Introduction
Articular cartilage is a highly organized soft
tissue. An articular cartilage defect is an area of damaged or
missing cartilage (1). Although
often caused by acute trauma, the defect may also occur as a result
of osteoarthritis, osteonecrosis, osteochondritis dissecans and
other pathologies (2). These
degenerative joint diseases affect more than a third of the world
population, and disorders of articular cartilage, in general,
account for more than half of all chronic conditions in individuals
aged 60 years and over (3).
Therefore, optimized treatment strategies for articular cartilage
lesions are of a high socio-economic importance. Symptomatic
cartilage defects require surgical treatment such as microfracture,
pridie drilling and abrasion arthroplasty, autologous chondrocyte
transplantation, and the transfer of both autologous or allogeneic
osteochondral transplants (4).
Although the development of surgical techniques in cartilage has
been extensively investigated (5,6),
there is currently no surgical method available for cartilage
injuries that can prevent its early onset (7). Tissue engineering offers promising
new approaches that have the potential to provide such
therapies.
Mesenchymal stem cells (MSCs), which can be easily
isolated in a non-invasive and abundant manner from various tissues
such as the bone marrow, bone, adipose tissue, muscle, synovium,
periosteum, perichondrium and many adult tissues (8), are capable of self-renewal and
differentiation into a variety of cell lineages, including
chondrocytes, osteoblasts and adipocytes (9,10).
MSCs have been identified in healthy and diseased cartilage, and
appear to retain at least some potential to regenerate cartilage
in vivo (11,12). Due to these advantages, MSCs are
attractive targets for manipulation in the goal of cartilage
regeneration.
Chondrocyte proliferation and differentiation toward
hypertrophy are the main challenges for cartilage regeneration from
MSCs (13,14). Various factors for chondrogenesis
of MSCs have been developed, including parathyroid hormone-related
protein (PTHrP) (15). PTHrP,
first identified as a factor involved in humoral hypocalcemia of
malignancy (16), maintains the
function of proliferating chondrocytes and inhibits chondrocyte
differentiation toward hypertrophy in the growth plate through the
PTHrP-Indian hedgehog (IHH) axis (17,18). This anti-hypertrophic activity has
been shown to result from binding of the N-terminus of PTHrP to its
cell surface receptor (PTH1R), activating Sox9 (19). PTHrP also stimulates proliferation
of endochondral chondrocytes and inhibits apoptosis, partly via
induction of Bcl-2 (20).
Therefore, PTHrP may be a therapeutic factor in the production of
MSC-derived tissue-engineered cartilage for use in cartilage
repair.
Collagen is the ubiquitous component of the tight
network of glycoproteins, collagen IV and proteoglycans in basement
membranes (21), and is widely
dispersed in the articular site (22). Therefore, collagen could be a
potential target for PTHrP which could be retained and enriched at
the injured site, enhancing the efficacy of cartilage regeneration.
A polypeptide TKKTLRT named collagen-binding domain (CBD) peptide
was derived from von Willebrand factor (vWF). Epidermal growth
factor, transforming growth factor-1 and basic fibroblast growth
factor have been added along with a collagen-binding peptide and
the results showed that CBD could specifically bind to native
collagen and the modified growth factors achieved better repair
ability compared to the native growth factors at the same
concentration (23–25). Here, we constructed a
collagen-based PTHrP-targeting system, and the effect on
chondrogenesis was tested by in vitro pellet assay in bone
marrow-derived (BM)-MSCs.
Materials and methods
Isolation and expansion of BM-MSCs
MSCs were isolated from fresh bone marrow samples
obtained from patients undergoing total hip replacement or iliac
bone graft harvest, as described elsewhere (26,27). Briefly, cells were fractionated on
a Ficoll-Paque Plus density-gradient (GE Healthcare), and the
low-density cell fraction was washed and seeded in expansion medium
consisting of high-glucose Dulbecco’s modified Eagle’s medium/F12
(DMEM/F12) and 10% FBS (Gibco-BRL, Carlsbad, CA, USA). Nonadherent
material was removed after 24–48 h. For expansion, cells were
replated at a density of 5×103 cells/cm2 and
used at passage 3.
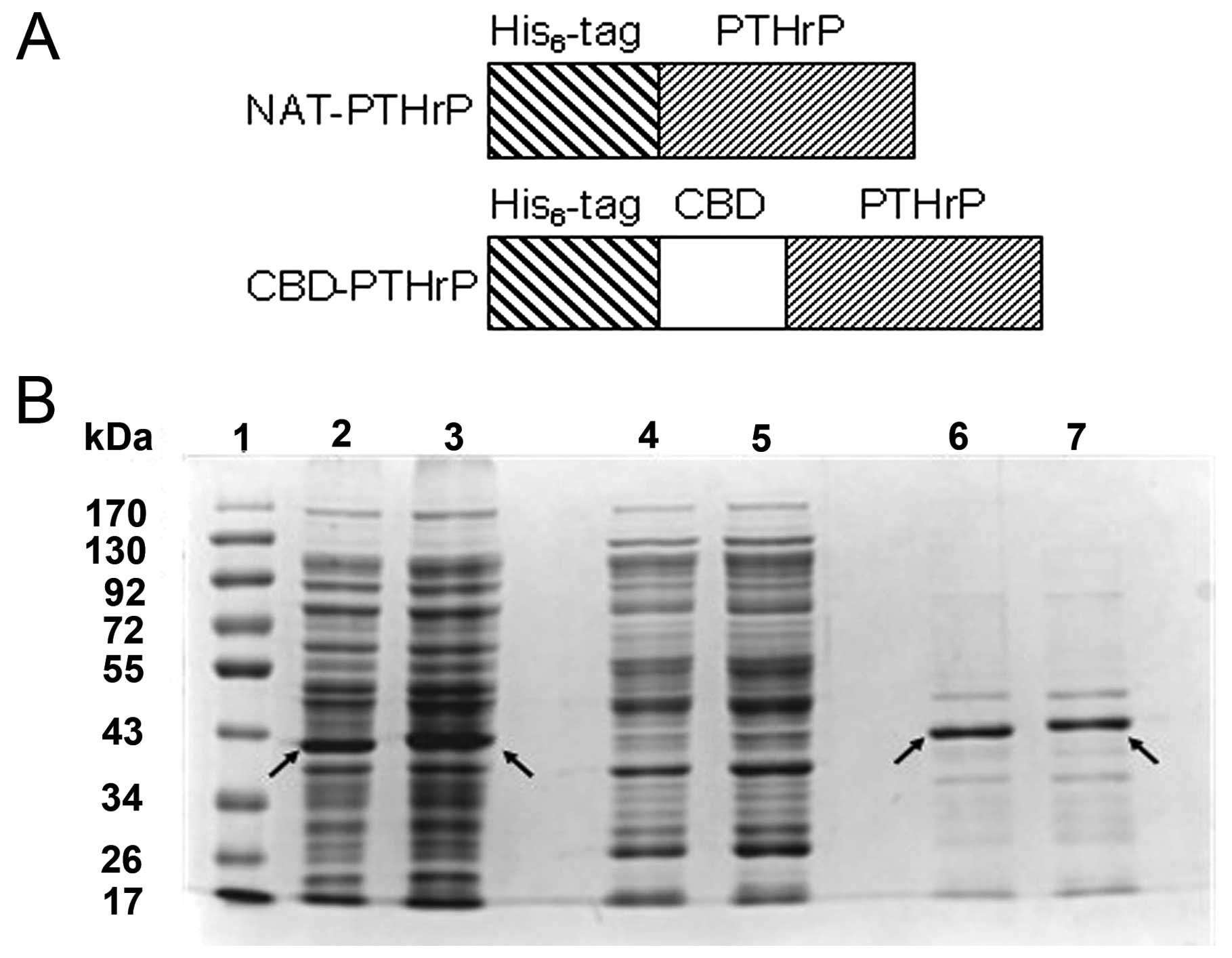
Engineering and preparation of NAT-PTHrP
and CBD-PTHrP
Engineering and preparation of NAT-PTHrP and
CBD-PTHrP were performed as previously described (28). Briefly, the gene of CBD together
with the linker domain was synthesized and then inserted into
pET-32a (Novage, USA). The vector was named pET-32a-CBD. A human
PTHrP DNA encoding a mature form was inserted into the pET-32a and
pET-32a-CBD vector, and the recombinant expression plasimd was
named pET-CBD-PTHrP (with CBD) and pET-NAT-PTHrP (without CBD).
They both contained a 6X His purification tag for purification and
detection in the subsequent experiments. Both of the plasmids were
transformed into Escherichia coli Rosetta (DE3) for
expressing the protein. E. coli was induced with 0.2 mM
isopropyl b-D-thiogalactopyranoside (IPTG) at 25°C for 1 h. The
recombinant fusion proteins were isolated as soluble bodies. The
purity and yields of recombinant proteins were analyzed by 10%
SDS-polyacrylamide gel electrophoresis (SDS-PAGE).
Collagen binding assay
The comparison of binding ability of these 2 factors
to collagen was studied using modified solid phase binding assays
as previously described (29).
Collagen member (0.1 mg) (Zenghai Bio, Shandong, China) was
neutralized and added to 96-well plates (1 mg/well), and washed 3
times. Wells were blocked with 200 μl bovine serum albumin
(BSA) (2.5 mg/ml) in phosphate-buffered saline (PBS) plus 0.1%
Tween-20 for 1 h at room temperature (RT). After washing the wells
once with PBS, 50-μl aliquots of the recombinant proteins
diluted in PBS were added for 1 h at 37°C with a series of
concentrations of 0.156-10 mM. Wells were washed 2 times with PBS,
and 50-μl aliquots of mouse anti-polyhistidine monoclonal
antibody (1:1,000 dilution) were added for 1 h at RT. After 3
washes as above, 100-μl aliquots of sheep
anti-mouse-alkaline phosphatase antibody (1:10,000 dilutions) were
added for 1 h at RT, followed by 3 washes as above. Bound proteins
were detected with 100 μl/well 2 mg/ml p-nitrophenyl
phosphate hexahydrate (p-NPP; Ameresco) in alkaline phosphatase
buffer (100 mM Tris-HC, 100 mM NaCl, 10 mM MgCl2, pH
9.6) for 10 min. The reactions were stopped with 0.2 M NaOH (100
μl/well). One hundred-microliter solutions were then
transported to a new 96-well plate. The plate was read in an ELISA
reader at a wavelength of 405 nm. All binding assays were carried
out in duplicate, and values showed <15% difference for the same
plate.
Induction of in vitro chondrogenesis
To induce chondrogenesis, in vitro pellet
cultures were carried out using 2.5×105 BM-MSCs at
passage 3 in chondrogenic differentiation medium (Cyagen, USA).
From the 14th day of culture, subsets of pellets were additionally
treated with PTHrP (100 ng/ml) and CBD-PTHrP (100 ng/ml), and
following 2 additional weeks of in vitro culture in their
respective media, the pellets were harvested for analysis. For
pellet cultures, 0.5 ml of the cell suspension was aliquoted into
15-ml polypropylene centrifuge tubes, and spun in a bench top
centrifuge at 150 × g for 5 min. Tubes were incubated in 5%
CO2 atmosphere for up to 4 weeks. Caps of tubes were
loosened in order to allow air exchange. The medium was changed
every 2 days.
RNA isolated and real-time PCR
analysis
RNA was isolated using the standard guanidine
isothiocyanate TRIzol reagent (Invitrogen). Isolated RNA samples
were converted to cDNA using Rotor-Gene SYBR Green RT-PCR kit
(Qiagen) and oligo(dT) primers. All PCR reactions were performed
using ABI system in standard 25-μl reaction volumes
containing 5 μl RNA, 0.5 μl of 100 mM sense and 0.5
μl of 100 mM antisense primer, 12.5 μl Rotor-Gene
SYBR-Green PCR Master Mix (Qiagen) and 6.5 μl RNA-free
ddH2O. The expression of the following genes were
examined: collagen type I (COL1A1), collagen type II (COL2A1),
collagen type X (COL10A1), Sox-9 and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) used as a housekeeping gene. The primers used
for amplification were: collagen type I, 5′-CCGCCGCTTCACCTACAGC-3′
and 5′-TTTTGTATTCAATCACTGTCTTGCC-3′; collagen type II,
5′-CCGAATAGCAGGTTCACGTACA-3′ and 5′-CGATAACAGTCTTGCCCCACTT-3′;
collagen type X, 5′-AAAGGCCCACTACCCAACAC-3′ and
5′-CTTCCGTAGCCTGGTTTTCC-3′; Sox-9, 5′-CACACAGCTCACTCGACCTTG-3′ and
5′-TTCGGTTATTTTTAGGATCATCTCG-3′.
Collagen extraction and western
blotting
Three pellets were homogenized and subjected to
pepsin digestion overnight at 4°C (0.5 M acetic acid, 0.2 M NaCl
and 2.5 mg/ml of pepsin). The pH was then adjusted to a neutral pH
7.0 with 1 M Tris Base prior to extraction of the collagens with
4.5 M NaCl (overnight at 4°C). The following day, the extracted
collagens were pelleted by centrifugation at 16,000 × g at 4°C for
30 min and subsequently precipitated with 400 μl of
precipitation buffer (0.4 M NaCl and 0.1 M Tris Base, pH 7.4) and
1,200 g of ethanol/sample for 4 h at −20°C. The precipitated
collagens were pelleted by centrifugation at 16,000 × g for 30 min
at 4°C and resolved in 50 μl of RIPA lysis buffer (1% Triton
X-100, 150 mM NaCl and 50 mM Tris, pH 8.0). The proteins (20
μg) were separated by SDS-PAGE and electronically
transferred onto a polyvinylidene difluoride membrane (Millipore,
Bedford, MA, USA). After blocking, the membranes were incubated
with the recommended dilution primary antibodies against collagen
I, collagen II, collagen X, Sox-9 (Millipore) and GAPDH (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), followed by incubation
with peroxidase-conjugated secondary antibodies (Abcam, Cambridge,
MA, USA). Peroxidase-labeled bands were visualized using an ECL kit
(Millipore).
Histological analysis
After 4 weeks of culture, pellets were fixed in 4%
paraformaldehyde solution for 4 h, dehydrated with 100% ethanol,
washed with xylene, and embedded in paraffin. Sections (4
μm) were cut from paraffin blocks and coated on APES-treated
glass slides. Safranin-O staining for proteoglycan and
immunohistochemistry for collagen types I, II, X and Sox-9
(Millipore) were then performed. For Safranin-O staining, sections
were deparaffinized with xylene and ethanol, aqueous Safranin-O
(0.1%) (Sigma, USA) was applied for 20 min, and then sections were
washed with distilled water. For immunohistochemistry, sections
were deparaffinized in xylene, treated with a graded series of
alcohol [100, 95 and 80% ethanol/double-distilled H2O
(v/v)], and rehydrated in PBS (pH 7.4). Endogenous peroxide was
blocked with 3% H2O2 for 10 min. After PBS
washes, slides were blocked with 5% normal goat serum in PBS for 15
min at RT followed by incubation with primary anti-collagen I
(1:100), anti-collagen II (1:500), anti-collagen X (1:400) or
anti-Sox-9 (1:400) antibody in blocking solution overnight at 4°C.
All slides were subsequently incubated with a 1:200 dilution of
biotin-conjugated goat anti-mouse, or goat anti-rabbit secondary
antibody for 15 min at 37°C and the streptavidinbiotin complex at
37°C for 15 min. The immunoreaction was visualized using
diaminobenzidine (DAB) peroxide solution, and cellular nuclei were
counterstained with hematoxylin. All specimens were evaluated using
an Olympus BX600 microscope and a Spot Fiex camera. Control samples
exposed to the secondary antibody alone showed no specific
staining.
Statistical analysis
Data are expressed as the means ± SD. Statistical
analysis was performed using the Student’s test for comparing 2
groups and by ANOVA for multiple group comparisons. P<0.05 was
taken to indicate a statistically significant result. The
Statistics Analysis System was used for all statistical
analyses.
Results
CBD-PTHrP expression and
purification
Western blotting showed that E. coli
expressed the recombinant proteins CBD-PTHrP and NAT-PTHrP when
induced by IPTG (Fig. 1). The
total soluble protein was purified by 6X His purification tag, and
the purified proteins were then diluted in PBS for subsequent
experiments.
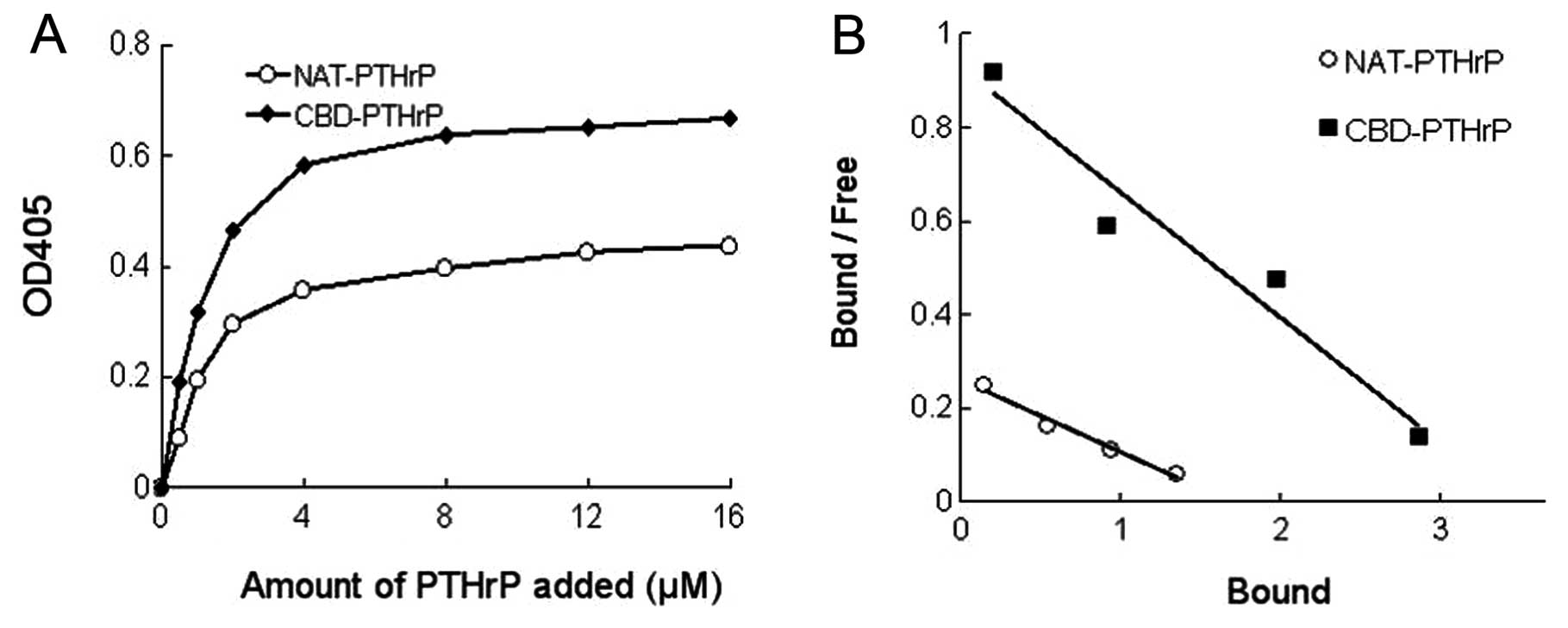
CBD-PTHrP specifically binds to
collagen
The binding abilities of NAT-PTHrP and CBD-PTHrP to
collagen were then studied in vitro through collagen-based
ELISAs. As shown in Fig. 2A, at
each concentration, the OD405 value in the CBD-PTHrP group was
higher than that in the NAT-PTHrP group, suggesting that more
proteins bound to collagen during the ELISA assay.
Based on the binding curve, the dissociation
constant Kd values for the 2 types of PTHrP binding to collagen (1
mg) were calculated by Scatchard analysis (Fig 2B). At each concentration, the ratio
of bound counts to unbound counts was plotted against the amount of
bound protein. The slope of the resulting straight line was −1/Kd.
The Kd value for the binding of NAT-PTHrP and CBD-PTHrP to 1 mg
collagen was 0.725 and 0.291 μM, respectively. The lower Kd
value indicated that the protein had a higher binding ability to
collagen. Thus, the results clearly demonstrated that CBD-PTHrP
possessed stronger collagen-binding capacity vs. NAT-NGF.
Expression of COL1A1, COL2A1, COL10A1 and
Sox-9 as determined by qRT-PCR and western blotting
In BM-MSCs, the expression levels of COL1A1, COL2A1,
COL10A1 and Sox-9 were determined by qRT-PCR and western blotting
at the mRNA and protein levels, respectively. The expression of
COL1A1 decreased by 45.38% (P<0.01) and 40.81% (P<0.01) after
treatment with 100 ng/ml NAT-PTHrP and CBD-PTHrP, respectively
(Fig. 3A). Meantime, Sox-9 mRNA
(Fig. 3D), the master gene of
chondrogenesis, increased from 1 to 2.3- (P<0.01) and 2.5-fold
(P<0.01) when compared to the untreated control following
treatment with 100 ng/ml NAT-PTHrP and CBD-PTHrP. The expression of
COL2A1 dramatically increased to 4.2-fold (P<0.01) following 100
ng/ml of NAT-PTHrP and 4.7-fold (P<0.01) following 100 ng/ml of
CBD-PTHrP (Fig. 2B). COL10A1, the
marker of hypertrophic chondrocytes, decreased 25.86% (P<0.05)
following 100 ng/ml of NAT-PTHrP and 24.79% (P<0.05) following
100 ng/ml of CBD-PTHrP (Fig. 3C).
Western blotting provided similar results as the qRT-PCR. As shown
in Fig. 3E, the protein levels of
COL2A1 and Sox-9 were significantly increased after treatment with
100 ng/ml NAT-PTHrP and CBD-PTHrP compared with the untreated
group. Meanwhile, the protein expression of COL1A1 and COL10A1 was
dramatically inhibited by NAT-PTHrP and CBD-PTHrP. There were no
significant differences between NAT-PTHrP and CBD-PTHrP at either
the mRNA or the protein level.
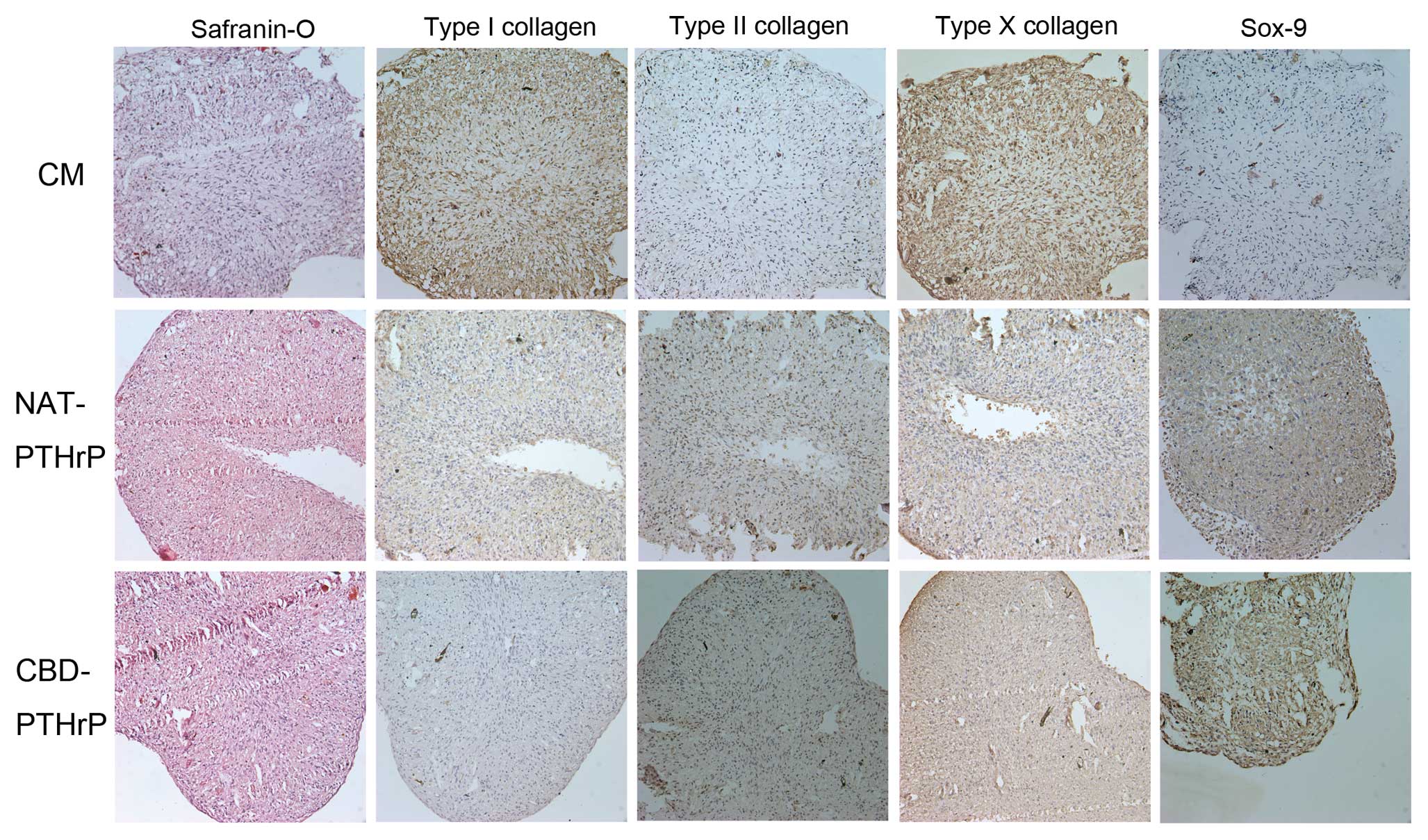
Histological findings
Histological findings of Safranin-O and
immunohistochemistry for type I, II and X collagens and Sox-9
generally mirrored changes detected by qRT-PCR and western blotting
with minor variations. Safranin-O staining showed an increase in
the metachromatic staining after NAT-PTHrP and CBD-PTHrP treatment.
Type II collagen expression markedly increased in both NAT-PTHrP
and CBD-PTHrP treatment groups while type I collagen staining
decreased in both treatment groups. Type X collagen expression
decreased after NAT-PTHrP and CBD-PTHrP treatment. Sox-9 protein
expression increased dramatically after NAT-PTHrP and CBD-PTHrP
treatment in BM-MSCs (Fig.
4).
Discussion
MSCs are an attractive option for cartilage regenera
tion because of their abilities to proliferate and their easy
accessibility (8–10). However, chondrocyte proliferation
and chondrocyte hypertrophy are the main challenges for cartilage
regeneration from MSCs (13,14). Previous studies suggest that PTHrP
may circumvent these problems by promoting chondrogenesis and
suppressing hypertrophy in the growth plate through the
PTHrP-Indian hedgehog (IHH) axis (17–20). During chondrocyte induction from
MSCs, the cells secrete PTHrP during the early phase of
differentiation, until days 14–21, when the mRNA levels for PTHrP
declined, whereas those for IHH were upregulated for the remaining
weeks of culture (30). PTHrP has
been shown to severely reduce type X collagen expression, AP
activity, and cell enlargement of lower sternal chondrocytes from
immature chicken, and these molecules are soluble factors produced
by articular chondrocytes (31).
These results make PTHrP an attractive candidate for chondrocyte
induction.
Previous studies have used the PTHrP protein for the
purpose of cartilage chondrocyte induction (15). However, simple absorption of PTHrP
to the collagen scaffold would allow the diffusion of PTHrP into
extracellular fluids, and would rapidly lose its activity. In
addition, overexpression of PTHrP by a gene transfer method was
also used and induced an arthritic phenotype in articular
chondrocytes (32). In the
present study, we first engineered PTHrP to construct a
collagen-targeting system. Safranin-O staining showed that the
recombined protein CBD-PTHrP increased meta-chromatic staining
after treatment (Fig. 4A). At the
same time, CBD-PTHrP treatment increased the expression of COL2A1
and Sox-9 and decreased the expression of COL1A1 and COL10A1 at the
mRNA and protein levels (Figs. 3
and 4). These results were
slightly different than those of Kafienah et al (33) and Kim et al (15). Kafienah et al (33) found that type II collagen
expression was unchanged and expression of type I and X collagen
was suppressed while Kim et al (15) found that type X collagen
expression was gradually decreased, type I collagen expression was
suppressed and type II collagen expression was increased after
treating chondrogenic cultures of human MSCs with PTHrP.
Tissue engineering of skin, bone, vascular and nerve
offers a promising means of producing 3-dimensional neocartilages
for clinical treatment (34), and
various factors for tissue engineering have been developed, such as
bFGF, NGF and PDGF (35–37). However, in clinical practice,
factors simply delivered in solution are difficult to be retained
at the injured site due to their rapid diffusion in extracellular
fluids. Many attempts have been made to overcome the difficulties
in using factors as a therapeutic agent. In order to maintain
adequate factor concentration, multiple injections are needed.
However, this would increase the cost and surgical risks, and may
even have adverse effects. Immobilization of bFGF on
heparin-Sepharose beads prolonged the storage and release (38). Recently, the most abundant
component of extracellular matrices, collagen, has been widely used
in drug delivery (39,24). Meanwhile, in tissue engineering,
many types of collagen-based scaffolds have been fabricated, and
they have shown good characteristics in wound repair (40,41). During the process, collagen was
found to play an important role in providing a cell anchorage site,
mechanical stability and structural guidance. They also provided
the interface to respond to physiological and biological changes,
and to remodel the extracellular matrix to integrate with the
surrounding native tissue (42,43). Moreover, collagen is commonly used
as an attractive targeted site for exogenous peptide growth
factors. Targeted growth factors on collagen may not only retain
its activity and control its diffusion but may also repair injured
tissues locally. Nerve growth factor-β, platelet-derived growth
factor and basic fibroblast growth factor have been added with a
collagen-binding peptide, and the results have shown that CBD
specifically binds to native collagen, and the modified growth
factors achieve better repair compared to the native growth factors
at the same concentration (28,44,45). In the present study, CBD which is
a peptide of 7 amino acids was first used to engineer PTHrP to
specially target the PTHrP to collagen. Our results showed that
CBD-PTHrP has a higher binding ability to collagen than NAT-PTHrP
(Fig. 2). The ability of
CBD-PTHrP to induce chondrogenesis in MSCs was also measured by an
in vitro pellet assay. As shown in Figs. 3 and 4, CBD-PTHrP induced chondrogenesis and
inhibited chondrocyte differentiation toward hypertrophy as well as
NAT-PTHrP. In future studies, the collagen-based scaffolds of bone
will be used to determine the ability of CBD-PTHrP to produce
MSC-derived tissue-engineered cartilage in vivo.
In conclusion, our study suggests that CBD-PTHrP is
an attractive recombined protein for use in cartilage tissue
engineering from MSCs. It demonstrated that CBD-PTHrP has a higher
binding ability to collagen than NAT-PTHrP and significantly
enhances chondrogenesis and suppresses hyper-trophy in BM-MSCs.
Further investigations are warranted to confirm the ability of
CBD-PTHrP to produce MSC-derived tissue-engineered cartilage in
vivo based on collagen-based scaffolds of bone.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (nos. 21002018 and
81071498).
References
|
1.
|
Cremer MA, Rosloniec EF and Kang AH: The
cartilage collagens: a review of heir structure, organization, and
role in the pathogenesis of experimental arthritis in animals and
in human rheumatic disease. J Mol Med. 76:275–288. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Madry H, van Dijk CN and Mueller-Gerbl M:
The basic science of the subchondral bone. Knee Surg Sports
Traumatol Arthrosc. 18:419–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Jackson DW, Simon TM and Aberman HM:
Symptomatic articular cartilage degeneration: the impact in the new
millennium. Clin Orthop Relat Res. (Suppl): S14–S25. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Gomoll AH, Farr J, Gillogly SD, Kercher JS
and Minas T: Surgical management of articular cartilage defects of
the knee. Instr Course Lect. 60:461–483. 2011.PubMed/NCBI
|
|
5.
|
Agnesi F, Amrami KK, Frigo CA and Kaufman
KR: Comparison of cartilage thickness with radiologic grade of knee
osteoarthritis. Skeletal Radiol. 37:639–643. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Bae WC, Temple MM, Amiel D, Coutts RD,
Niederauer GG and Sah RL: Indentation testing of human cartilage:
sensitivity to articular surface degeneration. Arthritis Rheum.
48:3382–3394. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Magnussen RA, Dunn WR, Carey JL and
Spindler KP: Treatment of focal articular cartilage defects in the
knee: a systematic review. Clin Orthop Relat Res. 466:952–962.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Cucchiarini M and Madry H: Gene therapy
for cartilage defects. J Gene Med. 7:1495–1509. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Charbord P: Bone marrow mesenchymal stem
cells: historical overview and concepts. Hum Gene Ther.
21:1045–1056. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Pittenger MF, Mackay AM, Beck SC, et al:
Multilineage potential of adult human mesenchymal stem cells.
Science. 284:143–147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Grogan SP, Miyaki S, Asahara H, D’Lima DD
and Lotz MK: Mesenchymal progenitor cell markers in human articular
cartilage: normal distribution and changes in osteoarthritis.
Arthritis Res Ther. 11:R852009. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Koelling S, Kruegel J, Irmer M, Path JR,
Sadowski B, Miro X and Miosge N: Migratory chondrogenic progenitor
cells from repair tissue during the later stages of human
osteoarthritis. Cell Stem Cell. 4:324–335. 2009. View Article : Google Scholar
|
|
13.
|
Barry F, Boynton RE, Liu B and Murphy JM:
Chondrogenic differentiation of mesenchymal stem cells from bone
marrow; differentiation-dependent gene expression of matrix
components. Exp Cell Res. 268:189–200. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Pelttari K, Winter A, Steck E, et al:
Premature induction of hypertrophy during in vitro chondrogenesis
of human mesenchymal stem cells correlates with calcification and
vascular invasion after ectopic transplantation in SCID mice.
Arthritis Rheum. 54:3254–3266. 2006. View Article : Google Scholar
|
|
15.
|
Kim YJ, Kim HJ and Im GI: PTHrP promotes
chondrogenesis and suppresses hypertrophy from both bone
marrow-derived and adipose tissue-derived MSCs. Biochem Biophys Res
Commun. 373:104–108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Suva LJ, Winslow GA, Wettenhall RE, et al:
A parathyroid hormone-related protein implicated in malignant
hypercalcemia: cloning and expression. Science. 237:893–896. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Kronenberg HM: PTHrP and skeletal
development. Ann NY Acad Sci. 1068:1–13. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Kobayashi T, Soegiarto DW, Yang Y, et al:
Indian hedgehog stimulates periarticular chondrocyte
differentiation to regulate growth plate length independently of
PTHrP. J Clin Invest. 115:1734–1742. 2005. View Article : Google Scholar
|
|
19.
|
Huang W, Chung UI, Kronenberg HM and de
Crombrugghe B: The chondrogenic transcription factor Sox9 is a
target of signaling by the parathyroid hormone-related peptide in
the growth plate of endochondral bones. Proc Natl Acad Sci USA.
98:160–165. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Amling M, Neff L, Tanaka S, et al: Bcl-2
lies downstream of parathyroid hormone-related peptide in a
signaling pathway that regulates chondrocyte maturation during
skeletal development. J Cell Biol. 136:205–213. 1997. View Article : Google Scholar
|
|
21.
|
Yurchenco PD, Smirnov S and Mathus T:
Analysis of basement membrane self-assembly and cellular
interactions with native and recombinant glycoproteins. Methods
Cell Biol. 69:111–144. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Eyre DR: Collagen of articular cartilage.
Clin Orthop Relat Res. (427 Suppl): S118–S122. 2004. View Article : Google Scholar
|
|
23.
|
Andrades JA, Han B, Becerra J, Sorgente N,
Hall FL and Nimni ME: A recombinant human TGF-beta1 fusion protein
with collagen-binding domain promotes migration, growth, and
differentiation of bone marrow mesenchymal cells. Exp Cell Res.
250:485–498. 1999. View Article : Google Scholar
|
|
24.
|
Nishi N, Matsushita O, Yuube K, Miyanaka
H, Okabe A and Wada F: Collagen-binding growth factors: production
and characterization of functional fusion proteins having a
collagen-binding domain. Proc Natl Acad Sci USA. 95:7018–7023.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Ishikawa T, Terai H, Yamamoto T, Harada K
and Kitajima T: Delivery of a growth factor fusion protein having
collagen-binding activity to wound tissues. Artif Organs.
27:147–154. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Im GI, Shin YW and Lee KB: Do adipose
tissue-derived mesenchymal stem cells have the same osteogenic and
chondrogenic potential as bone marrow-derived cells? Osteoarthritis
Cartilage. 13:845–853. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Im GI, Jung NH and Tae SK: Chondrogenic
differentiation of mesenchymal stem cells isolated from patients in
late adulthood: the optimal conditions of growth factors. Tissue
Eng. 12:527–536. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Sun WJ, Sun CK, Lin H, et al: The effect
of collagen-binding NGF-beta on the promotion of sciatic nerve
regeneration in a rat sciatic nerve crush injury model.
Biomaterials. 30:4649–4656. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Sun W, Lin H, Chen B, Zhao W, Zhao Y and
Dai J: Promotion of peripheral nerve growth by collagen scaffolds
loaded with collagen-targeting human nerve growth factor-beta. J
Biomed Mater Res A. 83:1054–1061. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Fischer J, Dickhut A, Rickert M and
Richter W: Human articular chondrocytes secrete parathyroid
hormone-related protein and inhibit hypertrophy of mesenchymal stem
cells in coculture during chondrogenesis. Arthritis Rheum.
62:2696–2706. 2010. View Article : Google Scholar
|
|
31.
|
Schmid TM and Linsenmayer TF:
Immunohistochemical localization of short chain cartilage collagen
(type X) in avian tissues. J Cell Biol. 100:598–605. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Wang D, Taboas JM and Tuan RS: PTHrP
overexpression partially inhibits a mechanical strain-induced
arthritic phenotype in chondrocytes. Osteoarthritis Cartilage.
19:213–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Kafienah W, Mistry S, Dickinson SC, Sims
TJ, Learmonth I and Hollander AP: Three-dimensional cartilage
tissue engineering using adult stem cells from osteoarthritis
patients. Arthritis Rheum. 56:177–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Caplan AI: Mesenchymal stem cells:
cell-based reconstructive therapy in orthopedics. Tissue Eng.
11:1198–1211. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Heldin CH and Westermark B: Mechanism of
action and in vivo role of platelet-derived growth factor. Physiol
Rev. 79:1283–1316. 1999.PubMed/NCBI
|
|
36.
|
Otto D, Unsicker K and Grothe C:
Pharmacological effects of nerve growth factor and fibroblast
growth factor applied to the transectioned sciatic nerve on neuron
death in adult rat dorsal root ganglia. Neurosci Lett. 83:156–160.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Ishihara M, Obara K, Ishizuka T, et al:
Controlled release of fibroblast growth factors and heparin from
photocrosslinked chitosan hydrogels and subsequent effect on in
vivo vascularization. J Biomed Mater Res A. 64:551–559. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Cai S, Liu Y, Zheng Shu X and Prestwich
GD: Injectable glycosaminoglycan hydrogels for controlled release
of human basic fibroblast growth factor. Biomaterials.
26:6054–6067. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Andrades JA, Wu LT, Hall FL, Nimni ME and
Becerra J: Engineering, expression, and renaturation of a
collagen-targeted human bFGF fusion protein. Growth Factors.
18:261–275. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Li X, Feng Q, Liu X, Dong W and Cui F:
Collagen-based implants reinforced by chitin fibres in a goat shank
bone defect model. Biomaterials. 27:1917–1923. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Park SN, Kim JK and Suh H: Evaluation of
antibiotic-loaded collagen-hyaluronic acid matrix as a skin
substitute. Biomaterials. 25:3689–3698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Rose FR and Oreffo RO: Bone tissue
engineering: hope vs. hype. Biochem Biophys Res Commun. 292:1–7.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Chapekar MS: Tissue engineering:
challenges and opportunities. J Biomed Mater Res. 53:617–620. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Zhao WX, Chen B, Li X, et al:
Vascularization and cellularization of collagen scaffolds
incorporated with two different collagen-targeting human basic
fibroblast growth factors. J Biomed Mater Res A. 82:630–636. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Lin H, Chen B, Sun W, Zhao W, Zhao Y and
Dai Y: The effect of collagen-targeting platelet-derived growth
factor on cellularization and vascularization of collagen
scaffolds. Biomaterials. 27:5708–5714. 2006. View Article : Google Scholar : PubMed/NCBI
|