Introduction
Myelodysplastic syndrome (MDS) is a heterogeneous
clonal disease that affects hematopoietic stem cells leading to
dysplasia and ineffective hematopoiesis in the bone marrow
(1). Low-risk MDS progresses
slowly in patients, who typically have superior prognoses. By
contrast, patients with high-risk MDS, particularly those with
MDS-refractory anemia with excess blast-1 and -2 (MDS-RAEB I/II),
have a 3-year disease-free survival rate of only 30% after
hematopoietic stem cell transplantation (HSCT) due to recurrence
(2). Therefore, it is important to
explore novel therapeutic methods for MDS. Guadecitabine (3) (a second-generation hypomethylating
agent), rigosertib (4) (a multiple
kinase inhibitor) and inhibitors of programmed cell death protein 1
(PD-1) (5)/programmed death ligand
1 (PD-L1) (6)/cytotoxic
T-lymphocyte-associated protein 4 (CTLA-4) (7) are among the agents used for treating
MDS.
Azacitidine (Vidaza, Pharmion) is a DNA
methyltransferase inhibitor that was first marketed in the United
States in July 2004. It is a cell cycle-specific drug acting on
cells in the S phase, and as a nucleoside analogue of cytidine, it
affects cellular DNA methylation by inhibiting DNA
methyltransferases; it may also incorporate into RNA and exert
direct cytotoxicity (8).
Azacitidine is mainly used for the clinical treatment of MDS with
refractory anemia (9). However,
patients with MDS treated with azacitidine monotherapy typically
have a high recurrence rate, low overall response rate (28-48%) and
prolonged response times (8-10 months) (10). The BCL-2 inhibitor venetoclax (VEN)
is used in combination with azacitidine for treating adult patients
with acute myeloid leukemia (AML) who are ineligible for intensive
induction chemotherapy in the USA (11). Previous clinical studies on VEN
have been extended to include hematological tumors, such as chronic
lymphocytic leukemia (CLL) and relapsed/refractory multiple myeloma
(R/R MM). Venetoclax monotherapy has demonstrated antimyeloma
activity in patients with R/R MM positive for t(11;14); the overall
response rate was 21% (12,13).
However, the full extent of the effects of the azacytidine/VEN
remains to elucidated.
In previous studies, VEN was shown to inhibit
mitochondrial reactive oxygen species (ROS) metabolism through
BCL-2; the characteristic feature of most functionally defined
leukemia stem cells is relatively low levels of reactive oxygen
species, and these LSCs exhibit abnormal overexpression of BCL-2
(14,15). It has also been reported that
regulating cellular mitochondrial ROS metabolism can affect the
efficacy of VEN (16,17). However, another study previously
found that VEN can exert inhibitory effects on mitochondrial
metabolism through the activating transcription factor 4
(ATF4)-pathway, independent of BCL-2(18). In addition, a study has suggested
that azacitidine can reduce the levels of myeloid cell leukemia-1
in older patients with AML (19),
which is an anti-apoptotic protein and a potential source of
resistance to VEN (20). In
particular, the effects of VEN on the efficacy of azacitidine
remain unclear.
VEN treatment has been shown to inhibit
mitochondrial reactive oxygen species metabolism in various cells,
such as primary leukemia cells, MCF7 breast cancer cells and the
CT26 colorectal cancer cell line (14,15,18).
It has been previously demonstrated that altering mitochondrial
glutamine metabolism can promote drug sensitivity to tumor cells,
such as HeLa and HCT116 cells (21). DNA damage can inhibit mitochondrial
glutamine metabolism. During glutamine metabolic inhibition, the
tricarboxylic acid cycle is blocked, which is essential for the DNA
damage response. This eventually leads to delayed DNA repair and
aggravates DNA damage in primary mouse embryonic fibroblasts
(22). Glutamine is important for
the maintenance of the cellular redox balance by removing ROS and
supporting the synthesis of glutathione (GSH), an antioxidant that
serves a key regulatory role against oxidative DNA damage (23). It has previously been shown that
increasing intracellular GSH levels can impair DNA methylation
(24). This phenomenon can be
explained by the fact that the GSH precursor cysteine is also
synthesized from the homocysteine pool necessary for the synthesis
of S-adenosine methionine, a co-factor for DNA and histone
methylation (25). However,
glutamine is a non-essential amino acid but can serve various
physiological functions. α-ketoglutarate is generated through
glutaminolysis and directly enters the tricarboxylic acid cycle in
the mitochondria, which feeds into the aerobic respiratory pathway
and anabolism (26). Furthermore,
glutamate is the metabolite of glutamine and is directly involved
in the synthesis of GSH (27).
In the present study, metabolic activity in the
mitochondria and cell cycle progression were assessed in an MDS
cell line after treatment with both VEN and azacitidine.
Materials and methods
Cell culture and treatment
The leukemic cell line SKM-1 (cat. no. CCL-95) was
purchased from American Type Culture Collection. SKM-1 cells were
cultured in an RPMI-1640 medium (cat. no. 10-040-CV; Corning, Inc.)
containing 10% FBS (cat. no. 04-001-1ACS; Biological Industries) at
37˚C in 5% CO2. SKM-1 cells were treated with 0-2 µM
venetoclax and 0-4 µM azacytidine or a combination of the two for
24, 48 and 72 h of treatment. In some specified experiments, 1 mM
GSH (cat. no. S4606; Selleck Chemicals) was added at the beginning
of venetoclax and azacytidine treatment at 37˚C for 24 and 48
h.
Reverse transcription-quantitative
(q)PCR
Total RNA was extracted using the TRIzol®
method (Invitrogen; Thermo Fisher Scientific, Inc.) and
reverse-transcribed into cDNA using Hifair® III 1st
Strand cDNA Synthesis SuperMix (cat. no. 11137ES10; Shanghai Yeasen
Biotechnology Co., Ltd.) following the manufacturer's instructions.
qPCR was used to analyze isocitrate dehydrogenase 2 (IDH2)
expression using the ABI 7500 system (Thermo Fisher Scientific,
Inc.) and the Hieff® qPCR SYBR Green Master Mix (cat.
no. 11201ES08; Shanghai Yeasen Biotechnology Co., Ltd.). The
thermocycling conditions used for qPCR were as follows: 95˚C for 5
min; followed by 95˚C for 10 sec and 60˚C for 30 sec for 40 cycles;
the melting curve stage was set to the instrument's default
settings. β-actin was used as the reference gene and
2-ΔΔCq method (28) was
used for data analysis. The primer sequences are listed below: IDH2
forward, 5'-CGCCACTATGCCGACAAAAG-3' and reverse,
5'-ACTGCCAGATAATACGGGTCA-3'; alanine-serine-cysteine transporter 2
(ASCT2) forward, 5'-GTGGCGCTGCGGAAGCT-3' and reverse,
5'-GGCGTACCACATGATCCAG-3' and β-actin forward,
5'-GTCATCACCATTGGCAATGAG-3' and reverse,
5'-CGTCATACTCCTGCTTGCTG'-3.
Flow cytometry
Cell cycle analysis was conducted using the fixed
cell staining method. The treated cells were first washed twice in
PBS, before ice-cold 70% ethanol was used to fix pelleted cells at
4˚C for 40 min. After fixation, cells were washed twice in PBS,
before being centrifuged at 850 x g at 4˚C for 5 min. RNAse (100
µg/ml; cat. no. 9001-99-4; Beijing Solarbio Science &
Technology Co., Ltd.) was then added to the cells and incubated at
37˚C for 15 min. In total, 1x106 cells/200 µl (50 µg/ml)
propidium iodide (PI; cat. no. P4170; MilliporeSigma) were added
and incubated at 4˚C for 15 min in the dark before the sample was
analyzed using Cytoflex LX (Beckman Coulter, Inc.). Data analysis
was conducted using FlowJo 10.8.1 (Becton Dickinson & Company).
An example of the gated strategy used is shown in Fig. S1.
Western blotting
Western blotting was performed to detect the protein
expression of IDH2 (1:1,000 dilution; cat. no. 60322; Cell
Signaling Technology, Inc.), ASCT2 (1:1,000 dilution; cat. no.
ab187692; Abcam), CCAAT/enhancer-binding protein homologous protein
(1:1,000 dilution; CHOP; cat. no. 5554; Cell Signaling Technology,
Inc.), Retinoblastoma protein (1:1,000 dilution; Rb; cat. no. 9309;
Cell Signaling Technology, Inc.) and GAPDH (1:5,000 dilution; cat.
no. 3683; Cell Signaling Technology, Inc.) following VEN and/or
azacitidine treatment. Prepare protein standard solutions at
concentrations of 0, 0.025, 0.05, 0.1, 0.2, 0.3, 0.4 and 0.5 mg/ml.
A total of 20 µl of each standard or lysate was added to each well
of a 96-well plate. Next, 200 µl BCA working solution was added to
each well and incubated at 37˚C for 30 min. The absorbance was
measured at 562 nm using a spectrophotometer (iMARK; Bio-Rad
Laboratories, Inc.), and the protein concentration of the samples
was calculated based on the standard curve. Proteins (20 µg/well)
were analyzed on a 12% gel using SDS-PAGE, and the proteins were
transferred onto PVDF membranes. The membranes were then blocked
with 5% BSA (cat. no. V900933; Vetec™; MilliporeSigma) at 26˚C for
1 h, before primary antibodies were added for incubation overnight
at 4˚C with shaking. The membranes were then washed with TBST (0.1%
Tween 20) for 5 min for four times. Subsequently, HRP-conjugated
goat anti-mouse (1:10,000 dilution; cat. no. 7076; Cell Signaling
Technology, Inc.) or anti-rabbit (1:10,000 dilution; cat. no. 7074;
Cell Signaling Technology, Inc.) IgG secondary antibodies were
added at 26˚C for 60 min. After washing with TBST for four times,
membranes were incubated for 3 min in Supersignal™ West Pico Plus
Chemiluminescent Substrate (cat. no. 34580; Thermo Fisher
Scientific, Inc.) and visualized using the iBright FL1000 Imaging
System (Thermo Fisher Scientific Inc.). iBright Analysis Software
(Desktop Version 5.1.0; Thermo Fisher Scientific Inc.) was used for
image quantification.
Cell Counting Kit-8 (CCK-8) assay
The cells (2,500 cells/100 µl) were first cultured
in 96-well plates for 24 h at 37˚C and 5% CO2. After the
aforementioned treatments for 12, 24, 48 and 72 h at 37˚C, cells
were washed with PBS, before 10 µl CCK-8 solution (cat. no. CA1210;
Beijing Solarbio Science & Technology Co., Ltd.) was added into
the wells and incubated for 4 h at 37˚C. Finally, the optical
density at 490 nm was measured using a microplate reader (iMARK;
Bio-Rad Laboratories, Inc.).
Metabolite measurement and
analysis
The glutamine concentration in the media was
measured using a colorimetric method with a Glutamine (Gln)
Colorimetric Assay Kit (cat. no. E-BC-K853-M; Elabscience
Biotechnology, Inc.) according to the manufacturer's instructions,
using a 50-kDa ultrafiltration filter (cat. no. UFC905096;
MilliporeSigma). The culture medium was centrifuged at 3,000 x g at
4˚C for 40 min. and the filtrate collected for analysis. Prior to
testing, the reagents were equilibrated to room temperature.
Working solutions and reaction working solutions were prepared
according to the manufacturer's instructions, kept on ice and then
the prepared reaction working solution was used within 1 h. A 0-2
mM glutamine standard solution was prepared. A total of 30 µl
working solution was added to each well of a 96-well plate, and
then 50 µl of the standard or test sample was added to each well.
The plate was incubated at 37˚C in the dark for 20 min, and then
140 µl reaction working solution was added to each well. The
absorbance was measured at 450 nm (OD A1) for each well using a
microplate reader. The plate was incubated at 37˚C in the dark for
an additional 30 min. The absorbance was measured at 450 nm (OD A2)
for each well using a microplate reader. The change in OD was then
calculated (ΔA=A2-A1). The ΔA values from the standard solutions
were used to construct a standard curve and the glutamine
concentration of each sample was calculated based on the standard
curve. In total, 1x106 to 2x106 cells/well of
a 6-well plate were seeded and glutamine concentration was
normalized to the number of cells in each well. Glutaminase
activity was measured using a glutaminase (GLS) activity assay kit
(AKAM007M; Beijing Box Biotechnology Co., Ltd.) and a
spectrophotometer at 630 nm.
Mitochondrial membrane potential (Δψm) was measured
using the fluorescent tetramethylrhodamine methyl (TMRM) ester
probe (Sigma-Aldrich; Merck KGaA). After drug treatment, the cells
were washed once with PBS, before TMRM at a working concentration
of 100 nM and 1 µg/ml Hoechst 33342 (cat. no. H3570; Invitrogen;
Thermo Fisher Scientific, Inc.) were added for nuclear staining.
Cells were incubated for 30 min at 37˚C, followed by another wash
with PBS. The cells were then plated on a confocal culture dish and
observed using a confocal microscope (LSM880; Carl Zeiss AG). The
TMRM excitation wavelength was set to 488 nm, whereas the emission
wavelength was set to 573 nm. The Hoechst excitation wavelength was
350 nm, with an emission wavelength of 461 nm.
Liquid chromatography (LC)-mass
spectrometry (MS)
Cells, washed with cold PBS, were treated with the
extraction buffer [5:3:2, methanol: acetonitrile: water; V/V].
After treatment with extraction buffer, the pellet was discarded
after centrifugation at 20,000 x g at 4˚C for 20 min. The
supernatant was analyzed using LC-MS.
A Millipore™ ZIC-pHILIC (2.1x150 mm, 5 µm) LC column
(cat. no. 150454; MilliporeSigma) was coupled to a Nexera XR system
(Shimadzu Corporation). In total, 20 µl were transferred into LC
vials containing glass inserts for analysis. Samples were then
subjected to an LC-MS analysis to detect and quantify known peaks.
The column oven temperature and flow rate were set to 25˚C and 100
µl/min, respectively. Mobile phase compositions were as follows: A,
10 mM ammonium carbonate in water (pH 9.0); and B, acetonitrile
hypergrade for LC/MS, ≤100% (cat. no. 100029; MilliporeSigma). The
following gradient elutions were used: 80-20% B, 0-30 min; 20-80%
B, 30-31 min; and hold at 80% B, 31-42 min. The LC system was
coupled to a Q Exactive™ HF mass spectrometer (Thermo Fisher
Scientific, Inc.) operating in heated electrospray ionization mode
(ESI) for LC-MS analysis. Negative ion mode was used in mass
spectrometry. The following default settings for ESI source were
used: Nebulizer gas pressure, 15 psi; drying gas flow rate, 7
l/min; and drying gas temperature, 300˚C. The capillary voltage
between the MS and nebulizer was ±3,500 V. All remaining ion
transport parameters were determined using the Target Mass (TM)
parameter, set by the operator. The TM was set as the closest value
rounded to the nearest 50 of the expected mass:charge ratio (m/z).
Target metabolites measured in the study: Glutathione (theoretical
m/z, 307.084355); succinate (theoretical m/z, 118.027157); and α-KG
(theoretical m/z, 146.022072).
Commercial standards, glutathione (cat. no.
YZ-140706), succinate (cat. no. SS9520) and α-KG (SK8210) (all
Beijing Solarbio Science & Technology Co., Ltd.), were run on
the system prior to analysis as a quality control. For the
preparation of standards, a 1-mM stock solution was first prepared
for each metabolite standard and stored at -80˚C. On the day of the
LC/MS run, the 1-mM stock solution and extraction solution was used
to prepare fresh standard curves at the following concentrations:
1, 10 and 100 nM, and 1, 10 and 30 µM. For each metabolite,
authentic standards were first run on the LC/MS instrument to
confirm that they produced stable peaks at the correct m/z ratio
and the retention time of the correct molecule. Peak areas were
normalized to the amount of protein used. The BCA method was used
to quantify protein content according to the manufacturer's
protocol.
Statistical analysis
Data are presented as the mean ± standard deviation
of three independent experimental repeats. An unpaired Student's
t-test was used to assess the differences between two groups,
one-way ANOVA was used for multiple comparisons followed by a
Holm-Šídák's multiple comparisons test. One-way ANOVA (parametric)
was used for multiple comparisons for the rest of the results.
Dunnett's test was used for each other treatment group compared
with the control group. Graphpad Prism 9.0 software (Graphpad
Software. Inc.; Dotmatics) was used for statistical analysis.
P<0.05 was considered to indicate a statistically significant
difference.
Results
VEN treatment inhibits mitochondrial
glutamine metabolism in the SKM-1 cell line
The potential effects of VEN treatment on metabolism
in SKM-1 cells were first evaluated. The SKM-1 cell line, which was
originally established from a patient with myelomonocytic leukemia,
has been frequently used for MDS research (29,30).
The clinical diagnosis and disease progression records of the
patient, who was initially diagnosed with MDS, refractory anemia
with an excess of blasts in transformation according to the
French-American-British criteria (31), are well documented (29,32).
Over the following 7 months, low-dose cytarabine therapy was
administered multiple times until the patient developed resistance
to treatment and subsequently developed AML (32). Therefore, SKM-I cell line was
considered suitable for the present study on MDS-RA. Different
concentrations of VEN were used for cell viability analysis, where
the results showed that VEN began to exert an inhibitory effect at
a concentration of 0.1 µM. In particular, there was little
difference in the inhibitory effects at concentrations of between
0.1 and 1 µM (Fig. 1A).
Furthermore, the extent of intracellular metabolism in the cells
treated with 0.1-1 µM VEN was assessed. The results showed that the
levels of α-ketoglutarate (α-KG) were increased but not
significantly so, suggesting that VEN can regulate α-KG metabolism
upstream of the IDH2 pathway (Fig.
1B). In addition, a significant accumulation of succinate
levels was also observed, suggesting that mitochondrial glutamine
metabolism was affected by VEN treatment (Fig. 1B). The consumption of glutamine and
the activity of glutaminase were next examined. The results
indicated that glutamine uptake was significantly inhibited by 0.1
µM VEN (Fig. 1C), whilst the
activity level of glutaminase remained almost unchanged (Fig. 1D). This suggests that VEN can
inhibit mitochondrial glutamine metabolism independent of
glutaminase activity.
VEN inhibits the mitochondrial
metabolism through ATF4 and ASCT2
According to the aforementioned results, the part of
mitochondrial metabolism inhibited by VEN appears to be independent
of glutaminase. Therefore, the expression levels of other key
markers involved in mitochondrial metabolism were next assessed.
Generic mitochondrial metabolic activity in the form of the
membrane potential was first measured after VEN treatment, where
the results showed that mitochondrial metabolism decreased to a
certain extent, but remained non-significant until the
concentration of VEN reached 2 µM (Fig. 2A and B). Western blotting results showed that
the expression levels of ATF4 and CHOP were increased 24 h after
VEN treatment, which was consistent with the previous results that
mitochondrial metabolic function was inhibited (16,33)
(Fig. 2C). In addition, the
expression level of IDH-2 was also increased, which was consistent
with the results shown in Fig. 1B,
as IDH-2 is a key metabolic enzyme catalyzing the interconversion
of isocitrate to α-KG (34).
Notably, there was a decrease in the expression of ASCT2, a key
transport protein required for glutamine uptake, at 24-h post
treatment (Fig. 2D). ASCT2
inhibition has been shown to prevent HCC1806 breast cancer cell
line proliferation by inhibiting glutamine uptake (35). This suggests that VEN may inhibit
the uptake of glutamine by inhibiting ASCT2 expression. During the
72-h treatment, ASCT2 transcription levels were first decreased at
24 h before recovery. The transcription levels of IDH2 continued to
increase; however, this was not statistically significant (Fig. 2D).
 | Figure 2Venetoclax affects ASCT2-mediated
glutamine uptake. (A) Representative TMRM fluorescence images of
each group, with red fluorescence representing mitochondria and
blue fluorescence representing the cell nuclei. Scale bar, 20 µm.
(B) Detection of mitochondrial membrane potential after treatment
with different concentrations of Venetoclax for 24 h. (C) After 24
h Venetoclax treatment in vitro, the expression of ATF4,
CHOP, ASCT2, IDH2 and Rb was measured through western blotting.
ATF4 and CHOP expression was markedly increased in the Venetoclax
group compared with that in the control group. (D) The
transcriptional levels of IDH2 and ASCT2 at different time points
after drug administration were detected using reverse
transcription-quantitative PCR (n=3). Results are presented as the
means ± standard deviation. *P<0.05 vs. 0 µM
venetoclax. **P<0.01 vs. 0 h post treatment. TMRM,
tetramethylrhodamine methyl ester; CHOP, CCAAT/enhancer-binding
protein homologous protein; ATF4, activating transcription factor
4; ASCT2, alanine-serine-cysteine transporter 2; IDH2, isocitrate
dehydrogenase 2; Rb, retinoblastoma. |
Low concentrations of VEN do not
promote the cytotoxic activity of azacytidine
The synergistic mechanism between azacitidine, a DNA
methyltransferase and VEN requires further evaluation. Therefore,
the optimal concentration of the combination of the two drugs was
first evaluated in vitro in the present study. At a lower
concentration of VEN (0.1 µM) and 2 µM of azacitidine, the
synergistic effect of VEN and azacitidine began to appear. By
contrast, when 0.5 and 1 µM azacitidine were used, the two drugs
could not produce a synergistic effect (Fig. 3A). When 0.5 and 1 µM VEN were used,
a synergistic cytotoxic effect of VEN and AZA was observed in all
concentration groups of AZA (Fig.
3B). Cell cycle progression after treatment with 0.1 µM VEN and
1 µM azacytidine was next tested, where the results showed that VEN
partially inhibited the inhibitory effect of azacitidine on S-phase
entrances (Fig. 3C and D). In addition, the proportion of cells
in the G2/M phase was significantly decreased in
response to the combination treatment compared with that in cells
treated with azacytidine alone (Fig.
3E). However, the use of 1 µM azacitidine had no effect on the
VEN-induced inhibition of mitochondrial glutamine metabolism
(Fig. 3F).
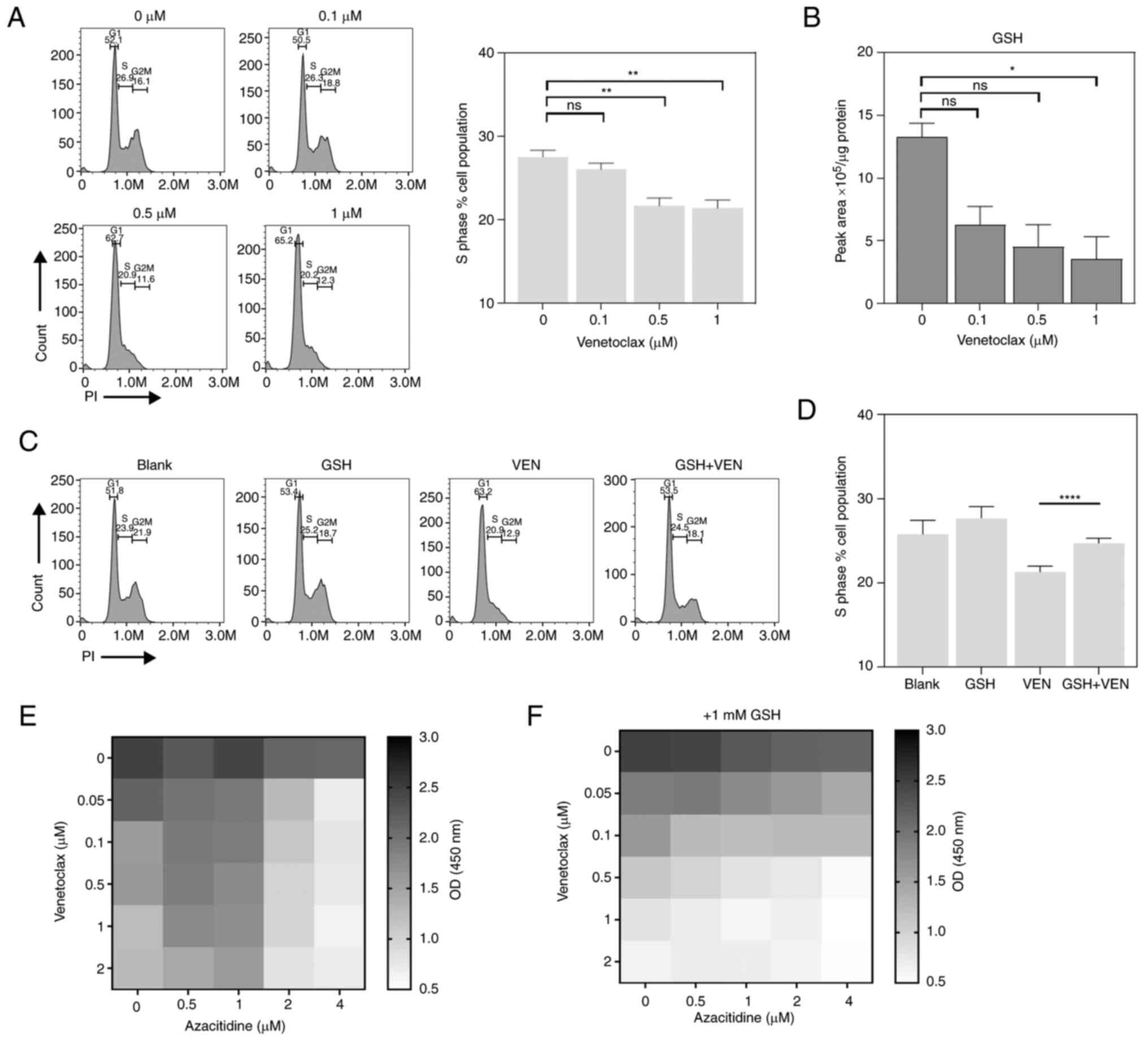
GSH can promote the synergistic effect
of VEN and azacytidine
According to the aforementioned results, it is
likely that azacytidine can ‘compensate’ for the effects of VEN on
DNA repair to some extent. In previous reports, azacitidine was
found to exert comparatively more potent cytotoxic effects on cells
in S phase (36,37). When VEN and azacitidine were
co-administered, there was a significant decrease in the proportion
of S phase cells compare with that in the azacitidine monotreatment
group. Further analysis of the proportion of S phase cells
following VEN monotherapy revealed that as the concentration of VEN
increased, the proportion of S phase cells also decreased, reaching
significance at 0.5 and 1 µM (Fig.
4A). The reduction in S-phase was positively associated with
the decrease in glutamine levels (Fig.
1C). The decrease in glutamine levels suggests that VEN may be
influencing cellular glutaminolysis, where glutamine generates
glutamate, which then maintains the intracellular redox state by
producing GSH (38). Therefore,
the level of intracellular GSH was further examined. The results
showed that as the concentration of VEN increased, there was a
corresponding decrease in intracellular GSH levels (Fig. 4B). This observation suggests that
replenishing GSH may contribute to the efficacy of VEN +
azacitidine treatment. Through supplementation experiments to
increase the GSH levels in the culture environment, it was found
that when 1 mM GSH was used, there was a significant recovery in
the proportion of S phase cells compared with that in the VEN
monotherapy group (Fig. 4C and
D). This may be due to VEN
affecting glutamine metabolism, thereby influencing the cell cycle.
When 1 mM glutathione was added to the culture medium, the
synergistic effect of azacitidine and VEN was also markedly
improved (Fig. 4E and F). Treatment using 0.1 µM VEN with 1 µM
azacitidine caused a notable inhibition of cell viability in SKM-1
cells in the presence of GSH compared with its absence.
Discussion
MDS frequently occurs in middle-aged and elderly
patients, of whom only ~10% are eligible for HSCT (39). The safety and efficacy of VEN +
azacitidine as the initial treatment of AML have been previously
proven (40). Tsao et al
(41) previously reported that VEN
combined with azacitidine exerted significant synergistic effects
and inhibited the proliferation of AML cells in vitro. In
another study, Pan et al (42) demonstrated that VEN rapidly killed
AML cells in a murine primary xenograft model. Bogenberger et
al (43) observed that VEN +
azacitidine therapy directly inhibited tumor cell proliferation in
patients with MDS. In addition, DiNardo et al (44) previously applied VEN + azacitidine
to treat patients with R/R AML and MDS, where the efficacy and
safety of VEN + azacitidine were identified. An objective response
was observed in 9 (21%) patients, including 2 complete responses.
However, the mechanism of this possible VEN/azacitidine synergism
remains unclear.
VEN is a selective inhibitor of BCL-2. In 2016, it
was approved by the United States Food and Drug Administration for
the treatment of CLL with chromosome 17p deletion (45). In recent years, the potential
application of VEN for the treatment of AML/MDS has been
extensively investigated in the USA and Europe, the consistency and
efficacy of which have been widely verified (46,47).
However, there have only been a small number of relevant reports
from Asian countries (48,49) and to the best of our knowledge, no
investigations involving patients of Han ethnicity in China have
been performed. BCL-2 inhibition is a novel targeted therapy for
the treatment of AML, which can activate the mitochondrial
apoptotic pathway in AML cells (42). In patients with high-risk MDS, high
BCL-2 expression has been reported (50). Previous studies have shown that
BCL-2 inhibitors are effective against high-risk MDS or secondary
AML (50,51).
Apart from targeting BCL-2, VEN has also been shown
to act on ATF4 upstream of altering mitochondrial metabolism
(18). Previous reports showed CD4
T cell function to depend on ATF4-mediated catabolic glycolysis and
glutaminolysis metabolism (52).
In a recent study, ATF4 was found to be highly expressed in a
variety of tumors, such as breast cancer (53) and melanoma (54), where it can promote tumor growth
through fibroblasts, as knockdown of ATF4 expression in fibroblasts
can significantly inhibit tumor angiogenesis and tumor growth in
melanoma and pancreatic tumors (54). This suggests that the ATF4 pathway
is a key factor mediating mitochondrial metabolism. In the present
study, VEN treatment was observed to not only promote ATF4
expression, but also inhibited ASCT2 and promoted IDH2 expression.
ATF4 is a factor that can promote tumor growth in breast cancer and
melanoma, but in the present results, ATF4 was highly expressed in
SKM-1 cells following VEN treatment. This suggests that there are
distinct molecular regulatory mechanisms between hematological
malignancies and their solid tumor counterparts. However, this also
suggests the potential of applying ATF4 inhibitors as a combination
therapy with VEN.
IDH2 catalyzes the reversible oxidative
decarboxylation of isocitrate into αKG in mitochondria whilst
reducing NADP+ to NADPH (55,56).
IDH2 is one of the genes showing the highest frequency of
mutations among other metabolic genes associated with human cancers
such as glioma (57) and
cholangiocarcinoma (58). IDH2 is
associated with cell metabolism and epigenetic regulation, where
IDH2 mutants have been reported to promote tumorigenesis
(59). IDH2 mutations
redirect carbon metabolites and oxidative phosphorylation towards
d-2-hydroxyglutarate (D-2HG) production, and elevated D-2HG may
somehow promote formation and progression of AML (60), although the mechanism by which
D-2HG promotes AML development remains unclear. In addition, lack
of the wild-type IDH1/2 enzyme can lead to downstream
vulnerabilities in gliomas (59),
which can improve the effects of small molecule inhibitors, such as
poly (ADP-Ribose) polymerase inhibitors, BCL-2 inhibitors and
biguanides (such as metformin, an antidiabetic drug that can
inhibit glutaminolysis) (61). The
sensitivity of cancer cells to radiotherapy and chemotherapy can
also be improved through knocking down wild-type IDH1
(62-64).
In a previous in vitro study on lung cancer cells, IDH2
knockdown by shRNA resulted in decreased HIF1α expression, leading
to the attenuation of lung cancer cell proliferation and tumor
growth (65). In the present
study, the expression of IDH2 was increased to some extent after
VEN treatment (Fig. 2C and
D), whereas the level of α-KG was
also increased to a certain extent, suggesting that VEN may
interfere with IDH2-related metabolism.
In addition to its impact on α-KG, it has been
reported that VEN can also affect mitochondrial metabolism
(16). In the present study, VEN
was found to inhibit the glutamine metabolic pathway. The products
regulated by the glutamine metabolism pathway, such as GSH, are
important for regulating the intracellular redox state (66). It has been previously shown that
the intracellular redox state serves an important role in
regulating the cell cycle (67,68).
Low-level cellular oxidation triggered by superoxide and hydrogen
peroxide activates proliferative cell signaling pathways, which are
necessary for physiological mitotic signal transduction (68). Oxidation events occurring during
the early G1 phase of the cell cycle are critical
regulatory steps for progression into the S phase (69). During the G1 phase,
cellular GSH levels are low, but the subsequent increases in total
GSH is necessary for cells to transition from G1 into
the S phase (70). The
intracellular levels of GSH were measured in the present study 24 h
following VEN treatment, which were observed to be significantly
decreased. This suggests that the application of VEN can lead to an
imbalance in the intracellular redox levels.
By GSH compensation, the synergistic effect of VEN +
azacytidine was enhanced. However, it is worth noting that
deficiency in GSH, which is an important antioxidant, can lead to
the excessive generation of ROS and subsequently cell apoptosis
(71). Previous studies have found
that GSH-mediated detoxification is involved in cisplatin
resistance in several types of tumors (72,73).
In addition, reducing GSH levels in cancer cells has been shown to
enhance the therapeutic effect of cisplatin and even reverse drug
resistance (74,75). Therefore, these results indirectly
suggest that an increase in the oxidative-reductive level of cells
does not induce VEN or AZA resistance.
In conclusion, results from the present study
suggest that VEN can inhibit glutamine metabolism, leading to a
reduction in intracellular GSH levels. This in turn causes cell
cycle arrest and affects the efficacy of azacitidine. The addition
of GSH was then found to promote the synergistic effects of VEN and
azacitidine in vitro. The present study provides a new
direction for the exploration into the synergistic effects of
azacitidine and VEN in clinical practice, by investigating the
impact of VEN on the cell cycle and glutamine metabolism in
MDS-RA.
Supplementary Material
Example of gating strategies used for
flow cytometry analysis. Gating strategy for cell cycle analysis in
Fig. 3C is shown. PI, propidium
iodide; FSC, forward scatter; SSC, side scatter.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Sanming Project of
Medicine in Shenzhen (grant no. SZSM201911004), Shenzhen Science
and Technology Plan Basic Research Project (grant nos.
JCYJ20180307150408596 and JCYJ20190809172403604) and Natural
Science Foundation of Guangdong Province (grant no.
2019A1515110703).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XX and DL contributed to the study conception and
design. Material preparation was performed by XW, data collection
was performed by XW and LY, and data analysis was performed by XW,
LY and BL. The first draft of the manuscript was written by XW and
LY and all authors commented on previous versions of the
manuscript. XW and XX confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Volpe VO, Garcia-Manero G and Komrokji RS:
Myelodysplastic Syndromes: A new decade. Clin Lymphoma Myeloma
Leuk. 22:1–16. 2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Montalban-Bravo G and Garcia-Manero G:
Myelodysplastic syndromes: 2018 update on diagnosis,
risk-stratification and management. Am J Hematol. 93:129–147.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Daher-Reyes GS, Merchan BM and Yee KWL:
Guadecitabine (SGI-110): An investigational drug for the treatment
of myelodysplastic syndrome and acute myeloid leukemia. Expert Opin
Investig Drugs. 28:835–849. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Navada SC and Silverman LR: The safety and
efficacy of rigosertib in the treatment of myelodysplastic
syndromes. Expert Rev Anticancer Ther. 16:805–810. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Haroun F, Solola SA, Nassereddine S and
Tabbara I: PD-1 signaling and inhibition in AML and MDS. Ann
Hematol. 96:1441–1448. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yang X, Ma L, Zhang X, Huang L and Wei J:
Targeting PD-1/PD-L1 pathway in myelodysplastic syndromes and acute
myeloid leukemia Exp Hematol. Oncol. 11(11)2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chien KS, Class CA, Montalban-Bravo G, Wei
Y, Sasaki K, Naqvi K, Ganan-Gomez I, Yang H, Soltysiak KA,
Kanagal-Shamanna R, et al: LILRB4 expression in chronic
myelomonocytic leukemia and myelodysplastic syndrome based on
response to hypomethylating agents. Leuk Lymphoma. 61:1493–1499.
2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Scott LJ: Azacitidine: A review in
myelodysplastic syndromes and acute myeloid leukaemia. Drugs.
76:889–900. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Salim O, Toptas T, Avsar E, Yucel OK,
Ozturk E, Ferhanoglu B, Geduk A, Mehtap O, Tombak A, Tiftik EN, et
al: Azacitidine versus decitabine in patients with refractory
anemia with excess blast-Results of multicenter study. Leuk Res.
45:82–89. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
de Lima M, Roboz GJ, Platzbecker U,
Craddock C and Ossenkoppele G: AML and the art of remission
maintenance. Blood Rev. 49(100829)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
DiNardo CD, Jonas BA, Pullarkat V, Thirman
MJ, Garcia JS, Wei AH, Konopleva M, Döhner H, Letai A, Fenaux P, et
al: Azacitidine and venetoclax in previously untreated acute
myeloid leukemia. N Engl J Med. 383:617–629. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kumar S, Kaufman JL, Gasparetto C, Mikhael
J, Vij R, Pegourie B, Benboubker L, Facon T, Amiot M, Moreau P, et
al: Efficacy of venetoclax as targeted therapy for
relapsed/refractory t(11;14) multiple myeloma. Blood.
130:2401–2409. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sidiqi MH, Al Saleh AS, Kumar SK, Leung N,
Jevremovic D, Muchtar E, Gonsalves WI, Kourelis TV, Warsame R,
Buadi FK, et al: Venetoclax for the treatment of multiple myeloma:
Outcomes outside of clinical trials. Am J Hematol. 96:1131–1136.
2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lagadinou ED, Sach A, Callahan K, Rossi
RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O'Dwyer
KM, et al: BCL-2 inhibition targets oxidative phosphorylation and
selectively eradicates quiescent human leukemia stem cells. Cell
Stem Cell. 12:329–341. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lucantoni F, Dussmann H, Llorente-Folch I
and Prehn JHM: BCL2 and BCL(X)L selective inhibitors decrease
mitochondrial ATP production in breast cancer cells and are
synthetically lethal when combined with 2-deoxy-D-glucose.
Oncotarget. 9:26046–26063. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sharon D, Cathelin S, Mirali S, Di Trani
JM, Yanofsky DJ, Keon KA, Rubinstein JL, Schimmer AD, Ketela T and
Chan SM: Inhibition of mitochondrial translation overcomes
venetoclax resistance in AML through activation of the integrated
stress response. Sci Transl Med. 11(eaax2863)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Guieze R, Liu VM, Rosebrock D, Jourdain
AA, Hernandez-Sanchez M, Martinez Zurita A, Sun J, Ten Hacken E,
Baranowski K, Thompson PA, et al: Mitochondrial Reprogramming
Underlies Resistance to BCL-2 inhibition in lymphoid malignancies.
Cancer Cell. 36:369–384 e13. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Roca-Portoles A, Rodriguez-Blanco G,
Sumpton D, Cloix C, Mullin M, Mackay GM, O'Neill K, Lemgruber L,
Luo X and Tait SWG: Venetoclax causes metabolic reprogramming
independent of BCL-2 inhibition. Cell Death Dis.
11(616)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
DiNardo CD, Pratz K, Pullarkat V, Jonas
BA, Arellano M, Becker PS, Frankfurt O, Konopleva M, Wei AH,
Kantarjian HM, et al: Venetoclax combined with decitabine or
azacitidine in treatment-naive, elderly patients with acute myeloid
leukemia. Blood. 133:7–17. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Phillips DC, Xiao Y, Lam LT, Litvinovich
E, Roberts-Rapp L, Souers AJ and Leverson JD: Loss in MCL-1
function sensitizes non-Hodgkin's lymphoma cell lines to the
BCL-2-selective inhibitor venetoclax (ABT-199). Blood Cancer J.
5(e368)2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hwang S, Yang S, Kim M, Hong Y, Kim B, Lee
EK and Jeong SM: Mitochondrial glutamine metabolism regulates
sensitivity of cancer cells after chemotherapy via amphiregulin.
Cell Death Discov. 7(395)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Jeong SM, Xiao C, Finley LW, Lahusen T,
Souza AL, Pierce K, Li YH, Wang X, Laurent G, German NJ, et al:
SIRT4 has tumor-suppressive activity and regulates the cellular
metabolic response to DNA damage by inhibiting mitochondrial
glutamine metabolism. Cancer Cell. 23:450–463. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Gerard-Monnier D and Chaudiere J:
Metabolism and antioxidant function of glutathione. Pathol Biol
(Paris). 44:77–85. 1996.PubMed/NCBI(In French).
|
|
24
|
Lertratanangkoon K, Wu CJ, Savaraj N and
Thomas ML: Alterations of DNA methylation by glutathione depletion.
Cancer Lett. 120:149–156. 1997.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hitchler MJ and Domann FE: An epigenetic
perspective on the free radical theory of development. Free Radic
Biol Med. 43:1023–1036. 2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yoo HC, Yu YC, Sung Y and Han JM:
Glutamine reliance in cell metabolism. Exp Mol Med. 52:1496–1516.
2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lu SC: Glutathione synthesis. Biochim
Biophys Acta. 1830:3143–3153. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Nakagawa T and Matozaki S: The SKM-1
leukemic cell line established from a patient with progression to
myelomonocytic leukemia in myelodysplastic syndrome
(MDS)-contribution to better understanding of MDS. Leuk Lymphoma.
17:335–339. 1995.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhou X, Kuang Y, Liang S and Wang L:
Metformin inhibits cell proliferation in SKM-1 cells via
AMPK-mediated cell cycle arrest. J Pharmacol Sci. 141:146–152.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Bennett JM, Catovsky D, Daniel MT,
Flandrin G, Galton DA, Gralnick HR and Sultan C: Proposals for the
classification of the myelodysplastic syndromes. Br J Haematol.
51:189–199. 1982.PubMed/NCBI
|
|
32
|
Nakagawa T, Saitoh S, Imoto S, Itoh M,
Tsutsumi M, Hikiji K, Nakao Y and Fujita T: Loss of multiple point
mutations of RAS genes associated with acquisition of chromosomal
abnormalities during disease progression in myelodysplastic
syndrome. Br J Haematol. 77:250–252. 1991.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chen X, Glytsou C, Zhou H, Narang S, Reyna
DE, Lopez A, Sakellaropoulos T, Gong Y, Kloetgen A, Yap YS, et al:
Targeting mitochondrial structure sensitizes acute myeloid leukemia
to venetoclax treatment. Cancer Discov. 9:890–909. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Guo J, Zhang R, Yang Z, Duan Z, Yin D and
Zhou Y: Biological roles and therapeutic applications of IDH2
mutations in human cancer. Front Oncol. 11(644857)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Schulte ML, Fu A, Zhao P, Li J, Geng L,
Smith ST, Kondo J, Coffey RJ, Johnson MO, Rathmell JC, et al:
Pharmacological blockade of ASCT2-dependent glutamine transport
leads to antitumor efficacy in preclinical models. Nat Med.
24:194–202. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Choi SH, Byun HM, Kwan JM, Issa JP and
Yang AS: Hydroxycarbamide in combination with azacitidine or
decitabine is antagonistic on DNA methylation inhibition. Br J
Haematol. 138:616–623. 2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Momparler RL: Pharmacology of
5-Aza-2'-deoxycytidine (decitabine). Semin Hematol. 42 (Suppl
2):S9–S16. 2005.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Jin L, Alesi GN and Kang S: Glutaminolysis
as a target for cancer therapy. Oncogene. 35:3619–3625.
2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Bartenstein M and Deeg HJ: Hematopoietic
stem cell transplantation for MDS. Hematol Oncol Clin North Am.
24:407–422. 2010.PubMed/NCBI View Article : Google Scholar
|
|
40
|
DiNardo CD, Pratz KW, Letai A, Jonas BA,
Wei AH, Thirman M, Arellano M, Frattini MG, Kantarjian H, Popovic
R, et al: Safety and preliminary efficacy of venetoclax with
decitabine or azacitidine in elderly patients with previously
untreated acute myeloid leukaemia: A non-randomised, open-label,
phase 1b study. Lancet Oncol. 19:216–228. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Tsao T, Shi Y, Kornblau S, Lu H, Konoplev
S, Antony A, Ruvolo V, Qiu YH, Zhang N, Coombes KR, et al:
Concomitant inhibition of DNA methyltransferase and BCL-2 protein
function synergistically induce mitochondrial apoptosis in acute
myelogenous leukemia cells. Ann Hematol. 91:1861–1870.
2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Pan R, Hogdal LJ, Benito JM, Bucci D, Han
L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, et al:
Selective BCL-2 inhibition by ABT-199 causes on-target cell death
in acute myeloid leukemia. Cancer Discov. 4:362–375.
2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Bogenberger JM, Delman D, Hansen N, Valdez
R, Fauble V, Mesa RA and Tibes R: Ex vivo activity of BCL-2 family
inhibitors ABT-199 and ABT-737 combined with 5-azacytidine in
myeloid malignancies. Leuk Lymphoma. 56:226–229. 2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
DiNardo CD, Rausch CR, Benton C, Kadia T,
Jain N, Pemmaraju N, Daver N, Covert W, Marx KR, Mace M, et al:
Clinical experience with the BCL2-inhibitor venetoclax in
combination therapy for relapsed and refractory acute myeloid
leukemia and related myeloid malignancies. Am J Hematol.
93:401–407. 2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Deeks ED: Venetoclax: First global
approval. Drugs. 76:979–987. 2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yang TT, Song XL, Zhao YM, Ye BD, Luo Y,
Xiao HW, Chen Y, Fu HR, Yu J, Liu LZ, et al: Outcome after
allogeneic hematopoietic stem cell transplantation following
Venetoclax-based therapy among AML and MDS patients. Ann Hematol.
101:2731–2741. 2022.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Masetti R, Baccelli F, Leardini D,
Gottardi F, Vendemini F, Di Gangi A, Becilli M, Lodi M, Tumino M,
Vinci L, et al: Venetoclax-based therapies in pediatric advanced
MDS and relapsed/refractory AML: A multicenter retrospective
analysis. Blood Adv. 7:4366–4370. 2023.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Chen Z, Zhen S, Zhang T, Shen Y, Pang A,
Yang D, Zhang R, Ma Q, He Y, Wei J, et al: Venetoclax plus
hypomethylating agents versus intensive chemotherapy for
hematological relapse of myeloid malignancies after allo-HSCT.
Front Oncol. 13(1137175)2023.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Chen X, Liu ZY, Zhang RL, Zhai WH, Ma QL,
Pang AM, Yang DL, He Y, Wei JL, Feng SZ, et al: Efficacy and safety
of Venetoclax in the treatment of 25 patients with recurrent
hematologic malignancies after an allogeneic hematopoietic stem
cell transplantation. Zhonghua Xue Ye Xue Za Zhi. 43:542–549.
2022.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
50
|
Jilg S, Reidel V, Muller-Thomas C, Konig
J, Schauwecker J, Hockendorf U, Huberle C, Gorka O, Schmidt B,
Burgkart R, et al: Blockade of BCL-2 proteins efficiently induces
apoptosis in progenitor cells of high-risk myelodysplastic
syndromes patients. Leukemia. 30:112–123. 2016.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Parker JE, Mufti GJ, Rasool F, Mijovic A,
Devereux S and Pagliuca A: The role of apoptosis, proliferation,
and the Bcl-2-related proteins in the myelodysplastic syndromes and
acute myeloid leukemia secondary to MDS. Blood. 96:3932–3938.
2000.PubMed/NCBI
|
|
52
|
Yang X, Xia R, Yue C, Zhai W, Du W, Yang
Q, Cao H, Chen X, Obando D, Zhu Y, et al: ATF4 Regulates CD4(+) T
cell immune responses through metabolic reprogramming. Cell Rep.
23:1754–1766. 2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Tang X, Lucas JE, Chen JL, LaMonte G, Wu
J, Wang MC, Koumenis C and Chi JT: Functional interaction between
responses to lactic acidosis and hypoxia regulates genomic
transcriptional outputs. Cancer Res. 72:491–502. 2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Verginadis II, Avgousti H, Monslow J,
Skoufos G, Chinga F, Kim K, Leli NM, Karagounis IV, Bell BI,
Velalopoulou A, et al: A stromal integrated stress response
activates perivascular cancer-associated fibroblasts to drive
angiogenesis and tumour progression. Nat Cell Biol. 24:940–953.
2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Yang H, Ye D, Guan KL and Xiong Y: IDH1
and IDH2 mutations in tumorigenesis: Mechanistic insights and
clinical perspectives. Clin Cancer Res. 18:5562–5571.
2012.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Mondesir J, Willekens C, Touat M and de
Botton S: IDH1 and IDH2 mutations as novel therapeutic targets:
Current perspectives. J Blood Med. 7:171–180. 2016.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Yan H, Parsons DW, Jin G, McLendon R,
Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ,
et al: IDH1 and IDH2 mutations in gliomas. N Engl J Med.
360:765–773. 2009.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Borger DR, Tanabe KK, Fan KC, Lopez HU,
Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M,
Liebman HM, et al: Frequent mutation of isocitrate dehydrogenase
(IDH)1 and IDH2 in cholangiocarcinoma identified through
broad-based tumor genotyping. Oncologist. 17:72–79. 2012.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Ohba S and Hirose Y: Association between
mutant IDHs and tumorigenesis in gliomas. Med Mol Morphol.
51:194–198. 2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Pardanani A, Patnaik MM, Lasho TL, Mai M,
Knudson RA, Finke C, Ketterling RP, McClure RF and Tefferi A:
Recurrent IDH mutations in high-risk myelodysplastic syndrome or
acute myeloid leukemia with isolated del(5q). Leukemia.
24:1370–1372. 2010.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Molenaar RJ, Coelen RJS, Khurshed M, Roos
E, Caan MWA, van Linde ME, Kouwenhoven M, Bramer JAM, Bovée JVMG,
Mathôt RA, et al: Study protocol of a phase IB/II clinical trial of
metformin and chloroquine in patients with IDH1-mutated or
IDH2-mutated solid tumours. BMJ Open. 7(e014961)2017.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Wahl DR, Dresser J, Wilder-Romans K,
Parsels JD, Zhao SG, Davis M, Zhao L, Kachman M, Wernisch S, Burant
CF, et al: Glioblastoma Therapy Can Be Augmented by Targeting
IDH1-Mediated NADPH Biosynthesis. Cancer Res. 77:960–970.
2017.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Mohrenz IV, Antonietti P, Pusch S, Capper
D, Balss J, Voigt S, Weissert S, Mukrowsky A, Frank J, Senft C, et
al: Isocitrate dehydrogenase 1 mutant R132H sensitizes glioma cells
to BCNU-induced oxidative stress and cell death. Apoptosis.
18:1416–1425. 2013.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Zarei M, Lal S, Parker SJ, Nevler A,
Vaziri-Gohar A, Dukleska K, Mambelli-Lisboa NC, Moffat C, Blanco
FF, Chand SNJ, et al: Posttranscriptional Upregulation of IDH1 by
HuR establishes a powerful survival phenotype in pancreatic cancer
cells. Cancer Res. 77:4460–4471. 2017.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Li J, He Y, Tan Z, Lu J, Li L, Song X, Shi
F, Xie L, You S, Luo X, et al: Wild-type IDH2 promotes the Warburg
effect and tumor growth through HIF1α in lung cancer. Theranostics.
8:4050–4061. 2018.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Voehringer DW: BCL-2 and glutathione:
Alterations in cellular redox state that regulate apoptosis
sensitivity. Free Radic Biol Med. 27:945–950. 1999.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Atzori L, Dypbukt JM, Sundqvist K,
Cotgreave I, Edman CC, Moldeus P and Grafström RC:
Growth-associated modifications of low-molecular-weight thiols and
protein sulfhydryls in human bronchial fibroblasts. J Cell Physiol.
143:165–171. 1990.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Davies KJ: The broad spectrum of responses
to oxidants in proliferating cells: A new paradigm for oxidative
stress. IUBMB Life. 48:41–47. 1999.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Menon SG, Sarsour EH, Spitz DR,
Higashikubo R, Sturm M, Zhang H and Goswami PC: Redox regulation of
the G1 to S phase transition in the mouse embryo fibroblast cell
cycle. Cancer Res. 63:2109–2117. 2003.PubMed/NCBI
|
|
70
|
Markovic J, Borras C, Ortega A, Sastre J,
Vina J and Pallardo FV: Glutathione is recruited into the nucleus
in early phases of cell proliferation. J Biol Chem.
282:20416–20424. 2007.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Circu ML and Aw TY: Glutathione and
modulation of cell apoptosis. Biochim Biophys Acta. 1823:1767–1777.
2012.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Silva MM, Rocha CRR, Kinker GS, Pelegrini
AL and Menck CFM: The balance between NRF2/GSH antioxidant mediated
pathway and DNA repair modulates cisplatin resistance in lung
cancer cells. Sci Rep. 9(17639)2019.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Zou M, Hu X, Xu B, Tong T, Jing Y, Xi L,
Zhou W, Lu J, Wang X, Yang X and Liao F: Glutathione S-transferase
isozyme alpha 1 is predominantly involved in the cisplatin
resistance of common types of solid cancer. Oncol Rep. 41:989–998.
2019.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Xu Y, Han X, Li Y, Min H, Zhao X, Zhang Y,
Qi Y, Shi J, Qi S, Bao Y and Nie G: Sulforaphane mediates
glutathione depletion via polymeric nanoparticles to restore
cisplatin chemosensitivity. ACS Nano. 13:13445–13455.
2019.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Ling X, Chen X, Riddell IA, Tao W, Wang J,
Hollett G, Lippard SJ, Farokhzad OC, Shi J and Wu J:
Glutathione-Scavenging Poly(disulfide amide) nanoparticles for the
effective delivery of Pt(IV) prodrugs and reversal of cisplatin
resistance. Nano Lett. 18:4618–4625. 2018.PubMed/NCBI View Article : Google Scholar
|