Introduction
The development of renal interstitial fibrosis is
closely associated with the progression of chronic kidney disease,
which eventually leads to renal failure. Increasing evidence
suggests that renal tubular epithelial-mesenchymal transition (EMT)
plays an important role in renal interstitial fibrosis (1), and that EMT is one of the cellular
and molecular mechanisms involved in renal interstitial fibrosis
(2). EMT is a phenomenon whereby
epithelial cells transform into mesenchymal cells under specific
physiological and pathological conditions. The transformation
begins with a reduction or loss of expression of the epithelial
cell marker, E-cadherin, followed by the disruption of tight
junctions, rearrangement of the cytoskeleton, the induction of
mesenchymal cell markers, such as α-smooth muscle actin (α-SMA),
and the destruction of the basement membrane, which allows for the
migration of transformed cells to the interstitial compartment
(3–5).
The involvement of EMT in fibrosis is
internationally controversial, and the controversy stems from the
fact that it is difficult for pathologists to observe EMT-like
morphological changes. This is mainly due to the fact that the EMT
process is extremely short, and is not easy to capture. However,
increasing evidence indicates that fibroblasts in the fibrotic
disease process are not only derived from mesenchymal stem cells in
bone marrow or the body organs, but they may also be the same cells
from the injured tissues following EMT (1,6).
Evidence indicates that epithelial cells are an important source of
muscle fibroblasts in the process of fibrosis (7). In vivo evidence of EMT has
been observed in animal models of obstructive nephropathy (8), diabetic nephropathy (9) and 5/6 nephrectomy remnant kidney
(10).
Transforming growth factor-β1 (TGF-β1) mediates
renal fibrosis by inducing the transformation of tubular epithelial
cells into myofibroblasts through EMT (10). TGF-β1 is formed as a ligand
receptor complex. At the cell surface, it is now well established
that the binding of TGF-β1 to its type II receptor (Tβ RII) can
activate the TGF-β receptor type I (Tβ RI)-kinase, resulting in the
phosphorylation of Smad2 and Smad3, 2 recepor-associated Smads
(R-Smads). Subsequently, phosphorylated Smad2 and Smad3 (p-Smad2
and p-Smad3) bind to the common Smad4 and form the Smad complex,
which translocates into the nucleus to regulate target gene
transcription (11).
miR-192 is a TGF-β-dependent microRNA, and during
renal fibrosis it is tightly regulated by TGF-β1 via Smad3
(12), and miR-192 is expressed
in mesangial cells and renal tubular epithelial cells (13,14). It has been demonstrated that
miR-192 regulates the expression of Smad-interacting protein 1
(SIP1) [also known as Zinc finger E-box-binding homeobox 2 (ZEB2)],
and that SIP1 plays a key role in the TGF-β1 signaling pathway. The
knockdown of SIP1 blocks the TGF-β1-mediated suppression of
E-cadherin, suggesting that through SIP1 modulation, TGFβ1
regulates the expression of E-cadherin, EMT and fibrosis (14). Taken together, these results
suggest that Smad3 and miR-192 are involved in TGF-β1-mediated
EMT.
Vascular endothelial growth factor (VEGF) is a
growth factor that is involved in angiogenesis and vasculogenesis.
In the kidneys, VEGF is mainly expressed in glomerular podocytes
and tubular epithelial cells (16). Previous studies have illustrated
that VEGF reduces the severity of disease in several experimental
models of kidney disease, such as anti-Thy1 glomerulonephritis,
thrombotic microangiopathy glomerulonephritis and anti-glomerular
basement membrane glomerular nephritis (17–19). Other studies have also documented
a reduction in VEGF expression in renal interstitial fibrosis
(20–22). In our previous study, we also
demonstrated that in mouse model of unilateral ureteral obstruction
(UUO), the expression of VEGF prevented renal interstitial fibrosis
and EMT, and that in human renal tubular epithelial cells, VEGF
directly inhibited TGF-β1-induced EMT (23).
The PI3K/Akt signaling pathway is one of the major
pathways in which VEGF plays a role (24–27). On the other hand, LY294002 is a
protein kinase inhibitor that blocks the phosphatidylinositol-3
kinase (PI3K) cell signaling pathway.
The data mentioned above suggest that VEGF protects
renal tubular epithelial cells and that Smad3 and miR-192 are
involved in the development of TGF-β1-induced renal tubular EMT.
Therefore, in this study, we aimed to further elucidate the
mechanisms by which VEGF suppresses EMT, as well as to investigate
the role of Smad3, miR-192, Smad2 and Smad4 in this process.
Materials and methods
Reagents
Rh-TGF-β1 was purchased from R&D Systems
(Minneapolis, MN, USA). Anti-α-SMA was obtained from Abcam
(Cambridge, UK); anti-E-cadherin, anti-Smad3, anti-p-Smad3,
anti-Smad2, anti-Smad4 and LY294002 were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA). The Hairpin-it™
miRNA quantitative PCR kit was purchased from Genepharma (Shanghai
GenePharma Co. Ltd, China) TRIzol reagent was obtained from Life
Technologies/BRL (Rockville, MD, USA). M-MLV reverse transcriptase,
dNTP, Oligo(dT) primer and Taq polymerase were purchased from
Promega (Madison, WI, USA). The BCA protein assay kits were
purchased from Beyotime (Haimen, China). The MTT kit was purchased
from Beijing Applygen Technologies Inc. (Beijing, China). DMEM-F12
and fetal bovine serum were purchased from HyClone Laboratories
Inc. (Logan, UT, USA). The VEGF ELISA kit was purchased from
R&D Systems.
Construction of human kidney cortex (HKC)
cells stably expressing VEGF
The full-length VEGF121 and VEGF165 sequences were
used to create the pBuDCE4.1 expression vectors. VEGF121 was under
the control of the CMV promoter and VEGF165 was under the control
of the EF-1α promoter. The vectors contained a Zeocin resistance
cassette. The constructs were sequenced and tested for Zeocin
resistance. The correct final construct was designated as Z7842-2.
The Z7842-2 plasmid was introduced into HKC cells by
electroporation, and the stable clones were selected in a gradual
increase in the concentration of Zeocin. The expression of VEGF was
detected by measuring the VEGF concentration in the supernatant by
ELISA. The detected concentration of VEGF in the Z7842-2 HKC
supernatant was 1210.061 pg/ml, which was 10.72-fold higher than
the control cells (p<0.05). The ELISA results confirmed that we
successfully established HKC cells stably overexpressing VEGF
(HKC-SOEV cells).
HKC culture
HKC cells were cultured in Dulbecco’s modified
Eagle’s medium/Ham’s F-12 (DMEM-F12) containing 10% fetal bovine
serum and grown at 37°C and 5% CO2 in a humidified
incubator. Cells were passaged with 0.25% trypsin digestion. The
medium was changed every day. When the cells reached 70–80%
confluence, the medium was replaced with serum-free medium for 24 h
and then the cells were divided into the indicated experimental
groups. All experiments were repeated 3 times. The cultured cells
were divided into the following experimental groups: group A,
normal untreated HKC cells; group B, normal HKC cells treated with
TGF-β1 (5 μg/l) for 24 or 48 h; group C, HKC-SOEV cells;
group D, HKC-SOEV cells treated with TGF-β1 (5 μg/l) for 24
or 48 h; and group E, HKC-SOEV cells co-treated with TGF-β1 (5
μg/l) and LY294002 (20 μmol/l) for 24 or 48 h.
Cell proliferation and toxicity
detection
HKC cells were incubated with various concentrations
of the PI3K-Akt signaling pathway inhibitor, LY294002 (0, 6.25,
12.5, 25, and 50 μmol/l), for 48 h and then cell
proliferation was analyzed by MTT assay. Cytotoxicity was analyzed
using the lactate dehydrogenase (LDH) release assay according to
the manufacturer’s instructions.
Quantitative PCR (real-time PCR)
RNA from the HKC cells was isolated using the TRIzol
One Step Isolation kit according to the manufacturer’s
instructions. The conditions for reverse transcription reaction
were as follows: 16°C for 30 min, followed by 42°C for 30 min and
85°C for 10 min. The real-time quantitative PCR reaction mix
comprised of real-time PCR buffer, miR-specific primer set (5
μM), miRNA RT product, Taq DNA polymerase (5 U/μl)
and ddH2O to a final volume of 20 μl. The
real-time quantitative PCR conditions were as follows: 95°C for 3
min followed by 40 cycles of 95°C for 12 sec and 62°C for 40–60
sec. By selecting SYBR detection dye, ROX can be used as a
calibration dye. Detection was set at 62°C. In order to reach the
set threshold cycle number (Ct value), the real-time quantitative
PCR fluorescent signal was measured in each reaction tube. U6 was
used as the internal control, and the miRNA results were measured
using the relative quantification method, in which
2−ΔΔCt indicates the relative fold of expression
compared to the control. ΔΔCt = (Ct of miRNA − Ct of
U6)treatment group − (Ct of miRNA − Ct of
U6)control group.
Western blot analysis
The cells in each group were washed with cold
phosphate-buffered saline (PBS) 3 times and then lysed in 100
μl RIPA buffer containing protease (100 μg/ml) and
phosphatase (1 mM) inhibitors at 4°C for 30 min. The samples were
then centrifuged at 12,000 rpm for 10 min. The protein
concentration was determined using a BCA protein assay kit, and
whole lysates were mixed with an equal amount of 2X SDS loading
buffer (125 mmol/l Tris-HCl, 4% SDS, 20% glycerol, 100 mmol/l DTT
and 0.2% bromophenol blue). Samples were heated at 100°C for 10 min
and then separated on SDS-polyacrylamide gels. The separated
proteins were then transferred onto a polyvinylidene difluoride
(PVDF) membrane. The membrane blots were first probed with a
primary antibody. Following incubation with horseradish
peroxidase-conjugated secondary antibody, autoradiograms were used
to visualize protein bands using an enhanced chemiluminescent
system. The signals were measured on a UVI Gel image analysis
system (UVI soft UVI band windows application V97.04), which
scanned and recorded the intensity of the optical density. The
primary antibodies that we used included anti-Smad3 (1:1,000),
anti-p-Smad3 (1:100), anti-Smad2 (1:1,000), anti-Smad4 (1:1,000),
anti-α-SMA (1:800), anti-E-cadherin (1:600) and anti-GAPDH
(1:10,000). The results were normalized to the signal intensity of
GAPDH, which was used as an internal control.
Confocal microscopy
Cells were washed with PBS and then fixed in 4%
formaldehyde for 10 min, followed by a wash with 0.2% PBS for 5
min. The cells were then incubated with sheep serum for 10 min and
0.5% Triton-100 at room temperature for 15 min. The cells were then
washed again in 0.2% PBS for 5 min and a rabbit anti-human
E-cadherin antibody was added at a dilution of 1:100 or rabbit
anti-human SMA at a dilution of 1:100. The cells were incubated
with the antibodies at 4°C overnight and then washed 3 times with
0.2% PBS for 5 min each the following day. They were then incubated
with goat anti-mouse rhodamine-labeled IgG at a dilution of 1:50
and goat anti-rabbit FITC-labeed IgG at a dilution of 1:50 at room
temperature for 1 h. The cells were then washed with 0.2% PBS 3
times for 5 min each. Finally, a fluorescent anti-quenching agent
in 5% PBS-glycerol was incubated with the cells. Microscopy was
performed with a Leica TCS SP2 AOBS confocal microscope (Leica
Micro-Systems, Heidelberg, Germany) using an excitation wavelength
of 488 nm from an Argon laser and an emission spectra between 520
and 570 nm. Collection parameters remained constant for all
samples.
Statistical analysis
SPSS v11.5 software (SPSS, Inc., Chicago, IL, USA)
was used for statistical analysis. All data are presented as the
means ± standard deviation (SD) and expressed using a significant
difference test with single analysis of variance (ANOVA). A p-value
<0.05 was considered to indicate a statistically significant
difference.
Results
Effect of LY294002 on HKC cell
proliferation and its toxicity
The results from the MTT and LDH assays demonstrated
that at concentrations <25 μmol/l, LY294002 had no
effects on HKC cell proliferation and did not induce significant
cytotoxic effects.
Comparison of the expression of Smad2 and
Smad4 upon the indicated treatments
The western blot analysis results indicated that
TGF-β1 and/or VEGF did not alter the expression of Smad2. TGF-β1
increased Smad4 expression, while VEGF reduced Smad4 expression.
The expression of Smad4 in the HKC-SOEV cells stimulated with
TGF-β1 for 48 h was lower than that in the other groups (p<0.05)
(Fig. 1).
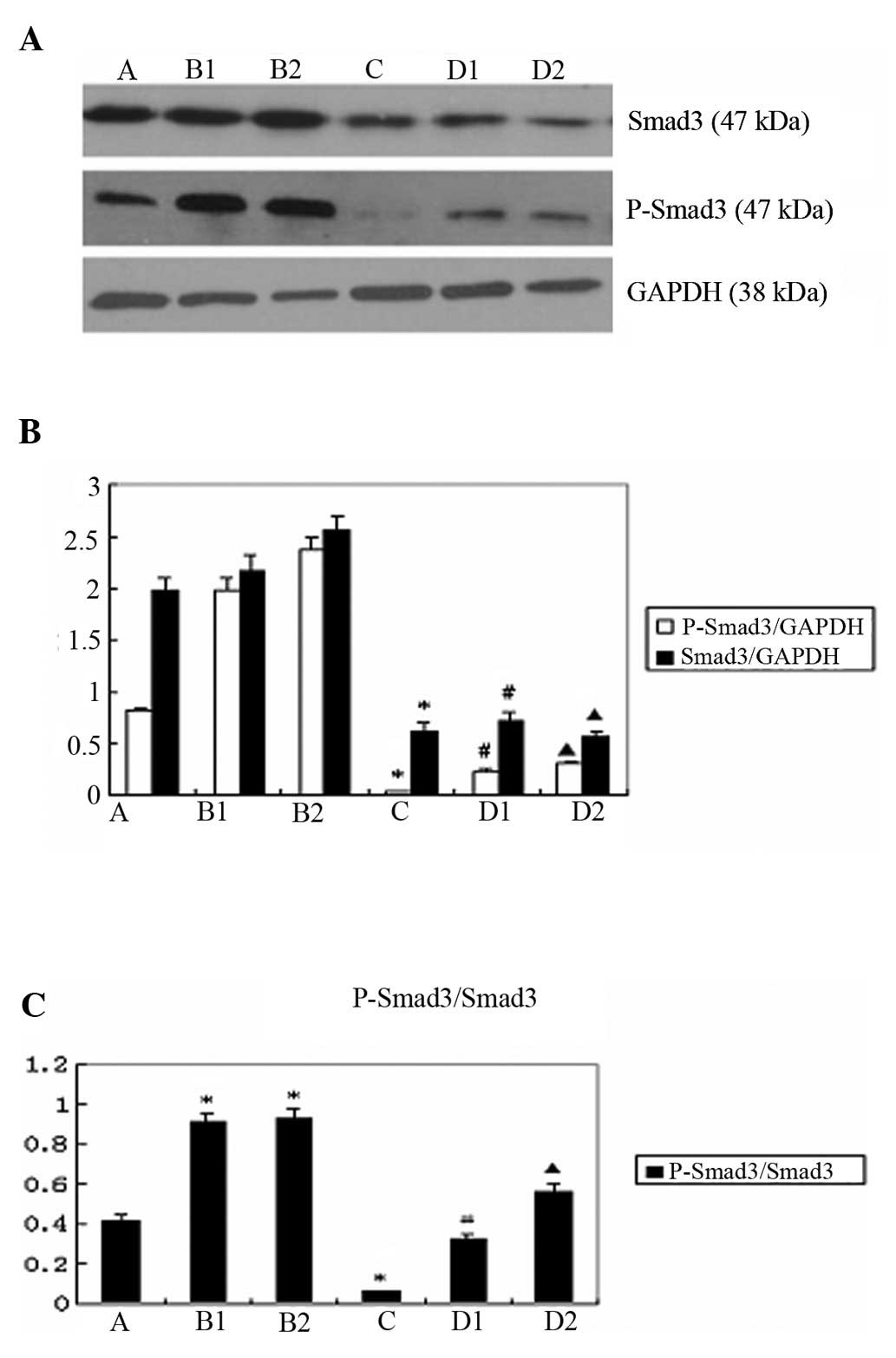
Comparison of the expression of p-Smad3
and Smad3 upon the indicated treatments
The western blot analysis results revealed that the
expression of p-Smad3 and Smad3 was lower in group D compared to
group B (p<0.05) (Fig. 2A and
B). The ratio of p-Smad3/Smad3 expression was significantly
lower in group D, compared to that in group B (p<0.05) (Fig. 2C).
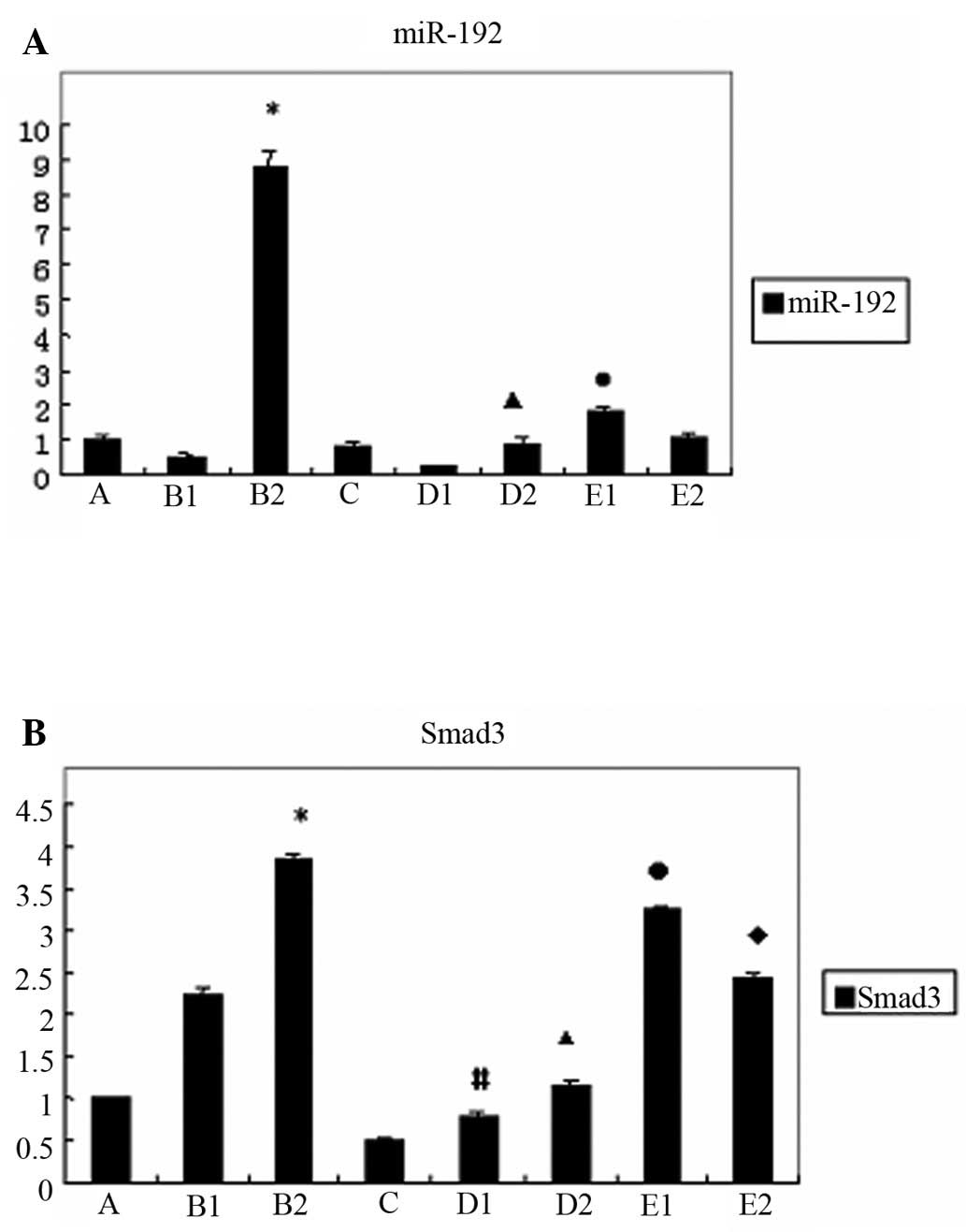
Gene expression of miR-192 and Smad3 in
the various HKC cell groups
We found that in normal HKC cells, the expression of
miR-192 was increased following treatment with TGF-β1 for 48 h
(p<0.05); however, in the HKC-SOEV cells stimulated with TGF-β1,
it was significantly lower (p<0.05). When the HKC-SOEV cells
were co-treated with TGF-β1 and LY294002 for 24 h, the expression
of miR-192 was higher compared to the cells treated with TGF-β1
alone (p<0.05). (Fig. 3A) The
treatment of normal HKC cells with TGF-β1 induced Smad3 expression,
as observed in group B (p<0.05). We also observed a reduction in
Smad3 expression in group C compared to group A. Moreover, the
expression of Smad3 in group D was lower compared to that in group
B (p<0.05). By contrast, in group E, the induction of Smad3
expression was clearly restored upon co-treatment with TGF-β1 and
LY294002 (p<0.05) (Fig.
3B).
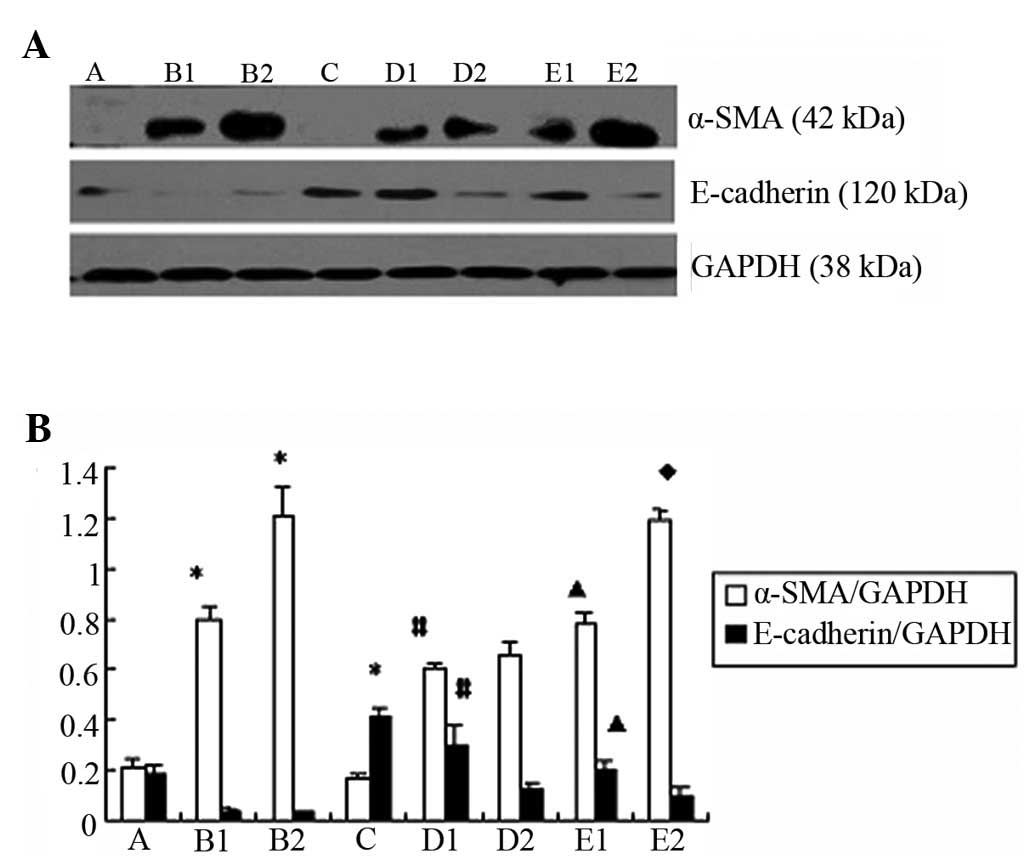
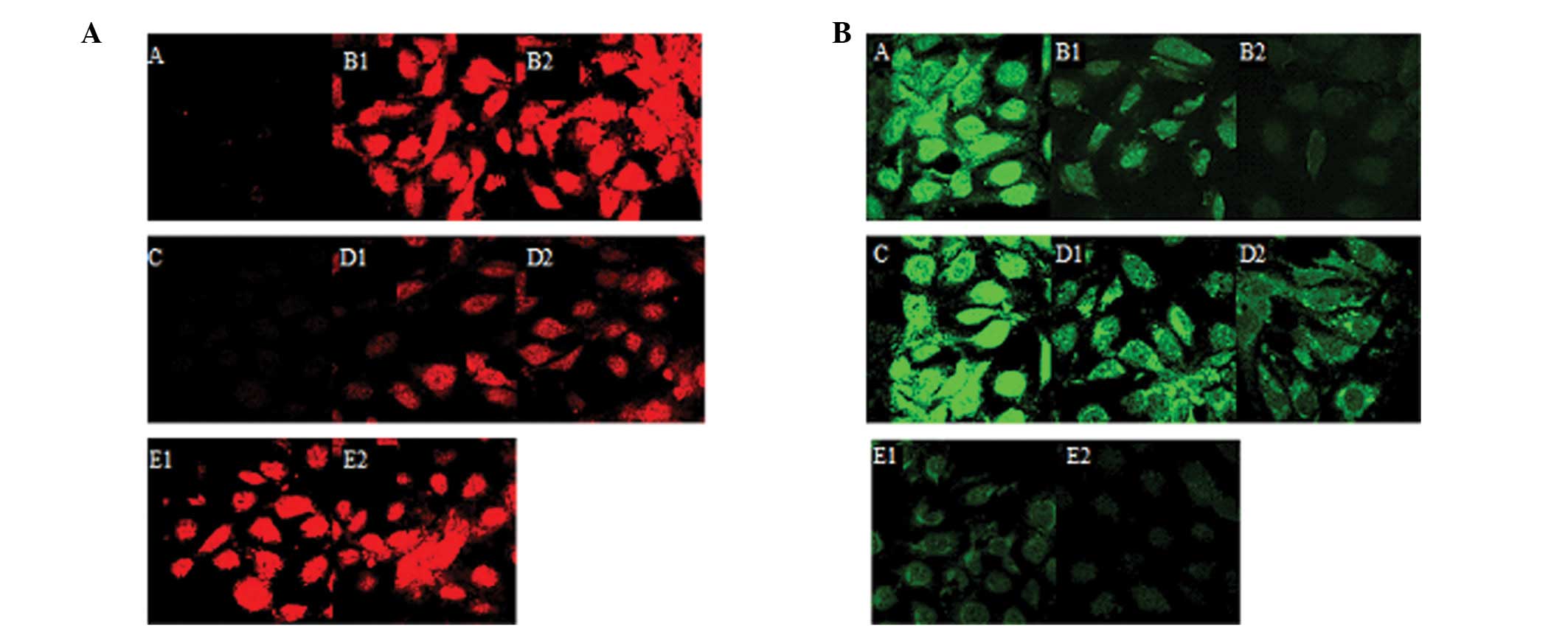
Comparison of the expression of
E-cadherin and α-SMA expression upon the indicated treatments
The western blot analysis and confocal microscopy
results indicated that the expression of α-SMA was significantly
reduced in group D compared to group B (p<0.05) (Figs. 4A and B and 5A). By contrast, the expression of
E-cadherin increased in group D compared to group B (p<0.05).
(Figs. 4A and B and 5B).
Discussion
Renal interstitial fibrosis is a common pathological
process that occurs during the development of a variety of chronic
kidney diseases, consequently leading to end-stage renal failure.
VEGF is a growth factor for endothelial cells and not only plays an
important role in maintaining the integrity of vascular endothelial
cells, but is also an important survival factor for renal tubular
epithelial cells and is involved in renal tubular formation
(28–30). Our previous studies confirmed that
VEGF inhibits TGF-β1-induced EMT (23,31).
TGF-β1-induced EMT has been widely documented in the
literature (32,33). In this study, we found that the
treatment of normal HKC cells with TGF-β1 resulted in a higher
level of α-SMA expression. In addition, the treatment had an
opposite effect on E-cadherin expression. Compared to HKC-SOEV
cells, the treatment of normal HKC cells with TGF-β1 suppressed
E-cadherin expression, but enhanced α-SMA expression. When the
HKC-SOEV cells were co-treated with TGF-β1 and LY294002, the effect
of VEGF was weakened. These results strongly suggest that VEGF
inhibits TGF-β1-induced EMT, which is consistent with the results
presented in our previous studies (23,31).
The TGF-β1/Smad signaling pathway is an important
mechanism through which TGF-β1 induces tubular EMT (3–5,34).
Smad2, Smad3 and Smad4 are direct mediators of the TGF-β signaling
pathway (35). In this study, we
found that TGF-β1 and/or VEGF did not alter the expression of
Smad2; however, TGF-β1 increased the expression of Smad4. To our
surprise, VEGF did not alter Smad4 alone, but reduced Smad4
expression in the cells treated with TGF-β1. This effect was more
pronounced when the HKC-SOEV cells were stimulated with TGF-β1 for
48 h. The mechanism involved however, remains unclear. Hence, the
action of Smad2 and Smad4 was scant in the procedure of EMT.
However, it is also important to determine the effect of VEGF on
Smad3. Smad3 has been shown to play a critical role in this pathway
(11,15,35). Our study verified that in the HKC
cells, treatment with TGF-β1 induced Smad3 expression, whereas in
the HKC-SOEV cells, the effect of TGF-β1 on Smad3 was reduced,
suggesting that VEGF suppresses the expression of Smad3. In
addition, treatment with TGF-β1 enhanced the p-Smad3/Smad3
expression ratio in normal HKC cells. However, in the HKC-SOEV
cells, the enhancement of this ratio was significantly reduced,
suggesting that VEGF suppresses Smad3 phosphorylation. Importantly,
treatment with LY294002 significantly diminished the effect of
VEGF, further illustrating that VEGF suppresses Smad3 and p-Smad3.
Our results also demonstrated that VEGF inhibited TGF-β1-mediated
EMT. Therefore, it is possible that VEGF inhibits EMT partly
through suppressing Smad3 expression and phosphorylation. The
change in Smad3 expression was much more pronounced than the change
in the expression of Smad2 and Smad4, which indicates that Smad3 is
the key modulator. These results are in agreement with those from a
previous study (15).
In a previous study, Perera reported that miR-192
promoter sequences contain a highly conserved Ets-1 binding site
(36); TGF-β1 can influence the
Ets-1 regulation of miR-192 expression (36). Our study demonstrated that
following treatment with TGF-β1 for 48 h, the expression of miR-192
significantly increased in HKC cells, indicating that in the human
tubular epithelium, TGF-β1 can also stimulate miR-192; these
results are consistent with those from a previous study (12). In our study, we observed that in
the HKC-SOEV cells, treatment with TGF-β1 for 48 h failed to induce
miR-192 expression, indicating that VEGF inhibits TGF-β1-induced
miR-192 expression. Treatment with the PI3K inhibitor, LY294002,
which impairs VEGF signal transduction, significantly increased the
expression of miR-192, further demonstrating that VEGF inhibited
the expression of miR-192. Our study results strongly suggest that
VEGF inhibits TGF-β1-induced EMT, and that TGF-β1 upregulates
miR-192 expression in the renal tubular epithelium; therefore, VEGF
suppresses EMT through the downregulation of miR-192.
Evidence supporting the interaction of Smad3 with
miR-192 also came from the findings that there are conserved Smad3
binding sites in the promoter region of miR-192 and that Smad3 is
able to interact with individual promoter regions as detected by
Chip assay (12). In a previous
study, it was demonstrated that in a mouse model of UUO, the
expression of miR-192 was significantly increased in wild-type mice
expressing Smad3, while it was downregulated in Smad3 gene knockout
mice (12). This indicates that
Smad3 and miR-192 are closely correlated. In our study, we
confirmed that VEGF suppressed Smad3 in the process of
TGF-β1-induced EMT. We found that VEGF significantly inhibited the
expression of miR-192. miR-192 is a specific microRNA targeting
Smad3, that is, miR-192 is the downstream target gene, and Smad3
may also positively regulate downstream specific miR-192 to mediate
renal fibrosis (11). Thus, we
inferred that VEGF repressed Smad3 and then reduced the expression
of miR-192, subsequently inhibiting EMT induced by TGF-β1. To the
best of our knowledge, this is the first study to report such an
observation.
In conclusion, the data presented in this study
indicate that VEGF inhibits the expression of Smad3 and
subsequently reduces miR-192 expression, thus suppressing EMT, and
thereby alleviating fibrosis. Therefore, our study provides a new
basis for the clinical application of VEGF in the treatment of
early chronic renal interstitial fibrosis.
Acknowledgements
This study was supported by a grant
from the National Natural Science Foundation of China (no.
30570854).
References
|
1
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wynn TA: Cellular and molecular mechanisms
of fibrosis. J Pathol. 214:199–210. 2008. View Article : Google Scholar
|
|
3
|
Roxburgh SA, Murphy M, Pollock CA and
Brazil DP: Recapitulation of embryological programmes in renal
fibrosis- the importance of epithelial cell plasticity and
developmental genes. Nephron Physiol. 103:139–148. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khew-Goodall Y and Wadham C: A perspective
on regulation of cell-cell adhesion and epithelial-mesenchymal
transition: known and novel. Cells Tissues Organs. 179:81–86. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grande MT and López-Novoa JM: Fibroblast
activation and myofibroblast generation in obstructive nephropathy.
Nat Rev Nephrol. 5:319–328. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Neilson EG: Setting a trap for tissue
fibrosis. Nat Med. 11:373–374. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Selman M and Pardo A: Role of epithelial
cells in idiopathic pulmonary fibrosis: from innocent targets to
serial killers. Proc Am Thorac Soc. 3:364–372. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Y: Epithelial to mesenchymal
transition in renal fibrogenesis: pathologic significance,
molecular mechanism, and therapeutic intervention. J Am Soc
Nephrol. 15:11–12. 2004.
|
|
9
|
Holian J, Qi W, Kelly DJ, Zhang Y, Mreich
E, Pollock CA and Chen XM: Role of Kruppel-like factor 6 in
transforming growth factorβ1-induced epithelial-mesenchymal
transition of proximal tubule cells. Am J Physiol Renal Physiol.
295:F1388–F1396. 2008.
|
|
10
|
Lan HY: Tubular epithelial-myofibroblast
transdifferentiation mechanisms in proximal tubule cells. Curr Opin
Nephrol Hypertens. 12:25–29. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lan HY: Diverse roles of TGF-β/Smads in
renal fibrosis and inflammation. Int J Biol Sci. 7:1056–1067.
2011.
|
|
12
|
Chung AC, Huang XR, Meng X and Lan HY:
miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J Am Soc
Nephrol. 21:1317–1325. 2010. View Article : Google Scholar
|
|
13
|
Wang B, Herman-Edelstein M, Koh P, Burns
W, Jandeleit-Dahm K, Watson A, Saleem M, Goodall GJ, Twigg SM,
Cooper ME and Kantharidis P: E-cadherin expression is regulated by
miR-192/215 by a mechanism that is independent of the profibrotic
effects of transforming growth factor-beta. Diabetes. 59:1794–1802.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kato M, Zhang J, Wang M, Lanting L, Yuan
H, Rossi JJ and Natarajan R: MicroRNA-192 in diabetic kidney
glomeruli and its function in TGF-beta-induced collagen expression
via inhibition of E-box repressors. Proc Natl Acad Sci USA.
104:3432–3437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sato M, Muragaki Y, Saika S, Roberts AB
and Ooshima A: Targeted disruption of TGF-beta1/Smad3 signaling
protects against renal tubulointerstitial fibrosis induced by
unilateral ureteral obstruction. J Clin Invest. 112:1486–1494.
2003. View Article : Google Scholar
|
|
16
|
Schrijvers BF, Flyvbjerg A and De Vriese
AS: The role of vascular endothelial growth factor (VEGF) in renal
pathophysiology. Kidney Int. 65:2003–2017. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wada Y, Morioka T, Oyanagi-Tanaka Y, Yao
J, Suzuki Y, Gejyo F, Arakawa M and Oite T: Impairment of vascular
regeneration precedes progressive glomerulosclerosis in anti-Thy1
glomerulonephritis. Kidney Int. 61:432–443. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim YG, Suga SI, Kang DH, Jefferson JA,
Mazzali M, Gordon KL, Matsui K, Breiteneder-Geleff S, Shankland SJ,
Hughes J, Kerjaschki D, Schreiner GF and Johnson RJ: Vascular
endothelial growth factor accelerates renal recovery in
experimental thrombotic microangiopathy. Kidney Int. 58:2390–2399.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shimizu A, Masuda Y, Mori T, Kitamura H,
Ishizaki M, Sugisaki Y and Fukuda Y: Vascular endothelial growth
factor165 resolves glomerular inflammation and accelerates
glomerular capillary repair in rat anti-glomerular basement
membrane glomerulonephritis. J Am Soc Nephrol. 15:2655–2665. 2004.
View Article : Google Scholar
|
|
20
|
Song YR, You SJ, Lee YM, Chin HJ, Chae DW,
Oh YK, Joo KW, Han JS and Na KY: Activation of hypoxia-inducible
factor attenuates renal injury in rat remnant kidney. Nephrol Dial
Transplant. 25:77–85. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun D, Feng J, Dai C, Sun L, Jin T, Ma J
and Wang L: Role of peritubular capillary loss and hypoxia in
progressive tubulointerstitial fibrosis in a rat model of
aristolochic acid nephropathy. Am J Nephrol. 26:363–371. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Burt LE, Forbes MS, Thornhill BA, Kiley SC
and Chevalier RL: Renal vascular endothelial growth factor in
neonatal obstructive nephropathy. I Endogenous VEGF. Am J Physiol
Renal Physiol. 292:F158–F167. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lian YG, Zhou QG, Zhang YJ and Zheng FL:
VEGF ameliorates tubulointerstitial fibrosis in unilateral ureteral
obstruction mice via inhibition of epithelial-mesenchymal
transition. Acta Pharmacol Sin. 32:1513–1521. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Holmes K, Roberts OL, Thomas AM and Cross
MJ: Vascular endothelial growth factor receptor-2: structure,
function, intracellular signalling and therapeutic inhibition. Cell
Signal. 19:2003–2012. 2007. View Article : Google Scholar
|
|
25
|
Kowanetz M and Ferrara N: Vascular
endothelial growth factor signaling pathways: therapeutic
perspective. Clin Cancer Res. 12:5018–5022. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cross MJ, Dixelius J, Matsumoto T and
Claesson-Welsh L: VEGF-receptor signal transduction. Trends Biochem
Sci. 28:488–494. 2003. View Article : Google Scholar
|
|
27
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Karihaloo A, Karumanchi SA, Cantley WL,
Venkatesha S, Cantley LG and Kale S: Vascular endothelial growth
factor induces branching morphogenesis/tubulogenesis in renal
epithelial cells in a neuropilin-dependent fashion. Mol Cell Biol.
25:7441–7448. 2005. View Article : Google Scholar
|
|
29
|
Kang DH, Joly AH, Oh SW, Hugo C,
Kerjaschki D, Gordon KL, Mazzali M, Jefferson JA, Hughes J, Madsen
KM, Schreiner GF and Johnson RJ: Impaired angiogenesis in the
remnant kidney model: I. Potential role of vascular endothelial
growth factor and thrombospondin-1. J Am Soc Nephrol. 12:1434–1447.
2001.PubMed/NCBI
|
|
30
|
Katavetin P, Miyata T, Inagi R, Tanaka T,
Sassa R, Ingelfinger JR, Fujita T and Nangaku M: High glucose
blunts vascular endothelial growth factor response to hypoxia via
the oxidative stress-regulated hypoxia-inducible
factor/hypoxia-responsible element pathway. J Am Soc Nephrol.
17:1405–1413. 2006. View Article : Google Scholar
|
|
31
|
Zhou QG, Zheng FL and Hou FF: Inhibition
of tubulointerstitial fibrosis by pentoxifylline is associated with
improvement of vascular endothelial growth factor expression. Acta
Pharmacol Sin. 30:98–106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zeisberg M, Hanai J, Sugimoto H, Mammoto
T, Charytan D, Strutz F and Kalluri R: BMP-7 counteracts
TGF-beta1-induced epithelial-to-mesenchymal transition and reverses
chronic renal injury. Nat Med. 9:964–968. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu Y, Wan J, Jiang D and Wu X: BMP-7
counteracts TGF-beta1-induced epithelial-to mesenchymal transition
in human renal proximal tubular epithelial cells. J Nephrol.
22:403–410. 2009.PubMed/NCBI
|
|
34
|
Liu Y: Renal fibrosis: new insights into
the pathogenesis and therapeutics. Kidney Int. 69:213–217. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brown KA, Pietenpol JA and Moses HL: A
tale of two proteins: differential roles and regulation of Smad2
and Smad3 in TGF-beta signaling. J Cell Biochem. 101:9–33. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Perera RJ: A microarray-based method to
profile global microRNA expression in human and mouse. Methods Mol
Biol. 382:137–148. 2007. View Article : Google Scholar : PubMed/NCBI
|