Introduction
Multiple myeloma is an age-related malignant disease
characterized by the clonal proliferation of B-cell lineage plasma
cells resulting in the production of monoclonal proteins in serum
and/or urine, and leads to destructive bony lesions, defective
hematopoiesis and renal failure (1,2).
Although increasing numbers of therapeutics are becoming available
to treat this disease with the potential for significant symptom
palliation, the induction of effective antibody responses, and
prolongation of disease-free survival, available therapeutic
approaches including peripheral blood stem cell transplantation
(3) and newer agents, such as
bortezomib, thalidomide and lenalidomide (4–6),
have had only a modest impact on patient overall survival in
several randomized trials. Multiple myeloma remains invariably
incurable. Therefore, a more effective treatment is urgently
required for this disease (7).
Natural products are fertile source for revealing
novel lead molecules. In fact, 67% of anticancer drugs are natural
products or natural product derivatives (8). Among them, chalcones represent an
important group of natural compounds, which are absorbed in the
daily diet and appear to be promising cancer chemopreventive agents
(9). Isobavachalcone (IBC), a
naturally occurring chalcone compound derived from the seeds of
Psoralea corylifolia L., has long been used in traditional
Chinese medicine as anthelmintic, antibacterial, aphrodisiac,
astringent and antiplatelet agent (10–12). Recently, IBC has also been shown
to have anticancer activity (13,14). IBC has been shown to exert an
inhibitory effect against skin tumor promotion in vivo in
mouse skin carcinogenesis (15),
and to induce apoptosis in neuroblastoma (16). More importantly, IBC has been
shown to be less toxic to normal cells, indicating that IBC is a
promising candidate for cancer therapy.
In this study, we assessed the anti-myeloma effect
of IBC. We found that IBC induces apoptosis and autophagy in
myeloma cells, in which autophagy plays a protective role.
Moreover, we found that chloroquine (CQ), the only clinically used
authophagy inhibitor, at its non-toxic concentration can markedly
enhance IBC-induced cell death in myeloma cells but not in normal
peripheral blood mononuclear cells (PBMCs). We further demonstrate
that the mitochondrial pathway and the protelytic activation of
protein kinase Cδ (PKCδ) contribute to this combined effect. To our
knowledge, this is the first report to show that IBC has
anti-myeloma activity. The combination treatment of CQ and IBC
warrants further investigation in preclinical or clinical
studies.
Materials and methods
Cell culture and reagents
H929 multiple myeloma cells were cultured in
RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO), supplemented with
10% fetal bovine serum (HyClone, Logan, UT). IBC was obtained from
the Tauto Biotech Co., Ltd., Shanghai China, with >98% purity as
judged by HPLC analysis. IBC were dissolved in dimethyl sulfoxide
(DMSO). CQ was purchased from Sigma. 3-Methyladenine (3-MA) was
obtained from Sigma and dissolved in distilled water. Bafilomycin A
was obtained from LC laboratories (Woburn, MA) and dissolved in
DMSO as 1 mM stock solution. Rottlerin (Biomol, Plymouth, PA) was
prepared in DMSO as 2 mM stock solution.
Isolation of PBMCs
Heparinized venous blood was obtained from two
healthy human volunteers with their mutual consent. Mononuclear
cells were isolated in a Ficoll-Hypaque (Pharmacia, Piscataway, NJ)
density gradient using standard procedures which separated PBMCs
from whole blood. The buffy coat containing PBMCs was removed
carefully following centrifugation and washed twice in RPMI-1640
medium containing 10% FBS. Cell viability determined by trypan blue
exclusion was above 90%. Cells (1x105) were incubated in
100 μl medium in round-bottom 96-well plates and were
cultured in the presence of CQ and/or IBC for 48 h and cell
proliferation was determined by the Cell Counting kit-8 (CCK-8)
assay as described previously (17).
Apoptosis assay
Apoptosis was measured by the Annexin V Fluos
apoptosis detection kit (Roche Molecular Biochemicals, Mannheim,
Germany) following the manufacturer’s instructions. Annexin
V-positive and PI-negative cells were considered to be in the early
apoptotic phase and those having positive staining both for Annexin
V and PI were deemed to undergo late apoptosis or necrosis.
Western blot analysis
The cells were washed with PBS and lysed with lysis
buffer (62.5 mM Tris-HCl, pH 6.8, 100 mM DTT, 2% SDS, 10%
glycerol). Cell lysates were centrifuged at 20,000 x g for 10 min
at 4°C, and proteins in the supernatants were quantified. Protein
extracts were equally loaded to an 8% to 15% SDS-polyacrylamide
gel, electrophoresed, and transferred to nitrocellulose membrane
(Amersham Bioscience, Buckinghamshire, UK). The blots were stained
with 0.2% Ponceau S red to ensure equal protein loading. After
blocking with 5% non-fat milk in PBS for 1 h at room temperature,
the membranes were probed with antibodies against cleaved caspase-3
and -9 (Cell Signaling Technology, Inc., Beverly, MA), Bcl-2, PKCδ
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and poly
(ADP-ribose) polymerase (PARP) (from BD Pharmingen™), LC3 (Sigma).
The signals were detected by the chemiluminescence phototope-HRP
kit (Cell Signaling Technology, Inc.) according to the
manufacturer’s instructions. Where necessary, the blots were
stripped and reprobed with anti-actin antibody (Oncogene, Fremont,
CA) as the internal control. All experiments were repeated three
times with similar results.
RNA interference and transfection
The retrovirus vector for beclin-1 protein
suppression by short hairpin RNA (shRNA) interference was generated
as described previously (18).
Briefly, retrovirus with two target shRNAs (B-sh2 and B-sh3) and
non-target control shRNA (NC)-containing plasmids were packaged in
HEK293T cells by co-transfecting with pSIREN-RetroQ, pEQPAM
(containing gag-pol, produced by Dr Lishan Su, UNC Chapel Hill,
USA) and VSVG (Clontech; T-334350). After transfection for 48 h,
the viral supernatant was collected, filter-sterilized and added
into H929 cells (2x105 cells/well) in 6-well plates with
medium containing 8 μg/ml of polybrene (TR-1003-G;
Millipore) and 0.75 μg/ml of puromycin (540411; Calbiochem)
for the selection of stably transfected cells after another 48
h.
Mitochondrial membrane potential (ΔΨm)
assessment
After washing twice with PBS, approximately
106 cells were incubated (37°C, 30 min) with 10
μg/ml rhodamine 123 (Rh123), a cationic lipophilic
fluorochrome absorbed by the mitochondria in proportion to the ΔΨm.
Then, 50 μg/ml PI, a membrane impermeable DNA-binding dye,
was added to the cells. Fluorescence intensities were determined by
flow cytometry (BD Biosciences). A total of 10,000 cells were
analyzed in each sample. All data were collected, stored and
analyzed by LYSIS II software (BD Biosciences).
Statistical analysis
The Student’s t-test was used to evaluate the
differences between two groups. A P-value <0.05 was considered
to indicate a statistically significant difference.
Results
IBC inhibits the proliferation of myeloma
H929 cells via apoptosis and autophagy induction
To examine the antitumor activity of IBC on myeloma
cells, H929 cells were treated with IBC at 0, 10, 20 and 40
μM for two days, and the cell proliferation and viability
were determined by trypan blue exclusion assay. As shown in
Fig. 1, IBC inhibited the
proliferation of H929 cells in a dose- and time-dependent manner
(Fig. 1A), and approximately 15%
cells lost viability upon treatment with IBC at 20 μM (50%
inhibition) for two days (Fig.
1B). The characterization of IBC-induced cell death was further
investigated. Following treatment with 10–40 μM IBC for 48
h, the 17- and 19-kDa cleaved fragments of caspase-3, the 37-kDa
cleaved caspase-9 and the 85-kDa fragments of PARP appeared in a
dose- and time-dependent manner, indicating the involvement of
apoptosis (Fig. 1C). We also
determined the expression level of LC-3, a reliable marker of
autophagosome formation. Tracking the conversion of LC3-I to LC3-II
is indicative of autophagic activity. Of note, IBC treatment also
increased the expression of LC-3 II in a dose- and time-dependent
manner (Fig. 1D). These data
suggest that IBC induces both apoptosis and autophagy in H929
cells.
Knockdown of beclin-1 or co-treatment
with autophagy inhibitors, 3-MA, bafilomycin A or CQ, enhances
IBC-induced cell death
Autophagy can either be protective or induce
programmed cell death. In most cases, autophagy acts as a survival
mechanism and it has been shown that the inhibition of
chemotherapy-induced autophagy frequently augments cell death
(19). In order to examine the
role of autophagy in IBC-induced cell death, two different
approaches were used to inhibit autophagy. Firstly, beclin-1, an
important autophagy-related protein, was silenced by RNA
interference in H929 cells. The target sequence, sh3, effectively
downregulated the expression of beclin-1 (H929B-sh3)
(Fig. 2A). Compared with the
vector-transfected H929 cells (H929NC), a significantly
increased cell death was observed in the H929B-sh3 cells
upon treatment with IBC (Fig.
2B). Secondly, when used in combination with IBC, 3-MA, a well
established autophagy inhibitor which inhibits hVps34, a class III
PI-3K (Fig. 2C) or bafilomycin A,
a specific vacuole-type H+-ATPase, which has been
reported to disrupt the progression of autophagy at the later stage
by inhibiting the fusion between autophagosomes and lysosomes
(Fig. 2D), or CQ, which blocks
lysosome and autophagosome fusion and lysosomal protein
degradation, significantly enhanced IBC-induced cell death in H929
cells (Fig. 2E). Of note, CQ used
at 20 μM for 48 h was non-cytotoxic. Taken together, these
data suggest that autophagy inhibition potentiates IBC-induced cell
death.
Mitochondrial cell death pathway involved
in CQ plus IBC-induced cell death in H929 cells
CQ has been widely used as an autophagy inhibitor to
enhance the efficacy of cancer therapeutic drugs and many clinical
trials have used CQ for this purpose (20,21). We then investigated the mechanism
underlying the CQ plus IBC-enhanced cell death. It is known that
the mitochondrial pathway plays a central role in determining cell
survival or death in response to diverse stimuli and that the
collapse of ΔΨm is associated with mitochondrial dysfunction. We
therefore assessed the effect of CQ plus IBC on mitochondrial ΔΨm
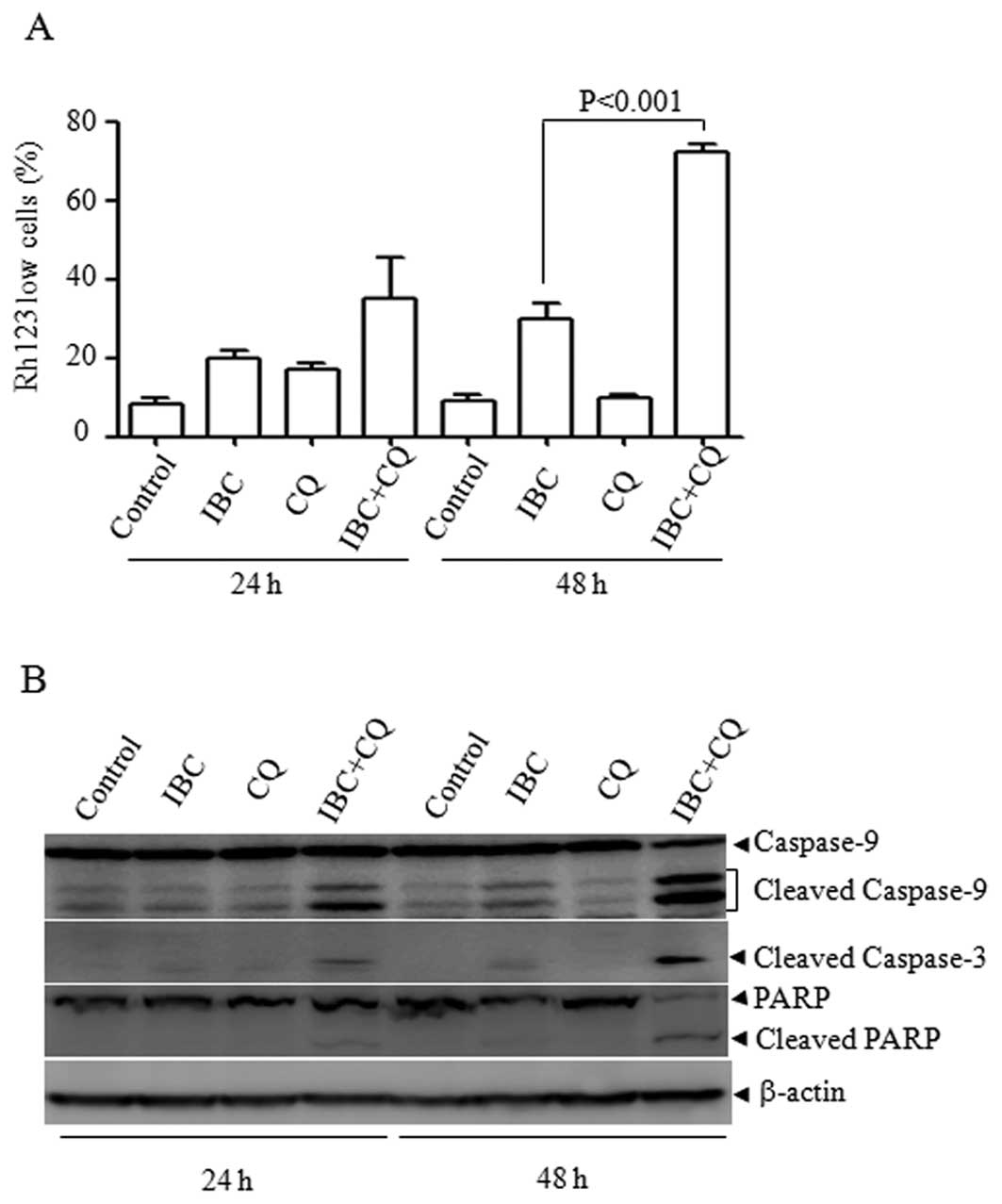
by Rh123/PI double staining assay. As depicted in Fig. 3A, compared with the single
treatment of IBC, CQ plus IBC significantly increased the
percentages of low Rh123-stained cells at 48 h (P<0.001),
suggesting that the dissipation of the ΔΨm was associated with CQ
plus IBC-induced cell death. Consistent with the collapse of ΔΨm,
CQ plus IBC led to the obvious activation of caspase-9, an
initiator caspase in the mitochondrial pathway. Accordingly,
caspase-3 was also activated and PARP was cleaved into 85-kDa
fragments (Fig. 3B). These data
suggest that the mitochondrial pathway contributes to CQ plus
IBC-induced cell death.
Protelytic activation of PKCδ is involved
in CQ plus IBC-induced cell death
PKCδ activation has been shown to play a critical
role in the mitochondrial cell death pathway and a previous report
showed that CQ can activate PKCδ (22). Thus, we investigated whether PKCδ
is also involved in the combined action of CQ plus IBC. Of note, CQ
plus IBC led to the protelytic cleavage of PKCδ into a 41-kDa
catalytic fragment, which was associated with cell death (Fig. 4A). To evaluate the possible role
of PKCδ in CQ plus IBC-induced cell death, we treated the H929
cells with CQ plus IBC in the presence or absence of rottlerin, a
specific PKCδ inhibitor for 30 h. As shown in Fig. 4B, rottlerin at 2 μM
significantly inhibited CQ plus IBC-induced cell death, as
evidenced by the decrease in the number of Annexin V-positive cells
(upper panel). Accordingly, the CQ plus IBC-induced proteolytic
cleavage of PKCδ, the cleavage of caspase-3 and PARP were also
inhibited (lower panel). These data indicate that PKCδ plays an
important role in CQ plus IBC-induced cell death.
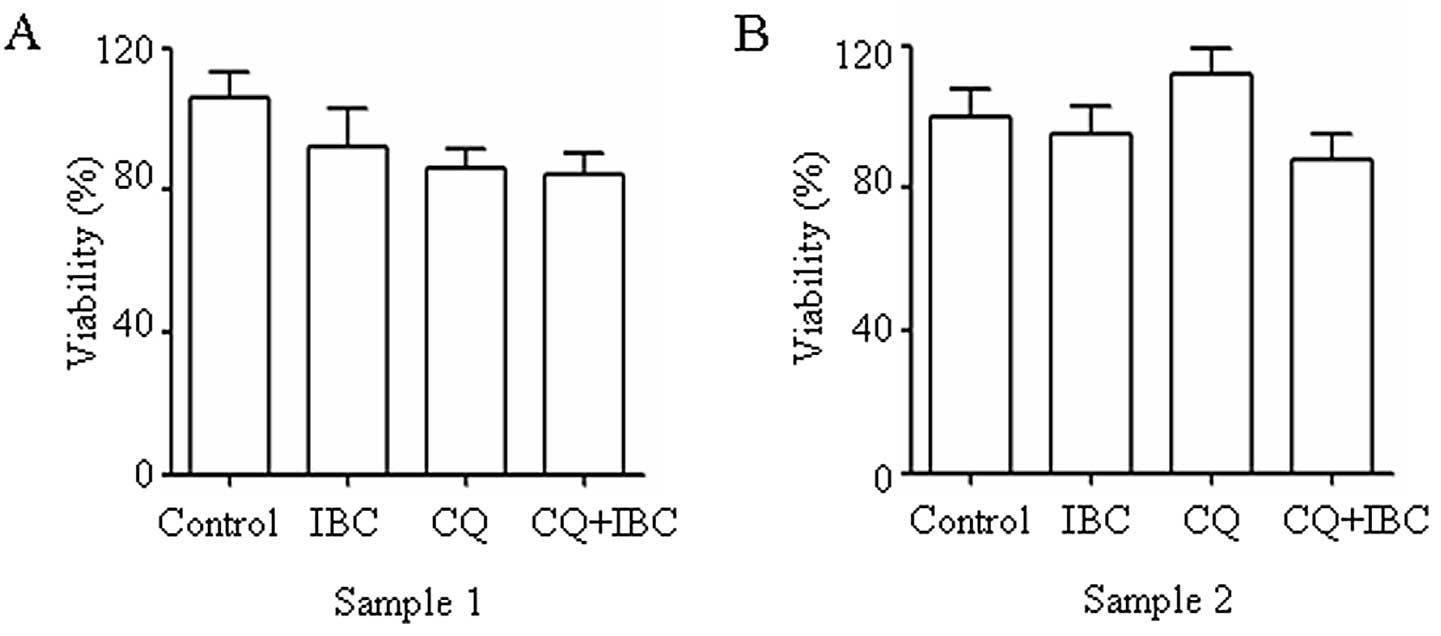
CQ plus IBC exhibits low toxicity to
normal PBMCs
To evaluate the possible effect of CQ plus IBC on
normal cells, two samples of PBMCs (Fig. 5) were used. At the concentration
used, CQ plus IBC did not significantly reduce cell viability in
normal PBMCs. In combination with the results showing the cell
death-enhancing effect of CQ plus IBC on H929 cells, these data
indicate that myeloma cells are more sensitive than PBMCs to CQ
plus IBC-induced cell death.
Discussion
Despite recent advances in the understanding of the
disease pathogenesis and the introduction of velcade, thalidomide
and other agents, multiple myeloma remains incurable. Therefore,
the development of a novel therapeutic strategy to improve the
outcome is urgently required. In this study, we demonstrate that
IBC has anti-myeloma activity and that autophagy inhibition
selectively augments the pro-apoptotic activity of IBC in myeloma
cells but not in normal PBMCs cells. Furthermore, we demonstrate
that the cell death-enhancing effect of CQ plus IBC is associated
with the mitochondrial pathway and the protelytic activation of
PKCδ.
Autophagy is a highly conserved catabolic programme
for degrading proteins and organelles. The role of autophagy in
cell death is controversial. Autophagy can act as a survival or a
cell death mechanism (23).
Accumulating data have shown that many chemotherapy drugs can
induce apoptosis accompanied by the activation of autophagy, which
provides a protective mechanism for cancer cells. Therefore,
combination treatment with autophagy inhibitors is an attracting
strategy to enhance the efficacy of chemotherapy (24). As IBC-induced cell death involves
both apoptosis and autophagy, we hypothesized that autophagy
inhibition may enhance the cell killing effect of IBC. In support
of this notion, the autophagy inhibition achieved by knocking down
beclin-1, a specific autophagy-promoting protein, or by using the
autophagy inhibitors, 3-MA, bafilomycin A and CQ, significantly
enhanced IBC-induced cell death. These data suggest that the
inhibition of autophagy in IBC-treated myeloma cells may switch the
pro-survival effect of autophagy to apoptosis.
Mitochondria are the main regulators of apoptotic
cell death. Anticancer drugs including IBC may damage the
mitochondria by increasing the permeability of the outer
mitochondrial membrane, which is associated with the collapse of
ΔΨm (25). The mitochondria have
also been shown to be the central organelles integrating autophagy
and apoptosis (26). Based on
these findings, we examined the role of mitochondria in CQ plus
IBC-induced cell death. Compared with the single treatment of IBC
or CQ, an obvious collapse of ΔΨm was observed following CQ plus
IBC treatment, which was paralleled with the increase in apoptosis.
These data indicate that the inhibition of autophagy enhances the
dysfunction of the mitochondria, which in turn augments the
apoptotic cell death. We then investigated the possible mechanisms
underlying mitochondrial dysfunction mediated by the co-treatment
of CQ and IBC. Due to its important role in the regulation of the
mitochondrial cell death pathway, PKCδ, a ubiquitously expressed
member of the novel PKC family, was examined (27,28). Indeed, paralleled with the loss of
ΔΨm, CQ plus IBC caused the proteolytic activation of PKCδ and
rottlerin, an inhibitor of PKCδ, significantly preventing CQ plus
IBC-induced cell death, indicating the important contribution of
PKCδ to cell death. On the contrary, Ni et al (29) reported that, PKCδ is commonly
expressed in myeloma cells and plays an important role in plasma
cell survival. This discrepancy may be explained by the fact that
the PKCδ catalytic fragment has pro-apoptotic activity, whereas the
holoenzyme induces the opposite effect. We do not rule out other
possibilities. For example, the CQ-induced autophagy inhibition may
eventually disrupt the lysosome and lead to the release of lysosome
proteases into the cytosol, which would eventually lead to the
dysfunction of the mitochondria.
Combination therapy is an important strategy to
improve the outcome of multiple myeloma. Our results showed that CQ
plus IBC may be a valuable regimen for the treatment of multiple
myeloma. Firstly, IBC has potential value in clinical applications.
IBC displayed a satisfactory selectivity between cancer cells and
normal cells. The safety of IBC is also indirectly reflected by the
fact that IBC is an active ingredient from the seeds of Psoralea
corylifolia L. and the crude drug has been used in certain
Chinese traditional medicines, such as ‘Qing E Wan’ for the
treatment of senile osteoporosis and arthralgia. Secondly, CQ is a
well known 4-aminoquinoline class of drug that is widely used as an
anti-malaria (30),
anti-inflammatory and antiviral agent (31). Recently, CQ has been studied for
its potential in the enhancement of radiation therapy,
chemotherapy, and combination therapy for cancer (21,32). The published data suggest that the
combined modalities of CQ with other therapeutics are very
promising for increasing therapeutic efficacy and decreasing
undesirable side-effects. For example, CQ has been shown to
dramatically increase the killing power of Gleevec and SAHA against
chronic myeloma leukemia cells (33,34). More importantly, our results
showed that CQ plus IBC at a relatively low concentration caused
significant cell death in myeloma cells but not in normal PBMCs,
indicating that this combination is more selective to cancer
cells.
In conclusion, we demonstrate for the first time IBC
has anti-myeloma activity, and that CQ can markedly enhance
IBC-induced cell death in myeloma cells, in which the mitochondrial
pathway and PKCδ activation play important roles. As CQ and IBC
have been shown to be relatively specific to cancer cells, the
combination treatment of both agents at nontoxic or sub-toxic
concentration warrants further investigation in preclinical and
clinical studies.
Acknowledgements
The present study was supported in
part by grants from the National Basic Research Program of China
(973 Program) (no. 2010CB912104), the National Natural Science
Foundation of China (nos. 81172322, 81070433, 91013008), the
Science and Technology Committee of Shanghai (no. 11JC1406500), the
SMC Program of Shanghai Jiao Tong University.
References
|
1.
|
A MahindraT HideshimaKC AndersonMultiple
myeloma: biology of the diseaseBlood Rev24Suppl
1S5S11201010.1016/S0268-960X(10)70003-5
|
|
2.
|
MA DimopoulosE TerposMultiple myelomaAnn
Oncol21Suppl 7S143S150201010.1093/annonc/mdq370
|
|
3.
|
S GiraltStem cell transplantation for
multiple myeloma: current and future status (Review)Hematology Am
Soc Hematol Educ
Program2011191196201110.1182/asheducation-2011.1.19122160033
|
|
4.
|
AA Chanan-KhanJ San MiguelS JagannathH
LudwigAM DimopoulosNovel therapeutic agents for the management of
multiple myeloma patients with renal impairmentClin Cancer
Res1821452163201210.1158/1078-0432.CCR-11-049822328563
|
|
5.
|
JM MarizGV EstevesReview of second-line
therapy for multiple myeloma: focus on lenalidomideCurr Opin
Oncol24Suppl
2S3S11201210.1097/01.cco.0000410243.84074.dc22245806
|
|
6.
|
M KropffHG BaylonJ HillengassThalidomide
versus dexamethasone for the treatment of relapsed and/or
refractory multiple myeloma: results from OPTIMUM, a randomized
trialHaematologica97784791201110.3324/haematol.2011.04427122133776
|
|
7.
|
A MahindraJ LaubachN RajeN MunshiPG
RichardsonK AndersonLatest advances and current challenges in the
treatment of multiple myelomaNat Rev Clin
Oncol9135143201210.1038/nrclinonc.2012.1522349016
|
|
8.
|
MJ BalunasAD KinghornDrug discovery from
medicinal plantsLife
Sci78431441200510.1016/j.lfs.2005.09.01216198377
|
|
9.
|
B OrlikovaD TasdemirF GolaisM DicatoM
DiederichDietary chalcones with chemopreventive and
chemotherapeutic potentialGenes
Nutr6125147201110.1007/s12263-011-0210-521484163
|
|
10.
|
H HaraguchiJ InoueY TamuraK
MizutaniAntioxidative components of Psoralea corylifolia
(Leguminosae)Phytother Res16539544200210.1002/ptr.972
|
|
11.
|
I JantanYH Mohd YasinS JamilH SiratN
BasarEffect of prenylated flavonoids and chalcones isolated from
Artocarpus species on platelet aggregation in human whole
bloodJ Nat Med64365369201010.1007/s11418-010-0410-020349149
|
|
12.
|
V KueteB NgameniJG TangmouoEfflux pumps
are involved in the defense of Gram-negative bacteria against the
natural products isobavachalcone and diospyroneAntimicrob Agents
Chemother5417491752201010.1128/AAC.01533-0920160051
|
|
13.
|
H JingX ZhouX DongAbrogation of Akt
signaling by Isobavachalcone contributes to its anti-proliferative
effects towards human cancer cellsCancer
Lett294167177201010.1016/j.canlet.2010.01.03520167420
|
|
14.
|
E SzliszkaZP CzubaB MazurL SedekA
ParadyszW KrolChalcones enhance TRAIL-induced apoptosis in prostate
cancer cellsInt J Mol Sci11113200910.3390/ijms1101000120161998
|
|
15.
|
O OhnoT WatabeK NakamuraInhibitory effects
of bakuchiol, bavachin, and isobavachalcone isolated from Piper
longum on melanin production in B16 mouse melanoma cellsBiosci
Biotechnol Biochem7415041506201010.1271/bbb.10022120622433
|
|
16.
|
R NishimuraK TabataM
ArakawaIsobavachalcone, a chalcone constituent of Angelica
keiskei, induces apoptosis in neuroblastomaBiol Pharm
Bull3018781883200710.1248/bpb.30.187817917255
|
|
17.
|
Y ZhaoJX PuSX Huangent-Kaurane
diterpenoids from Isodon pharicusJ Nat
Prod72988993200910.1021/np9000366
|
|
18.
|
Y HuangJK HouTT ChenPML-RARalpha enhances
constitutive autophagic activity through inhibiting the Akt/mTOR
pathwayAutophagy711321144201110.4161/auto.7.10.1663621673516
|
|
19.
|
S ShenO KeppG KroemerThe end of autophagic
cell death?Autophagy813201210.4161/auto.8.1.16618
|
|
20.
|
W HanJ SunL FengAutophagy inhibition
enhances daunorubicin-induced apoptosis in K562 cellsPLoS
One6e28491201110.1371/journal.pone.002849122164300
|
|
21.
|
VR SolomonH LeeChloroquine and its
analogs: a new promise of an old drug for effective and safe cancer
therapiesEur J
Pharmacol625220233200910.1016/j.ejphar.2009.06.06319836374
|
|
22.
|
TH ChenPC ChangMC ChangYF LinHM
LeeChloroquine induces the expression of inducible nitric oxide
synthase in C6 glioma cellsPharmacol
Res51329336200510.1016/j.phrs.2004.10.00415683746
|
|
23.
|
D DentonS NicolsonS KumarCell death by
autophagy: facts and apparent artefactsCell Death
Differ198795201210.1038/cdd.2011.14622052193
|
|
24.
|
JD ManciasAC KimmelmanTargeting autophagy
addiction in cancerOncotarget213021306201122185891
|
|
25.
|
G KroemerL GalluzziC BrennerMitochondrial
membrane permeabilization in cell deathPhysiol
Rev8799163200710.1152/physrev.00013.200617237344
|
|
26.
|
D GozuacikA KimchiAutophagy as a cell
death and tumor suppressor
mechanismOncogene2328912906200410.1038/sj.onc.120752115077152
|
|
27.
|
SF SteinbergDistinctive activation
mechanisms and functions for protein kinase CdeltaBiochem
J384449459200410.1042/BJ2004070415491280
|
|
28.
|
DN JacksonDA FosterThe enigmatic protein
kinase Cdelta: complex roles in cell proliferation and
survivalFASEB J18627636200410.1096/fj.03-0979rev15054085
|
|
29.
|
H NiM ErginSS TibudanMF DenningKF IzbanS
AlkanProtein kinase C-delta is commonly expressed in multiple
myeloma cells and its downregulation by rottlerin causes
apoptosisBr J
Haematol121849856200310.1046/j.1365-2141.2003.04368.x12786795
|
|
30.
|
D RathoreTF McCutchanM SullivanS
KumarAnti-malarial drugs: current status and new developmentsExpert
Opin Investig
Drugs14871883200510.1517/13543784.14.7.87116022576
|
|
31.
|
RG CooperT MagwereChloroquine: novel uses
& manifestationsIndian J Med Res1273053162008
|
|
32.
|
P MaycotteS AryalCT CummingsJ ThorburnMJ
MorganA ThorburnChloroquine sensitizes breast cancer cells to
chemotherapy independent of
autophagyAutophagy8200212201210.4161/auto.8.2.1855422252008
|
|
33.
|
C BellodiMR LidonniciA HamiltonTargeting
autophagy potentiates tyrosine kinase inhibitor-induced cell death
in Philadelphia chromosome-positive cells, including primary CML
stem cellsJ Clin Invest11911091123200910.1172/JCI35660
|
|
34.
|
JS CarewST NawrockiCN KahueTargeting
autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistanceBlood110313322200710.1182/blood-2006-10-05026017363733
|