposure.
Pretreatment with B355252 prevented the glutamate-dependent
increase, resulting in the level of Fis1 in the cells remaining at
baseline levels. No significant change in Fis1 protein was observed
in the cytosolic fraction in the presence of glutamate with or
without B355252. In addition, treatment of the cells with B355252
alone did not alter the levels of p-Drp1 or Fis1 in the
mitochondria or cytosol of the HT22 cells.
B355252 prevents glutamate-induced
changes of mitochondrial fusion markers Opa1, Mfn1 and Mfn2
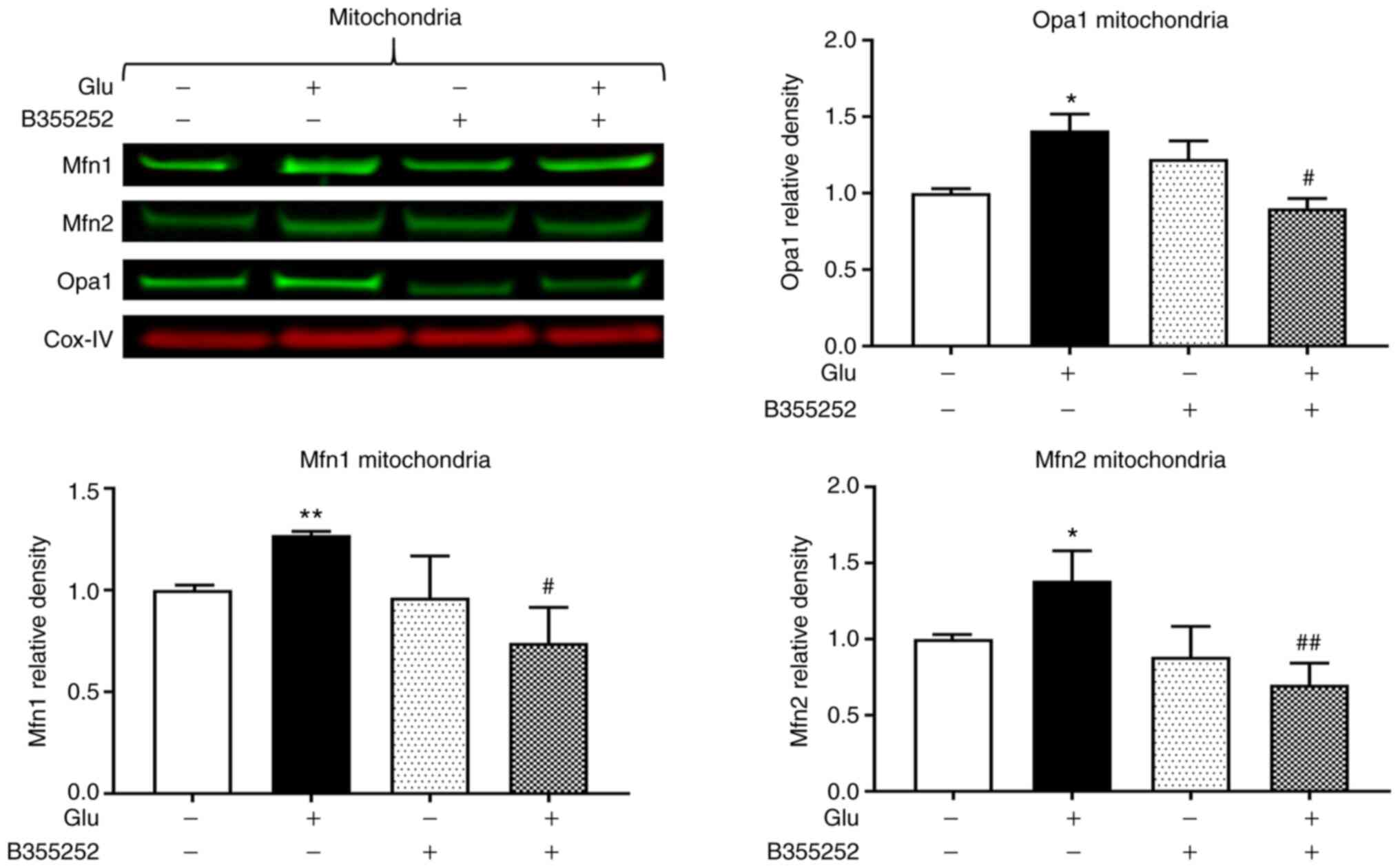
Western blotting revealed that B355252 caused small,
statistically insignificant changes in Opa1, Mfn1 and Mfn2 protein
levels in the mitochondria in comparison with those in untreated
control cells, and glutamate exposure significantly increased Opa1,
Mfn1 and Mfn2 expression by ~40% (P<0.05), 25% (P<0.01) and
40% (P<0.05), respectively (Fig.
3). Furthermore, B355252 pretreatment significantly decreased
the glutamate-induced elevations in Opa1 and Mfn1 proteins by 40%
(P<0.05) and in Mfn2 protein by 65% (P<0.01) relative to the
levels in cells treated with glutamate alone.
B355252 decreases glutamate-induced
mitochondrial fragmentation
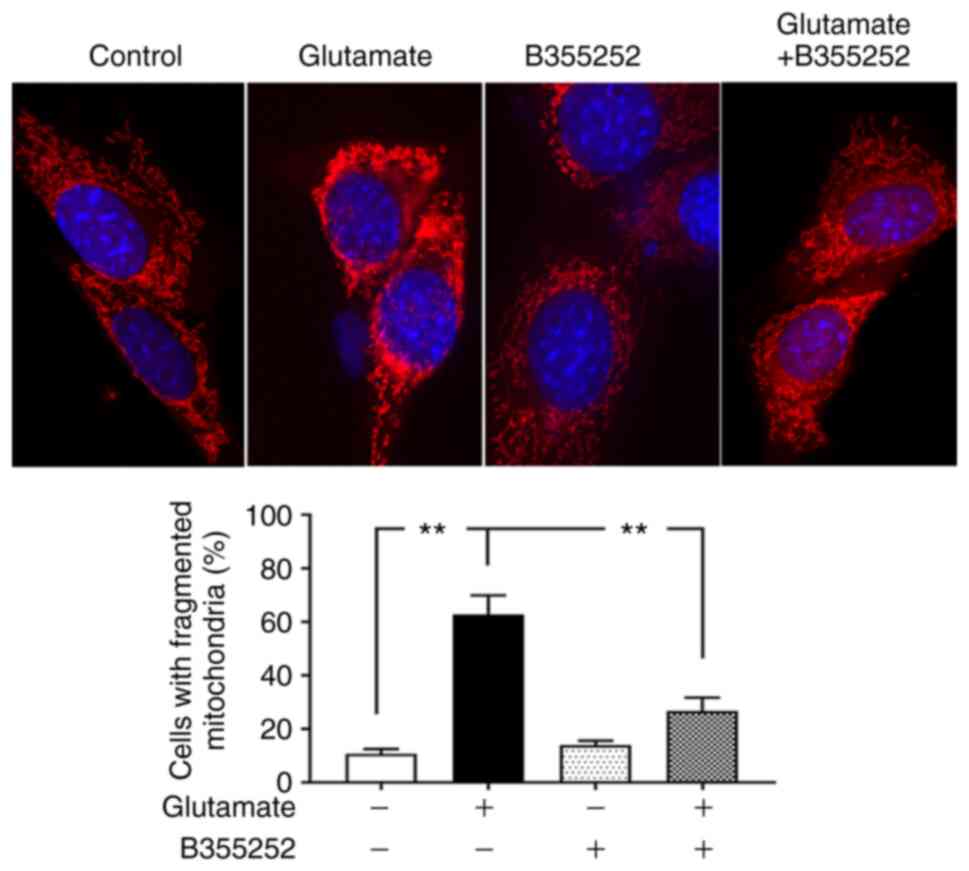
Based on the preceding results, morphological
imaging was performed to evaluate the dynamic changes occurring in
the mitochondria. Consistent with the protein levels of the
mitochondrial fission regulators p-Drp1 and Fis1, mitochondrial
imaging using MitoTracker Red revealed that marked morphological
changes were induced by glutamate in the mitochondria. As seen in
Fig. 4, the mitochondria in control
cells displayed rod and wire shapes with a reticular mitochondrial
network in the cell body. By contrast, the reticulitis of the
mitochondrial network was broken into prominent small, rounded
specks after glutamate exposure. However, pretreatment with B355252
protected the mitochondrial tubular network in glutamate-exposed
cells, while B355252 treatment alone had no adverse effect on the
HT22 cells. Counts of the numbers of cells that contain fragmented
mitochondria revealed that glutamate treatment resulted in
mitochondrial fragmentation in ~60% of the cells, a 6-fold increase
compared with that in the control group (P<0.01), whereas only
30% of cells with fragmented mitochondria was observed in B355252
pre-treated, glutamate exposed cells. These results suggest that
B355252 ameliorated glutamate-induced mitochondrial
fragmentation.
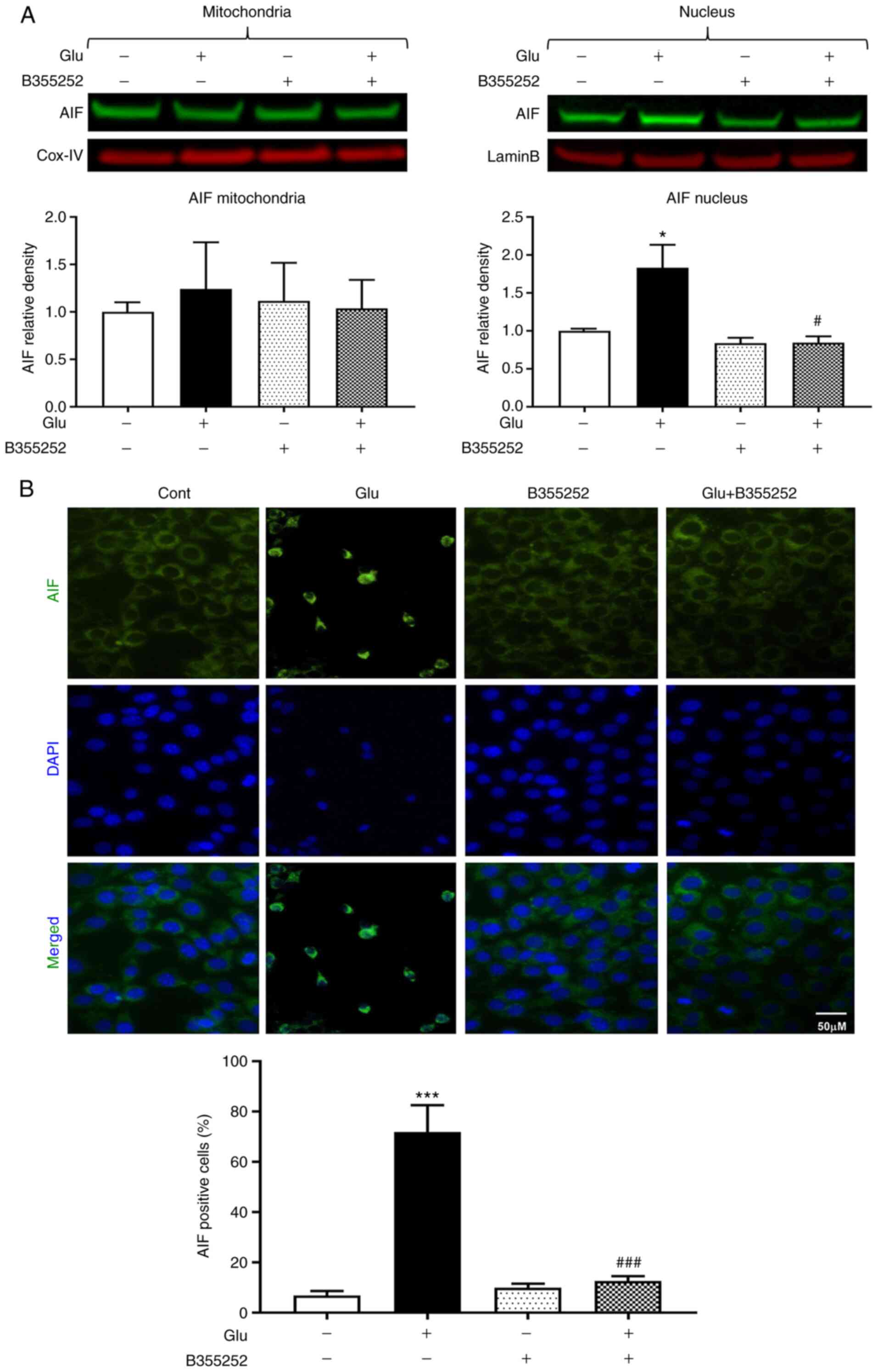
B355252 suppresses the
glutamate-induced elevation of proapoptotic AIF
Glutamate treatment significantly increased the AIF
content in HT22 cell nuclei nearly 2-fold compared with that in the
untreated control cells (P<0.05; Fig. 5A). However, the nuclear content of
AIF was markedly reduced to a level approaching that in the
untreated control cells following glutamate exposure with B355252
pretreatment; B355252 alone had no effect on the level of AIF in
the nucleus. The mitochondrial content of AIF was not significantly
altered by glutamate and/or B355252. These immunoblot findings were
further confirmed by AIF immunocytochemistry, which showed the
presence of ~70% AIF positive cells following glutamate treatment,
as measured by immunofluorescence localization in the nucleus. This
significant increase compared with the untreated control
(P<0.001) was significantly inhibited and maintained at control
levels by pre-exposure of the cells to B355252 (P<0.001;
Fig. 5B).
Discussion
Neurodegenerative diseases are a set of disorders
that typically lead to deterioration in the function of cells of
the CNS. The prevalence of these diseases is increasing owing to
numerous factors, including advances in medicine that have led to
increased lifespans, poor insight into the mechanisms responsible
for disease initiation and progression, and the absence of
effective therapies that reduce or prevent their incidence. The
identification of B355252, a phenoxythiophene compound from a
proprietary chemical library, as a novel agent that potentiates
neurotrophin-dependent neuronal cell signaling was reported in our
previous study (14-17).
B355252 has been shown to exhibit a wide range of pharmacological
actions, including the potentiation of NGF-initiated neurite
outgrowth and extension, and the promotion of cell survival and
neuroprotection in a serum-deprivation model (15) and an in vitro model of
Parkinson's disease (17).
Furthermore, the rescue of neuronal cells from glutamate-induced
oxidative mitochondrial injury by B355252 has been demonstrated to
occur through the inhibition of ROS production and the robust
blockade of [Ca2+]i influx (16). In the present study, the aim was to
define the role of B355252 in the attenuation of glutamate-induced
injury and dysfunction of the mitochondria. The results reveal that
B355252 protected HT22 neurons via the regulation of key
mitochondrial remodeling proteins, preservation of the
mitochondrial architecture, and prevention of the translocation of
proapoptotic AIF to the nucleus.
The viability of HT22 cells was assessed using
resazurin, a dye of low cytotoxicity that is reduced by viable
cells to resorufin. The resazurin assay reflects mitochondrial
dysfunction. Therefore, whether the decreased viability of
glutamate-exposed cells and preventive effects of B355252 indicated
by the resazurin assay results reflect the real cell survival
status is contentious. However, images of the cells clearly show a
reduction in cell density following exposure to glutamate and the
restoration of cell density when B355252 was applied prior to
glutamate treatment, indicating that B355252 prevented
glutamate-induced cell death.
Glutamate is a major excitatory neurotransmitter in
the CNS where it regulates the activity of two major classes of
postsynaptic receptors, namely ionotropic and metabotropic
glutamate receptors. At physiological concentrations, glutamate
plays a crucial role in memory, cognition and neuronal plasticity.
However, abnormally high concentrations of glutamate can lead to
neuronal dysfunction, excitotoxicity and cell death, mediated in
part by the overexcitation of postsynaptic glutamate receptors
(20). In accordance with previous
findings, the exposure of HT22 cells to increased extracellular
concentrations of glutamate led to a significant
concentration-dependent reduction in cell viability.
Previous studies have shown that the toxicity
exerted by glutamate in HT22 cells is largely mediated via the
induction of oxidative stress (16,21).
The formation of ROS is a consequence of glutamate-induced
oxidative stress, and mitochondria are a major source of ROS in
neuronal cells exposed to glutamate, acutely or chronically. ROS
are produced as a normal consequence of cellular metabolism and can
be harmful or beneficial, depending on homeostatic systems in the
cell. However, high concentrations of glutamate can overwhelm
antioxidant defense systems and result in the disruption of
cellular homeostasis by increasing the accumulation of ROS, and
decrease cell survival by a mechanism involving mitochondrial
hyperpolarization and dysfunction (18,22).
It is not known whether the protective effect of B355252 against
glutamate toxicity is due only to its antioxidant property. The
role of B355252 in ROS, Ca2+, glutathione (GSH) and ERK
kinase modulation was defined in our previous study (16). The inhibitory effect of B355252 on
the glutamate-induced increases in ROS and Ca2+ levels
and depletion of GSH appears similar to the antioxidant activity of
N-acetylcysteine (NAC) (23).
However, the changes observed using NAC, although significant, were
less pronounced compared with those induced by B355252 in naïve
cells. Therefore, the effectiveness of B355252 in the protection of
neurons is not likely to be limited to its role as an antioxidant,
and other mechanisms may be contributing to the protection
conferred against glutamate-induced injury. Given that mitochondria
are the major site of ROS and Ca2+ production during
glutamate-dependent injury, and glutamate has been reported to
affect the mitochondrial dynamics of neuronal cells (18,24),
the role of B355252 in the mitochondrial dynamics of HT22 cells was
investigated in the present study. The results support the
hypotheses that B355252 has a role in mitochondrial remodeling, and
that an additional mechanism other than antioxidation contributes
to the ability of the compound to completely protect neuronal cells
from glutamate-induced stress.
The association between glutamate toxicity and
mitochondrial fragmentation has previously been established. It has
been reported that mitochondrial dynamic events controlled by the
fission proteins Drp1 and Fis1, and the fusion proteins Opa1 and
Mfn1/2, are altered during glutamate-induced stress (18,24,25).
Drp1, a cytoplasmic GTPase is the master regulator of mitochondrial
fission. Mitochondrial fission occurs through a series of steps
involving the translocation of the activated form of Drp1 from the
cytoplasm to the outer mitochondrial membrane, where it drives
membrane fission (12). The
recruitment of Drp1 is facilitated by Fis1, a key receptor located
in the outer mitochondrial membrane. Other proteins that mobilize
Drp1 to the mitochondria independent of Fis1, such as Mff and the
mitochondrial dynamics proteins MiD49 and MiD51, have since been
identified (26,27). These proteins serve critical roles
in mitochondrial fission events.
Activation of Drp1 by phosphorylation leads to
division of the mitochondria to generate two daughter organelles
(28). Earlier studies into the
role of Drp1 showed that the overexpression of mutant Drp1 caused
abnormal mitochondrial morphology in mammalian cells (29,30),
while the inhibition of Drp1 via the overexpression of a
dominant-negative mutant prevented the loss of transmembrane
potential and release of cytochrome c, and protected against
cell death (31). Likewise, the
overexpression of Fis1 causes mitochondrial fragmentation in
mammalian cells, while Fis1 knockdown alleviates it (32,33).
Furthermore, a study of small molecule inhibitors of Drp1
activation revealed that they protected PC12 cells against ischemic
neuronal injury in an oxygen-glucose deprivation model via the
attenuation of mitochondrial dysfunction promoted by
[Ca2+]i uptake (34).
Drp1 can be phosphorylated at distinct serine
residues, including Ser618 by Cdk1/cyclin B, and Ser637 or 656 by
protein kinase A (35,36). Previous studies have demonstrated
that mitochondrial fission is promoted when Drp1 is phosphorylated
at Ser616 and inhibited when phosphorylation occurs at Ser637 or
Ser656 (37,38). In the present study, a significant
increase in phosphorylated Drp1 levels in the mitochondria of HT22
cells was observed using a p-Drp1 (Ser616)-targeting antibody
following glutamate exposure. Fis1 was also increased in the
mitochondria of cells treated with glutamate, suggesting that an
interaction between Drp1 and Fis1 leads to excessive mitochondrial
fission and contributes to glutamate-mediated cell death. B355252
decreased the glutamate-induced increase in the levels of p-Drp1 in
the mitochondria, and restored p-Drp1 levels in the cytoplasm.
Therefore, these effects of B355252 support the hypothesis that the
compound reduces mitochondrial fission by decreasing the
glutamate-induced activation of Drp1 and its recruitment to the
mitochondrial membrane, as well as restoring the Drp1 balance in
the cytoplasm. In the present study, a 7-fold upregulation of Drp1
accompanied by increased Fis1 expression in the mitochondrial
fraction was observed when cells were exposed to glutamate. These
increases are consistent with the observation that the percentage
of cells containing fragmented mitochondria increased significantly
in glutamate-treated cells. These data suggest that glutamate
activates the mitochondrial fission machinery that causes
mitochondrial fragmentation and eventually cell death. The
pretreatment of glutamate-exposed cells with B355252 completely
blocked the glutamate-induced increases of p-Drp1 and Fis1 in the
mitochondria and reduced mitochondrial fragmentation, suggesting
that the protective effect of B355252 against glutamate-induced
excitotoxicity is at least partially mediated through the
inhibition of mitochondrial fission. One limitation of the study
must be noted. Ideally the ratio of p-Drp1 to Drp1 should be
calculated to evaluate whether the observed changes in p-Drp1
reflect a change in phosphorylation per se or a change of
total Drp1 protein. Since total Drp1 was not detected alongside
with p-Drp1 in the cytosol, whether the observed change in p-Drp1
in the cytosol reflects a change in total Drp1 is not known.
Mitochondria constantly undergo cycles of fission
and fusion in order to maintain the morphology and functions of the
organelle (39). The fusion process
is controlled by Opa1, Mfn1 and Mfn2, three large GTPases belonging
to the dynamin family of proteins. Mfn1 and Mfn2 are located on the
outer mitochondrial membrane and are key regulators of outer
membrane fusion, while Opa1 is embedded in the inner membrane and
is involved in inner mitochondrial membrane fusion (40). Several lines of evidence demonstrate
that mitochondrial fusion serves an essential role in mammalian
development (41,42). Opa1 is a key regulator of neuronal
fate during excitotoxic cell death (43,44).
The association of glutamate-induced mitochondrial fragmentation
with reduced levels of mitochondrial fusion proteins has been
observed in various studies. For example, glutamate toxicity
reduces the levels of Opa1 and promotes neuronal cell death by
inducing mitochondrial morphology defects, whereas the upregulation
of Opa1 confers significant protection against
N-methyl-D-aspartate-induced neuronal death (45). Studies have also shown that Mfn2
deficiency leads to mitochondrial fragmentation and neuronal death,
and renders motor neurons vulnerable to glutamate excitotoxicity,
while the overexpression of Mfn2 prevents glutamate-induced
fragmentation of the mitochondria, and blocks the death of spinal
cord motor neurons and cortical neurons in vitro and in
vivo (46-50).
However, in the present study, 0.25- to 0.4-fold increases in the
levels of the pro-fusion proteins Opa1 and Mfn1/2 were observed in
the mitochondrial fraction after glutamate incubation. This could
be a protective reaction of the mitochondria to glutamate stress,
similar to the expression of certain pro-survival genes following
glutamate or ischemic stress. Nevertheless, even with the slight
increases of pro-fusion proteins, the mitochondria were destined
for fragmentation, as evidenced by mitochondrial imaging,
suggesting that glutamate predominantly induced mitochondrial
fission even in the presence of slightly elevated levels of fusion
proteins. Pretreatment with B355252 prevented the increases in
mitochondrial Opa1 and Mfn1/2 levels, in addition to blocking the
glutamate-induced increases p-Drp1 and Fis1, suggesting that
maintenance of the mitochondrial dynamic balance plays a key role
in the rescue of the cells by B355252.
A direct consequence of glutamate-induced toxicity
in cells is bioenergetic failure provoked by collapse of the
mitochondrial membrane potential. Dissipation of the membrane
potential triggers prolonged opening of mitochondrial permeability
transition pores (MPTPs) under the control of cyclophilin-D. The
formation of MPTPs leads to permeabilization of the mitochondria
and the influx of molecules into the mitochondrial matrix, which in
turn causes the mitochondria to swell and rupture, releasing
several apoptotic factors that promote cell death (51). AIF, a key signaling molecule of the
caspase-independent apoptotic pathway, is localized in the
mitochondrial intermembrane space. AIF is released into the
cytoplasm in response to mitochondrial damage, where its
interaction with pro-death protein partners, such as cyclophilin A,
initiates translocation into the nucleus, leading to DNA
fragmentation (52). Previous
studies have demonstrated the neuroprotective effects of AIF
inhibition in vitro and in vivo (53,54).
The results of the present study confirmed previous observations of
glutamate-mediated increases in the transfer of AIF from the
mitochondria to the nucleus in HT22 cells (55). Immunocytochemical staining revealed
a high number of AIF positive cells under acute glutamate
challenge. B355252 abrogated the nuclear translocation of AIF, and
was neuroprotective when applied prior to glutamate insult.
In conclusion, the phenoxythiophene compound B355252
conferred neuroprotective effects on HT22 cells in vitro by
attenuating glutamate-induced dysfunction of the mitochondrial
fission process. The glutamate-induced upregulation of Drp1 and
Fis1 was effectively blocked by B355252. In contrast with previous
studies, glutamate increased, rather than inhibited Opa1 and Mfn1/2
expression to promote mitochondrial dysfunction in the HT22 cell
model. B355252 inhibited the glutamate-induced effects on these
proteins and protected the cells against mitochondrial dysfunction.
In addition, B355252 prevented the translocation of AIF from the
mitochondria to the nucleus. These results suggest that B355252
exerts its neuroprotective effects through mitochondria-dependent
events. However, the molecular targets of B355252 and its
mechanisms of action remain to be elucidated.
Acknowledgements
The authors greatly appreciate Dr Hernán Navarro
from North Carolina Central University (Durham, North Carolina) for
proofreading and English editing.
Funding
YZ is supported by the Project of Innovation and
Entrepreneurship for Overseas Students of Ningxia Hui Autonomous
Region (2018) and Ningxia Natural Science Foundation (grant no.
2019AAC03236). Biomanufacturing Research Institute and Technology
Enterprise is partially funded by the Golden Leaf Foundation.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PAL and GI conceived and designed the experiments.
YZ and NSG performed the experiments. YZ and PAL analyzed the data.
YZ, PAL and GI wrote the manuscript. YZ, GI and PAL have confirmed
the authenticity of the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fernandes D and Carvalho AL: Mechanisms of
homeostatic plasticity in the excitatory synapse. J Neurochem.
139:973–996. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Dzubay JA and Jahr CE: The concentration
of synaptically released glutamate outside of the climbing
fiber-Purkinje cell synaptic cleft. J Neurosci. 19:5265–5274.
1999.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Choi DW: Glutamate neurotoxicity and
diseases of the nervous system. Neuron. 1:623–634. 1988.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lau A and Tymianski M: Glutamate
receptors, neurotoxicity and neurodegeneration. Pflugers Arch.
460:525–542. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lewerenz J and Maher P: Chronic glutamate
toxicity in neurodegenerative diseases-what is the evidence? Front
Neurosci. 9(469)2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mehta A, Prabhakar M, Kumar P, Deshmukh R
and Sharma PL: Excitotoxicity: Bridge to various triggers in
neurodegenerative disorders. Eur J Pharmacol. 698:6–18.
2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Jezek J, Cooper KF and Strich R: Reactive
oxygen species and mitochondrial dynamics: The Yin and Yang of
mitochondrial dysfunction and cancer progression. Antioxidants
(Basel). 7(13)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bock FJ and Tait SWG: Mitochondria as
multifaceted regulators of cell death. Nat Rev Mol Cell Biol.
21:85–100. 2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Vakifahmetoglu-Norberg H, Ouchida AT and
Norberg E: The role of mitochondria in metabolism and cell death.
Biochem Biophys Res Commun. 482:426–431. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Archer SL: Mitochondrial
dynamics-mitochondrial fission and fusion in human diseases. N Engl
J Med. 369:2236–2251. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Westermann B: Mitochondrial fusion and
fission in cell life and death. Nat Rev Mol Cell Biol. 11:872–884.
2010.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Tandler B, Hoppel CL and Mears JA:
Morphological pathways of mitochondrial division. Antioxidants
(Basel). 7(30)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Song Z, Ghochani M, McCaffery JM, Frey TG
and Chan DC: Mitofusins and OPA1 mediate sequential steps in
mitochondrial membrane fusion. Mol Biol Cell. 20:3525–3532.
2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yeyeodu ST, Witherspoon SM, Gilyazova N
and Ibeanu GC: A rapid, inexpensive high throughput screen method
for neurite outgrowth. Curr Chem Genomics. 4:74–83. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Williams AL, Dandepally SR, Gilyazova N,
Witherspoon SM and Ibeanu G: Microwave-assisted synthesis of
4-chloro-N-(naphthalen-1-ylmethyl)-5-(3-(piperazin-1-yl)phenoxy)thiophene-2-sulfo
namide (B-355252): A new potentiator of Nerve Growth Factor
(NGF)-induced neurite outgrowth. Tetrahedron. 66:9577–9581.
2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Gliyazova NS, Huh EY and Ibeanu GC: A
novel phenoxy thiophene sulphonamide molecule protects against
glutamate evoked oxidative injury in a neuronal cell model. BMC
Neurosci. 14(93)2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Gliyazova NS and Ibeanu GC: The chemical
molecule B355252 is neuroprotective in an in vitro model of
Parkinson's disease. Cell Mol Neurobiol. 36:109–122.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kumari S, Mehta SL and Li PA: Glutamate
induces mitochondrial dynamic imbalance and autophagy activation:
Preventive effects of selenium. PLoS One. 7(e39382)2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Mendelev N, Mehta SL, Witherspoon S, He Q,
Sexton JZ and Li PA: Upregulation of human selenoprotein H in
murine hippocampal neuronal cells promotes mitochondrial biogenesis
and functional performance. Mitochondrion. 11:76–82.
2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Sattler R and Tymianski M: Molecular
mechanisms of glutamate receptor-mediated excitotoxic neuronal cell
death. Mol Neurobiol. 24:107–129. 2001.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Stanciu M, Wang Y, Kentor R, Burke N,
Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW
and DeFranco DB: Persistent activation of ERK contributes to
glutamate-induced oxidative toxicity in a neuronal cell line and
primary cortical neuron cultures. J Biol Chem. 275:12200–12206.
2000.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Kang Y, Tiziani S, Park G, Kaul M and
Paternostro G: Cellular protection using Flt3 and PI3Kalpha
inhibitors demonstrates multiple mechanisms of oxidative glutamate
toxicity. Nat Commun. 5(3672)2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bavarsad Shahripour R, Harrigan MR and
Alexandrov AV: N-acetylcysteine (NAC) in neurological disorders:
Mechanisms of action and therapeutic opportunities. Brain Behav.
4:108–122. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
24
|
Ma YM, Ibeanu G, Wang LY, Zhang JZ, Chang
Y, Dong JD, Li PA and Jing L: Selenium suppresses glutamate-induced
cell death and prevents mitochondrial morphological dynamic
alterations in hippocampal HT22 neuronal cells. BMC Neurosci.
18(15)2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sanderson TH, Raghunayakula S and Kumar R:
Release of mitochondrial Opa1 following oxidative stress in HT22
cells. Mol Cell Neurosci. 64:116–122. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Otera H, Wang C, Cleland MM, Setoguchi K,
Yokota S, Youle RJ and Mihara K: Mff is an essential factor for
mitochondrial recruitment of Drp1 during mitochondrial fission in
mammalian cells. J Cell Biol. 91:1141–1158. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Palmer CS, Osellame LD, Laine D,
Koutsopoulos OS, Frazier AE and Ryan MT: MiD49 and MiD51, new
components of the mitochondrial fission machinery. EMBO Rep.
12:565–573. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Rosenbloom AB, Lee SH, To M, Lee A, Shin
JY and Bustamante C: Optimized two-color super resolution imaging
of Drp1 during mitochondrial fission with a slow-switching Dronpa
variant. Proc Natl Acad Sci USA. 111:13093–13098. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Smirnova E, Griparic L, Shurland DL and
van der Bliek AM: Dynamin-related protein Drp1 is required for
mitochondrial division in mammalian cells. Mol Biol Cell.
12:2245–2256. 2001.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Smirnova E, Shurland DL, Ryazantsev SN and
van der Bliek AM: A human dynamin-related protein controls the
distribution of mitochondria. J Cell Biol. 143:351–358.
1998.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Frank S, Gaume B, Bergmann-Leitner ES,
Leitner WW, Robert EG, Catez F, Smith CL and Youle RJ: The role of
dynamin-related protein 1, a mediator of mitochondrial fission, in
apoptosis. Dev Cell. 1:515–525. 2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Stojanovski D, Koutsopoulos OS, Okamoto K
and Ryan MT: Levels of human Fis1 at the mitochondrial outer
membrane regulate mitochondrial morphology. J Cell Sci.
117:1201–1210. 2004.PubMed/NCBI View Article : Google Scholar
|
|
33
|
James DI, Parone PA, Mattenberger Y and
Martinou JC: hFis1, a novel component of the mammalian
mitochondrial fission machinery. J Biol Chem. 278:36373–36379.
2003.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tian Y, Li B, Shi WZ, Chang MZ, Zhang GJ,
Di ZL and Liu Y: Dynamin-related protein 1 inhibitors protect
against ischemic toxicity through attenuating mitochondrial Ca2+
uptake from endoplasmic reticulum store in PC12 cells. Int J Mol
Sci. 15:3172–3185. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Cribbs JT and Strack S: Reversible
phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and
calcineurin regulates mitochondrial fission and cell death. EMBO
Rep. 8:939–944. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chang CR and Blackstone C: Cyclic
AMP-dependent protein kinase phosphorylation of Drp1 regulates its
GTPase activity and mitochondrial morphology. J Biol Chem.
282:21583–21587. 2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Taguchi N, Ishihara N, Jofuku A, Oka T and
Mihara K: Mitotic phosphorylation of dynamin-related GTPase Drp1
participates in mitochondrial fission. J Biol Chem.
282:11521–11529. 2007.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Otera H and Mihara K: Molecular mechanisms
and physiologic functions of mitochondrial dynamics. J Biochem.
149:241–251. 2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Bartolák-Suki E, Imsirovic J, Nishibori Y,
Krishnan R and Suki B: Regulation of mitochondrial structure and
dynamics by the cytoskeleton and mechanical factors. Int J Mol Sci.
18(1812)2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Meeusen S, McCaffery JM and Nunnari J:
Mitochondrial fusion intermediates revealed in vitro. Science.
305:1747–1752. 2004.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wai T, García-Prieto J, Baker MJ,
Merkwirth C, Benit P, Rustin P, Rupérez FJ, Barbas C, Ibañez B and
Langer T: Imbalanced OPA1 processing and mitochondrial
fragmentation cause heart failure in mice. Science.
350(aad0116)2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bertholet AM, Delerue T, Millet AM, Moulis
MF, David C, Daloyau M, Arnauné-Pelloquin L, Davezac N, Mils V,
Miquel MC, et al: Mitochondrial fusion/fission dynamics in
neurodegeneration and neuronal plasticity. Neurobiol Dis. 90:3–19.
2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kushnareva YE, Gerencser AA, Bossy B, Ju
WK, White AD, Waggoner J, Ellisman MH, Perkins G and Bossy-Wetzel
E: Loss of OPA1 disturbs cellular calcium homeostasis and
sensitizes for excitotoxicity. Cell Death Differ. 20:353–365.
2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Nguyen D, Alavi MV, Kim KY, Kang T, Scott
RT, Noh YH, Lindsey JD, Wissinger B, Ellisman MH, Weinreb RN, et
al: A new vicious cycle involving glutamate excitotoxicity,
oxidative stress and mitochondrial dynamics. Cell Death Dis.
2(e240)2011.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Jahani-Asl A, Poilon-Larose K, Xu W,
Maclaurin JG, Park DS, McBride HM and Slack RS: The mitochondrial
inner membrane GTPase, optic atrophy 1 (Opa1), restores
mitochondrial morphology and promotes neuronal survival following
excitotoxicity. J Biol Chem. 286:4772–4782. 2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Wang W, Zhang F, Li L, Tang F, Siedlak SL,
Fujioka H, Liu Y, Su B, Pi Y and Wang X: MFN2 couples glutamate
excitotoxicity and mitochondrial dysfunction in motor neurons. J
Biol Chem. 290:168–182. 2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Wang X, Su B, Siedlak SL, Moreira PI,
Fujioka H, Wang Y, Casadesus G and Zhu X: Amyloid-beta
overproduction causes abnormal mitochondrial dynamics via
differential modulation of mitochondrial fission/fusion proteins.
Proc Natl Acad Sci USA. 105:19318–19323. 2008.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Jahani-Asl A, Vheung EC, Neuspiel M,
MacLaurin JG, Fortin A, Park DS, McBride HM and Slack RS: Mitofusin
2 protects cerebellar granule neurons against injury-induced cell
death. J Biol Chem. 282:23788–23798. 2007.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Neuspiel M, Zunino R, Gangaraju S,
Rippstein P and McBride H: Activated mitofusin 2 signals
mitochondrial fusion, interferes with Bax activation, and reduces
susceptibility to radical induced depolarization. J Biol Chem.
280:25060–25070. 2005.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ju Wk, Lindsey JD, Angert M, Patel A and
Weinreb RN: Glutamate receptor activation triggers OPA1 release and
induces apoptotic cell death in ischemic rat retina. Mol Vis.
14:2629–2638. 2008.PubMed/NCBI
|
|
51
|
Halestrap AP: What is the mitochondrial
permeability transition pore? J Mol Cell Cardiol. 46:821–831.
2009.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Sevrioukova IF: Apoptosis-inducing factor:
Structure, function, and redox regulation. Antioxid Redox Signal.
14:2545–2579. 2011.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Jantas D, Greda A, Leskiewicz M, Grygier
B, Pilc A and Lason W: Neuroprotective effects of mGluR II and III
activators against staurosporine- and doxorubicin-induced cellular
injury in SH-SY5Y cells: New evidence for a mechanism involving
inhibition of AIF translocation. Neurochem Int. 88:124–137.
2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Piao CS, Loane DJ, Stoica BA, Li S,
Hanscom M, Cabatbat R, Blomgren K and Faden AI: Combined inhibition
of cell death induced by apoptosis inducing factor and caspases
provides additive neuroprotection in experimental traumatic brain
injury. Neurobiol Dis. 46:745–758. 2012.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Tobaben S, Grohm J, Seiler A, Conrad M,
Plesnila N and Culmsee C: Bid-mediated mitochondrial damage is a
key mechanism in glutamate-induced oxidative stress and
AIF-dependent cell death in immortalized HT-22 hippocampal neurons.
Cell Death Differ. 18:282–292. 2011.PubMed/NCBI View Article : Google Scholar
|