Introduction
Hypoxia is common in cerebral vascular diseases and
leads to brain damage. Endothelial cells are particularly
vulnerable to hypoxia-induced damage (1). Hypoxia has been reported to inhibit
vessel formation (2), resulting
in a series of pathophysiological changes in brain endothelial
cells (3). Endothelin-1 (ET-1) is
a potent vasoconstrictor released by endothelial cells. ET-1
functions as a paracrine regulator of vascular tone, and exerts
deleterious effects on water homeostasis, cerebral edema and brain
blood barrier (BBB) integrity. All these factors further contribute
to severe ischemic brain injury (4). The increase in ET-1 secretion by
endothelial cells is one of the major pathological changes induced
by hypoxia (3). A previous study
on brain endothelial cells revealed that, within 0.5 to 2 h of
hypoxia, the mRNA level of ET-1 was elevated (2). In addition, physiologically low
oxygen tension has been shown to increase endothelin secretion from
cultured human endothelial cells (5). As ET-1 functions by activating the
endothelin A receptor (ETAR) (6),
antagonists of ET-1 receptors may serve as good candidates for the
treatment of cerebral ischemia (7). PD155080 is a selective ETAR
antagonist. It has been reported to be effective in limiting tissue
damage induced by ET-1 in animal models of pathological vasospasm
and therefore, it may be beneficial for clinical use in
ET-implicated diseases (8).
However, the protective effects of PD155080 in in vitro
models, such as cultured endothelial cells, remain largely unknown.
Since the in vitro culture of cerebral microvascular
endothelial cells is important in the research of cerebral vascular
disease, particularly cerebral ischemia/hypoxia-related diseases
(9,10), in the present study, we
investigated the protective effects of PD155080 against
hypoxia-induced rat brain microvascular endothelial cell (BMEC)
injury in vitro.
Materials and methods
Isolation of BMECs
This study was approved by the Ethics Committee of
the 2nd Affiliated Hospital of Chongqing Medical University. The
BMECs were isolated from the cerebral cortex of Wistar rats and
cultured according to a previous report (11). Briefly, brain tissue was collected
from newborn (1–5 days old) Wistar rats, provided by the
Experimental Animal Center of Chongqing Medical University,
Chongqing, China. Following the removal of major vessels, cerebral
pia mater, brain stem and medullary substances, the tissue samples
were homogenized. The homogenate was filtered through a nylon
membrane (75 μm aperture). The filter residue was flushed with cold
Hank’s solution, and was centrifuged at 500 rpm for 3 min to
collect the microvascular fragments. The collections were
dissociated with 0.1% collagenase VII (Sigma-Aldrich Co., LLC, St.
Louis, MO, USA) at 37°C for 20 min, and centrifuged again at 800
rpm for 3 min. The liquid supernatant was discarded and the cell
pellet was resuspended in M199 complete culture medium (HyClone
Laboratories, Inc., Logan, UT, USA), which contained 15% fetal
bovine serum. The cells were cultured in 35-ml plastic flasks,
which were placed upside down immediately after cell implantation,
and incubated at 37°C, 5% CO2 for 2 h. The plastic
flasks were then turned upward. The medium was replaced 1 day after
the cells were plated, and again after 3 days and, thereafter,
until the cells became unified and formed a monostratal layer.
During the first 2–5 days of culture, the cultured cells were
observed under an inverted microscope once a day,. All the cells or
cell clusters that were not typically triangular or shuttle-shaped
with pale nuclei and a distinct cytomembrane were suspected as
contaminated cells and were eliminated using a cell scrapper. The
cells were dissociated with 0.25% parenzyme (Sigma-Aldrich Co.,
LLC) for passage cultures, and the third generation of cultured
cells was used in the experiments in this study.
Establishment of a hypoxic BMEC model in
vitro
The cultured BMECs were placed in a hypoxic,
hermetic container with 95% N2 and 5% CO2
(Chongqing Air Liquefaction Corporation, Ltd., Chongqing, China)
and were cultured at 37°C for 12 h.
Experimental groups
The BMECs were divided into the normal controls,
hypoxia and PD155080 groups. In the normal controls, the BMECs were
conventionally cultured without any stimulatory factors. In the
hypoxia group, the BMECs were cultured inside a container with 95%
N2 and 5% CO2 for 12 h. In the PD155080
group, the BMECs were first treated with PD155080 (1 μM) (Pfizer
Pharmaceutical Co, Ltd., New York, NY, USA) for 2 h, and were then
cultured in the same hypoxic environment as the hypoxia group for
12 h in the presence of PD155080.
Detection of biomarkers
Identification of BMECs and their
ultrastructures
In this study, the morphology of the endothelial
cells was observed using an inverted microscope, a transmission
electronic microscope and by immunocytochemical staining for factor
VIII-related antigen (Beijing Zhongshan Golden Bridge Biotechnology
Co., Ltd., Beijing, China). To assess the ultrastructure of the
cells, the BMECs were conventionally prepared for electron
microscopic observation following fixation with 2% glutaric
dialdehyde solution, and the changes to the BMEC ultrastructure
were observed using a transmission electronic microscope.
Measurement of cell viability by
methyl thiazolyl tetrazolium (MTT) assay
A total of 200 μl BMECs were plated in a 96-well
culture plate at a density of 3×104·ml−1, and
were cultured for the indicated periods of time. At the end of the
experiments, the culture medium was discarded and 200 μl M199 + 20
μl of 5 mg/ml MTT (Sigma-Aldrich Co., LLC) were added to each well.
The cells were then cultured for a further 4 h and centrifuged at
2,000 rmp for 5 min. The supernatant was discarded and 200 μl
dimethyl sulfoxide were added to each well. The absorbance (A) in
each well was measured with an automatic enzyme-labeled instrument
at a wavelength of 570 nm.
Calculation of the mortality rate (%)
of BMECs with trypan blue (TB) staining
The BMEC suspension of each group was stained with
TB (0.4%), and 200 BMECs from each group (stained and unstained),
were counted on blood cell counting plates under a light
microscope. Mortality rate was calculated as follows: mortality =
(number of dead BMECs/200) ×100%.
Measurement of lactate dehydrogenase
(LDH) activity
A total of 100 μl of liquid supernatant from each
group was collected at the indicated time points for each
experiment. The LDH activity was measured using an LDH kit (Nanjing
Jiancheng Bioengineering Institute, Nanjing, China) according to
the manufacturer’s instructions.
Determination of ET-1
concentration
The BMECs were plated in a 24-well plate at a
density of 3×104·ml−1 (n=6 per group). At the
end of each experiment, 100 μl of liquid supernatant from each
group were collected to determine the concentration of ET-1
according to the instructions provided with the ET-1 radioimmunity
kit (Dongya Immuno-technology Institute, Beijing, China).
Determination of ET-1 mRNA expression
by in situ hybridization
An ET-1 in situ hybridization kit was
purchased from Wuhan Boster Bio-Engineering Ltd., Co. (Wuhan,
China). Digoxin-labeled oligonucleotides were used as the probe.
The probe sequences were as follows: 5′-GATTATTTGCCCATG
ATCTTCTCTCTGCTGTTCGT-3′ and 5′-CCACCTGGACAT
CATCTGGGTCAACACTCCCGAGC-3′. The cells were cultured on coverslips
in 24-well plates. At the end of each experiment, the cells were
fixed with 4% paraformaldehyde and washed with PBS. The cells were
then immersed in pre-hybridization buffer at 37°C for 2 h, followed
by incubation in ET-1 oligonucleotide probe hybridization buffer at
43°C for 30 h and 1:400 anti-digoxin antibody at 37°C for 4 h.
Finally, the treated cells were measured using a
5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium
(BCIP/NBT) color-appearing system, using a hybridization buffer
without a probe as the negative control. The results of in
situ hybridization were analyzed using an image analysis system
(Beihang CM2000B; Beihang University, Beijing, China) by measuring
the absorbance (A) of each visual field under a microscope.
Determination of ET-1 mRNA expression
by reverse transcription PCR (RT-PCR)
a) the extraction of total RNA from BMECs
Total RNA from the BMECs in each experimental group
was extracted using TRIzol reagent (Sangon Biotech Co., Ltd.,
Shanghai, China) according to the manufacturer’s instructions. RNA
was dissolved with DEPC (Sigma-Aldrich Co. LLC) deionized water.
The amount and purity of the RNA in each tube were measured using a
UV-VIS spectrophotometer (Shanghai Precision Scientific Instrument
Co., Ltd, Shanghai, China), and the samples with an optical density
(OD) of ≥1.6 were reverse transcribed.
b) cDNA synthesis
cDNA was synthesized using the M-MuLV First-Strand
cDNA Synthesis kit (Sangon Biotech Co., Ltd.). Briefly, 2 μl total
RNA (1 μg/μl) from each group was mixed with 8 μl DEPC deionized
water and 1 μl random hexamer primer (0.2 μg/μl), and then vortexed
for 5 sec and incubated at 70°C for 5 min. Following incubation,
the mixture was immersed in an ice-bath for 30 sec and vortexed
again for 5 sec. Subsequently, 4 μl 5× reverse transcription
buffer, 1 μl RNAase inhibitor (20 U/μl) and 2 μl
deoxy-ribonucleoside triphosphate (10 mM) were mixed with the RNA,
and the mixture was incubated at 70°C for 5 min. Subsequently, 1 μl
M-MuLV reverse transcriptase (200 U/μl) was added followed by
sequential treatment in a water bath at 25°C for 10 min, 37°C for
60 min and 70°C for 10 min; the mixture was then cooled on ice.
c) PCR reaction
PCR reactions were performed using the Gene/PCR
System 2400 (PerkinElmer, Inc., Waltham, MA, USA). The sequences of
ET-1 were as follows: sense primer, 5′-CGTTGCTCCTGCTCCTCCTTGATGG-3′
and antisense primer, 5′-AAGATCCCAGCCAGCATGGAGAGCG-3′. Following
initial denaturation at 94°C for 5 min, an additional denaturation,
annealing and elongation were performed at 94°C for 1 min, 60°C for
1 min and 72°C for 2.5 min for 35 cycles; the final elongation was
performed at 72°C for 7 min.
d) Identification and measurement of the ET-1
mRNA expression level
The mixture of 4 μl amplification product and 2 μl
of sample loading buffer (6X) was loaded on a 1.5% agarose gel,
which was run with a 0.5× TBE electrophoresis buffer (the buffers
and the gel were produced by Sangon Biotech Co., Ltd). The gel was
analyzed using an ultraviolet image analysis system (Gucun
Electrical and Optical Instrument Factory, Shanghai, China), and
the integral OD was measured using β-actin mRNA as an internal
control. The ET-1 mRNA expression level was expressed as the ratio
of the OD of the samples to β-actin.
Statistical analysis
The experimental results were analyzed using SPSS
software, and are expressed as the means ± standard deviation
(means ± SD). Analysis of variance was used for comparisons between
the groups, and the differences between groups were considered
significant at P<0.05. All figures were created using GraphPad
Prism 6.01 sorftware (GraphPad Software, Inc., La Jolla, CA,
USA).
Results
Identification of BMECs
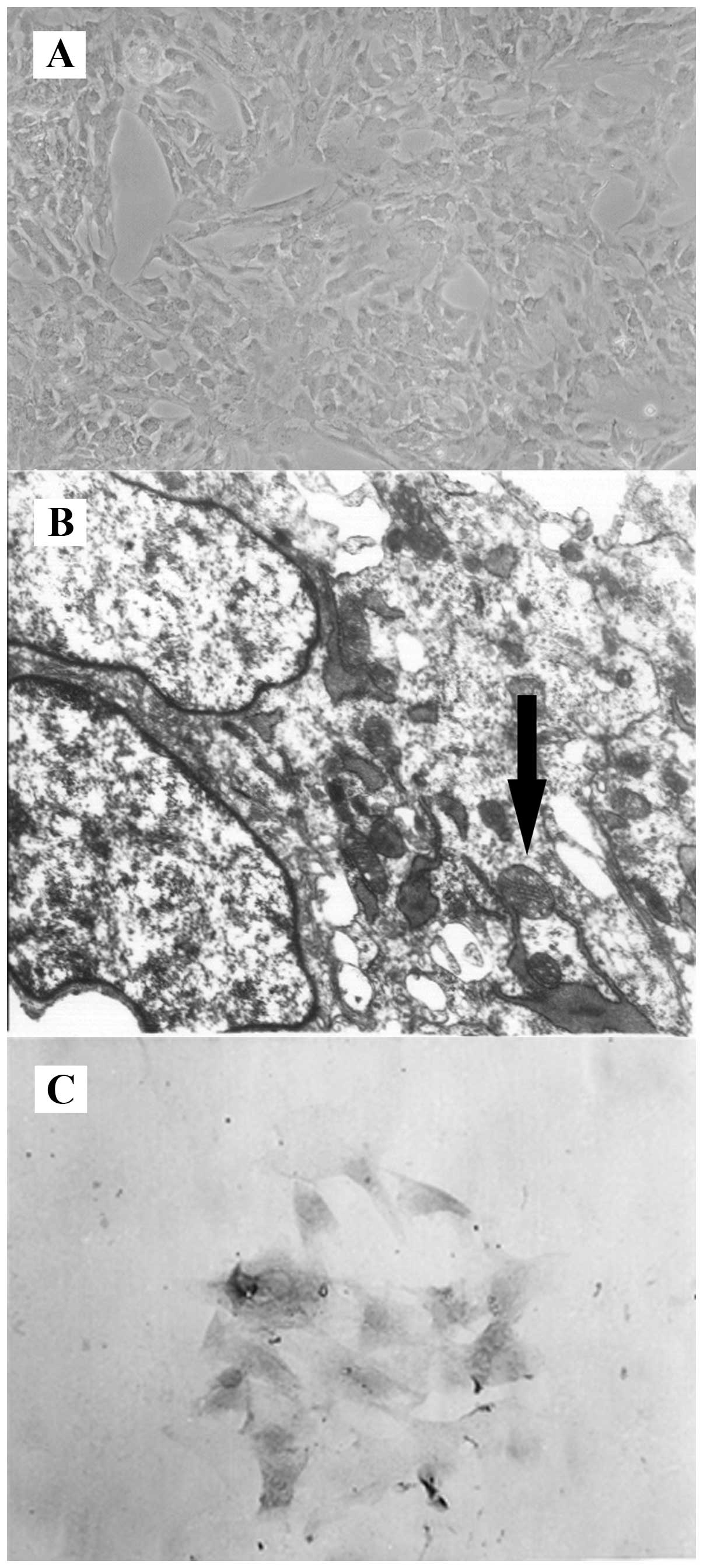
To identify the characteristics of the isolated
BMECs, observations were made under inverted and transmission
electron microscopes. Under the inverted microscope, the
endothelial cells were flaky, aggregated and closely arranged, and
presented a typical pavestone-like structure (Fig. 1A). Typical claviform Weibel-Palada
bodies were detected in the cytoplasm under the transmission
electron microscope (Fig. 1B). We
also performed immunocytochemical staining for factor VIII-related
antigen, a marker for endothelial cells. As shown in Fig. 1C, the cytoplasm and the areas
around the nuclear membrane were stained fuscous in endothelial
cells of both primary and third generations, and >95% of the
third generation cells were positive for factor VIII-related
antigen. These results suggested that the isolated cells were
vascular endothelial cells.
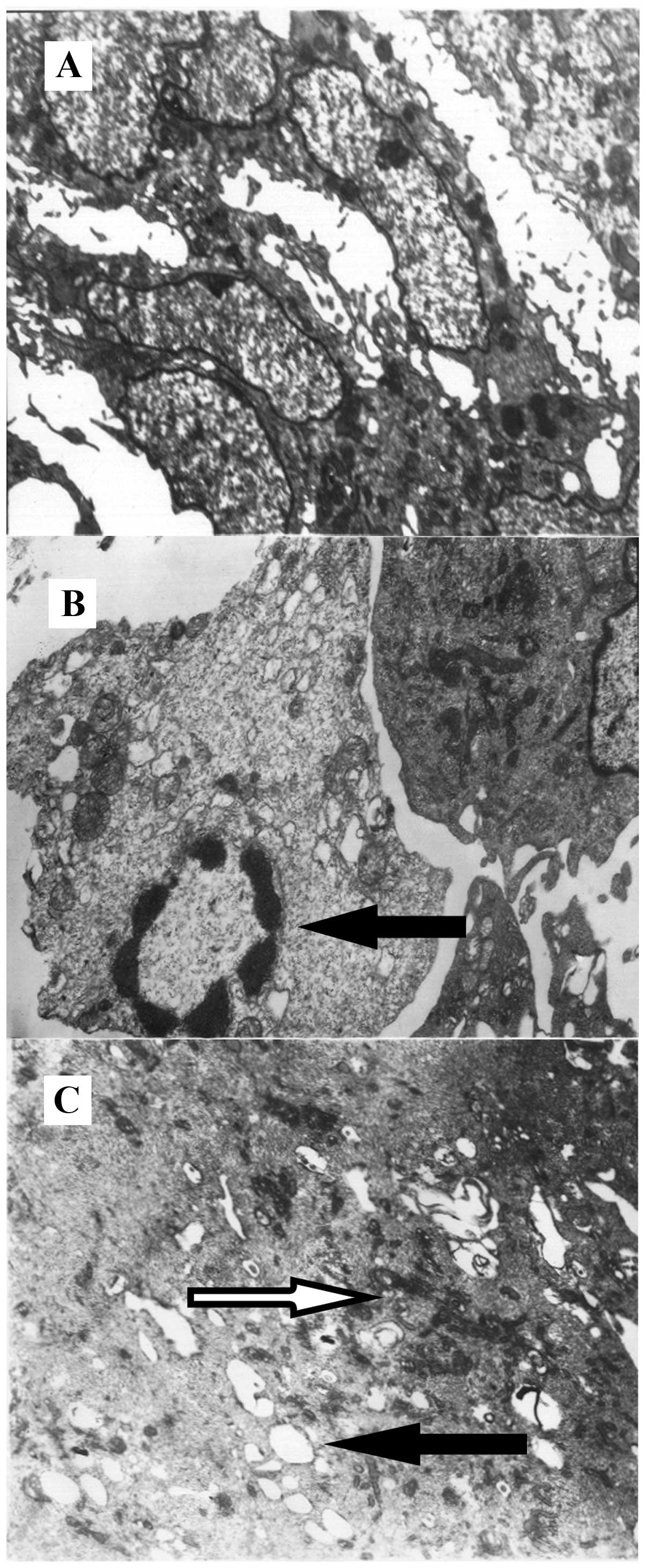
Ultrastructural changes of BMECs cultured
under hypoxic conditions
In order to observe the changes in the
ultrastructure of the BMECs cultured under different experimental
conditions, a transmission electron microscope was employed. The
normal BMECs (conveniontally cultured) had an oblong-oval shape, as
observed under an electron microscope, and were characterized by
dense endonuclear chromatin, typical Golgi apparatus, endoplasmic
reticulum and mitochondria, a high numer of perinuclear plasma, a
complete plasma membrane and a nuclear membrane (Fig. 2A). However, the BMECs cultured
under hypoxic conditions for 12 h were characterized by chromatin
margination, chromatin agglutination, plasma edema, an increased
number of intracellular liposomes and vacuoles, mitochondrial
swelling and the expansion of a rough surfaced endoplasmic
reticulum (Fig. 2B). Of note, the
BMECs cultured under hypoxic conditions with PD155080 showed only
mild mitochondrial swelling and the expansion of a rough surfaced
endoplasmic reticulum (Fig. 2C),
suggesting that PD155080 alleviated hypoxia-induced ultrastructural
BMEC injury.
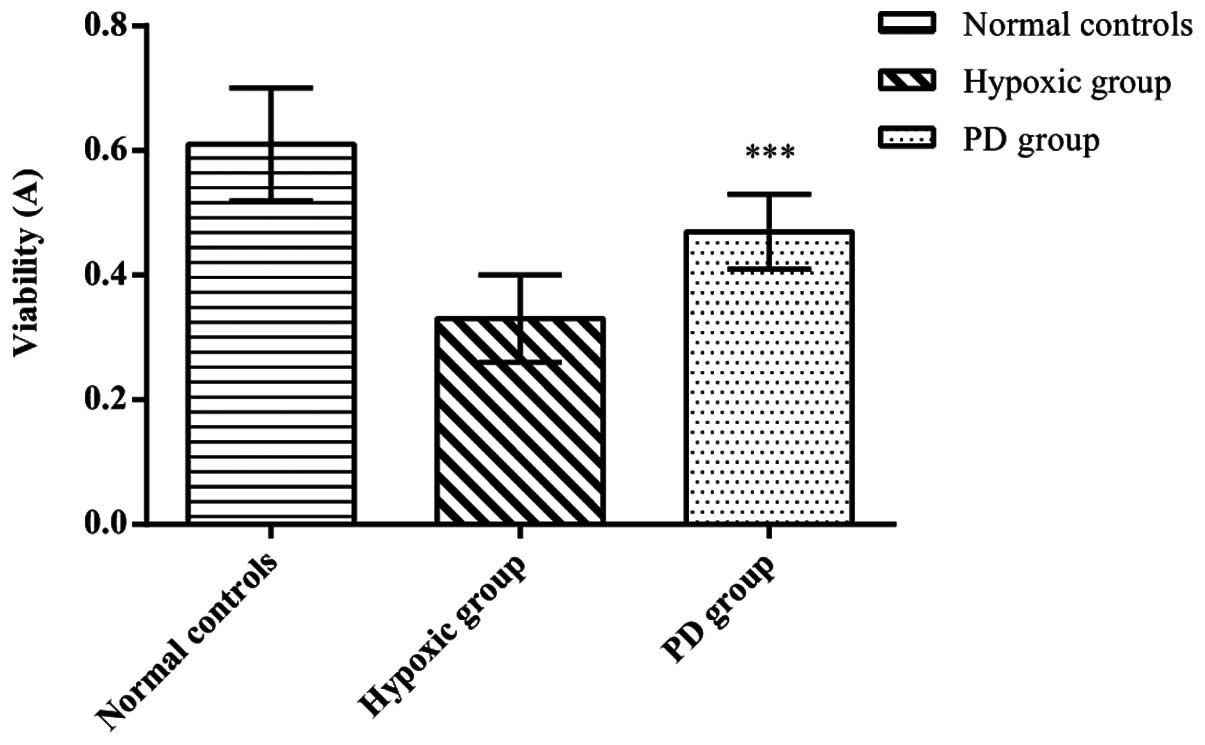
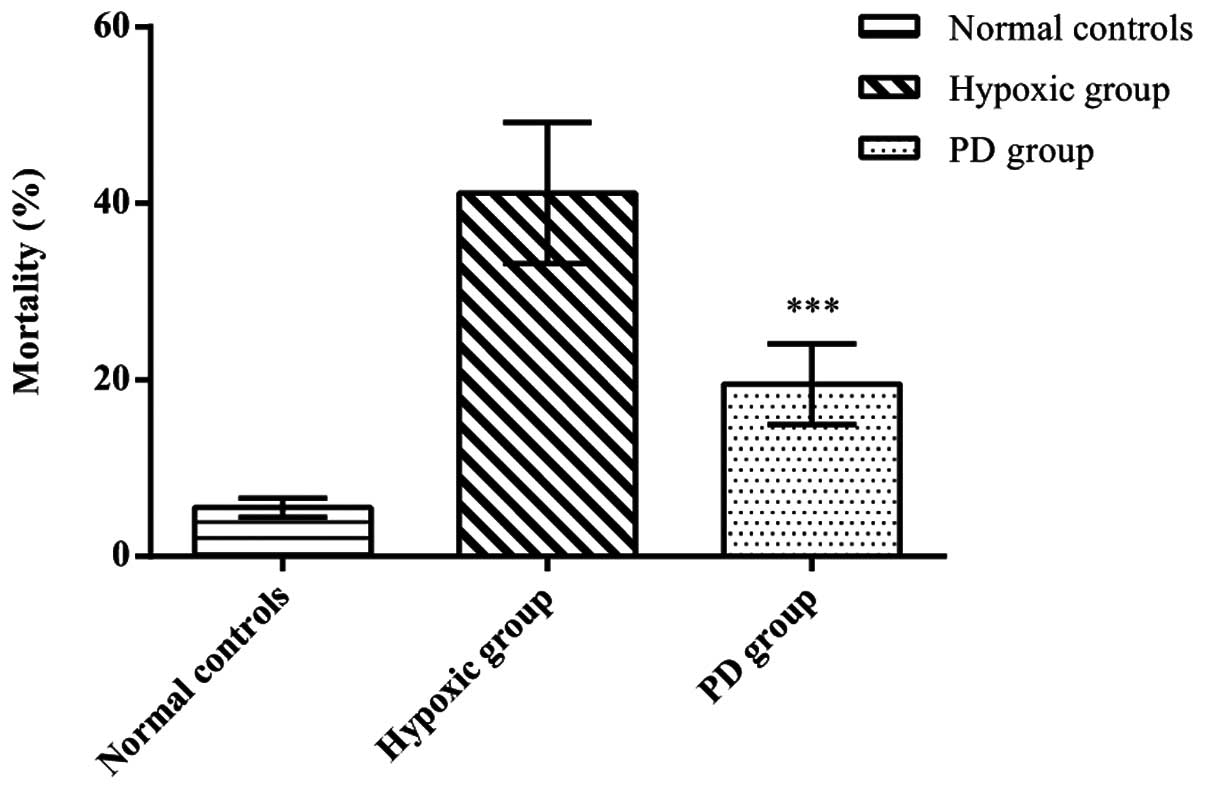
Effects of PD155080 on the viability and
mortality of BMECs
To assess the effects of PD155080 on BMEC viability
and mortality, we calculated the viability and mortality of BMECs
in the 3 experimental groups. Compared to the normal controls
(viability, 0.61±0.09; mortality, 5.5±1.05), the cerebral
microvascular endothelial cells in the hypoxic group (viability,
0.33±0.07; mortality, 41.17±8.01) showed a significantly decreased
viability and an increased mortality (p<0.01). In the presence
of PD155080, the cell viability was significantly increased and the
mortality significantly decreased (viability, 0.47±0.06; mortality,
19.50±4.59) compared to the hypoxic group (p<0.01) (Figs. 3 and 4). These results suggested that PD155080
inhibited hypoxia-induced damage to BMECs, and protected the BMECs
from hypoxia-induced cell death.
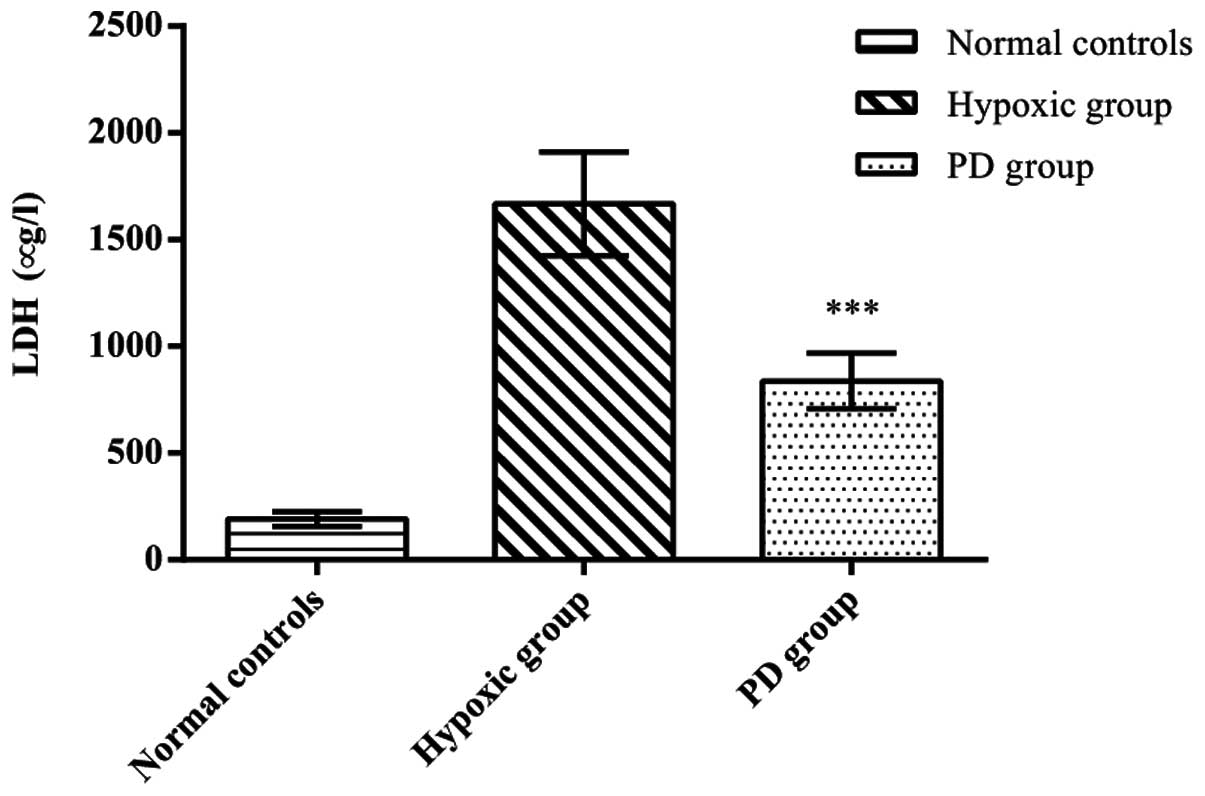
Effects of PD155080 on LDH release
In order to determine to what extent PD155080
protects BMECs from hypoxia, we measured the release of LDH from
the BMECs. A significant increase in the release of LDH was
detected in the BMECs from the hypoxic group (LDH concentration,
1,667.33±244.31) compared with the normal controls (LDH
concentration, 191.71±34.26) (p<0.01), whereas the release of
LDH in the PD155080 group (LDH concentration, 837.50±130.16) showed
a marked reduction compared to the hypoxic group (p<0.05)
(Fig. 5). These results further
demonstrated that treatment with PD155080 reversed the
hypoxia-induced cellular damage.
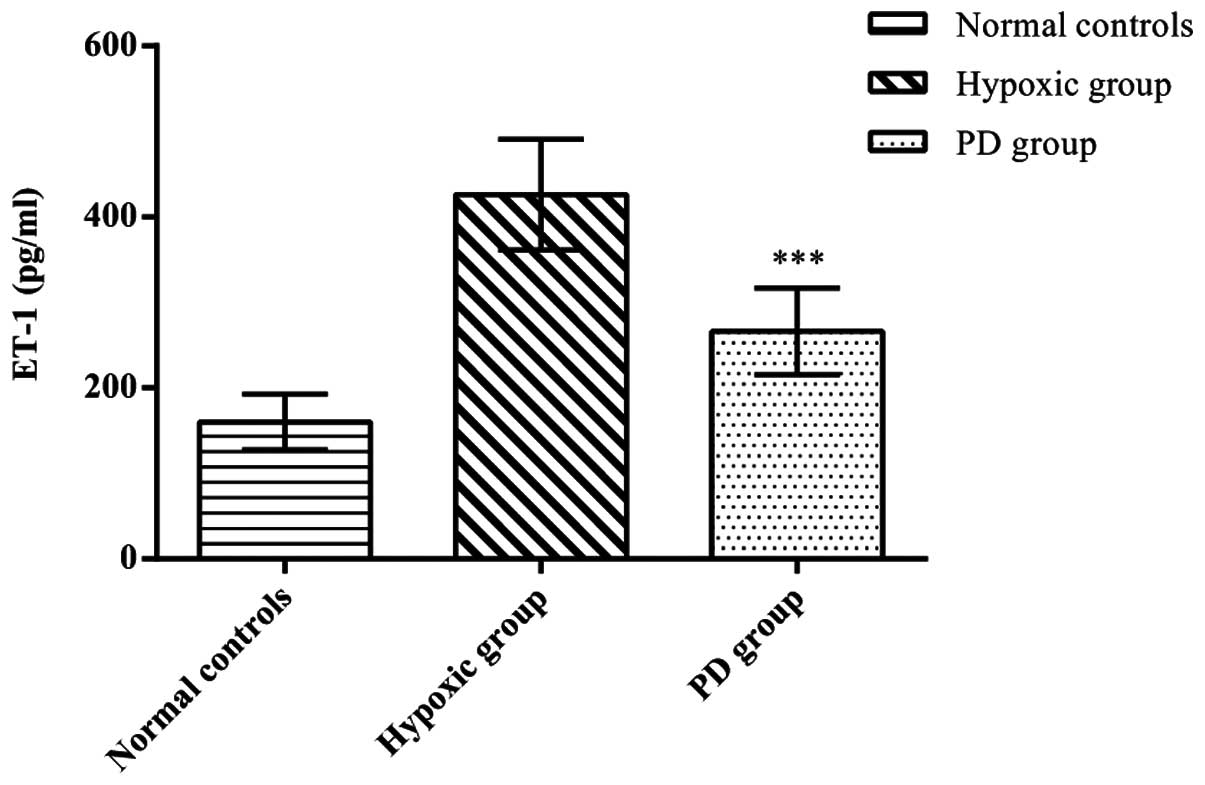
Effects of PD155080 on the secretion of
ET-1 by BMECs
To clarify the effects of PD155080 on ET-1 secretion
by BMECs, we measured the ET-1 levels in the BMECs. We found that
hypoxia increased ET-1 secretion from the BMECs compared with the
control group (p<0.01), whereas the administration of PD155080
significantly reduced hypoxia-induced ET-1 secretion from the BMECs
compared to those in the hypoxic group (p<0.01) (normal
controls, 160.19±32.34; hypoxic group, 426.13±64.80; PD155080
group, 266.40±50.45; p<0.01) (Fig.
6).
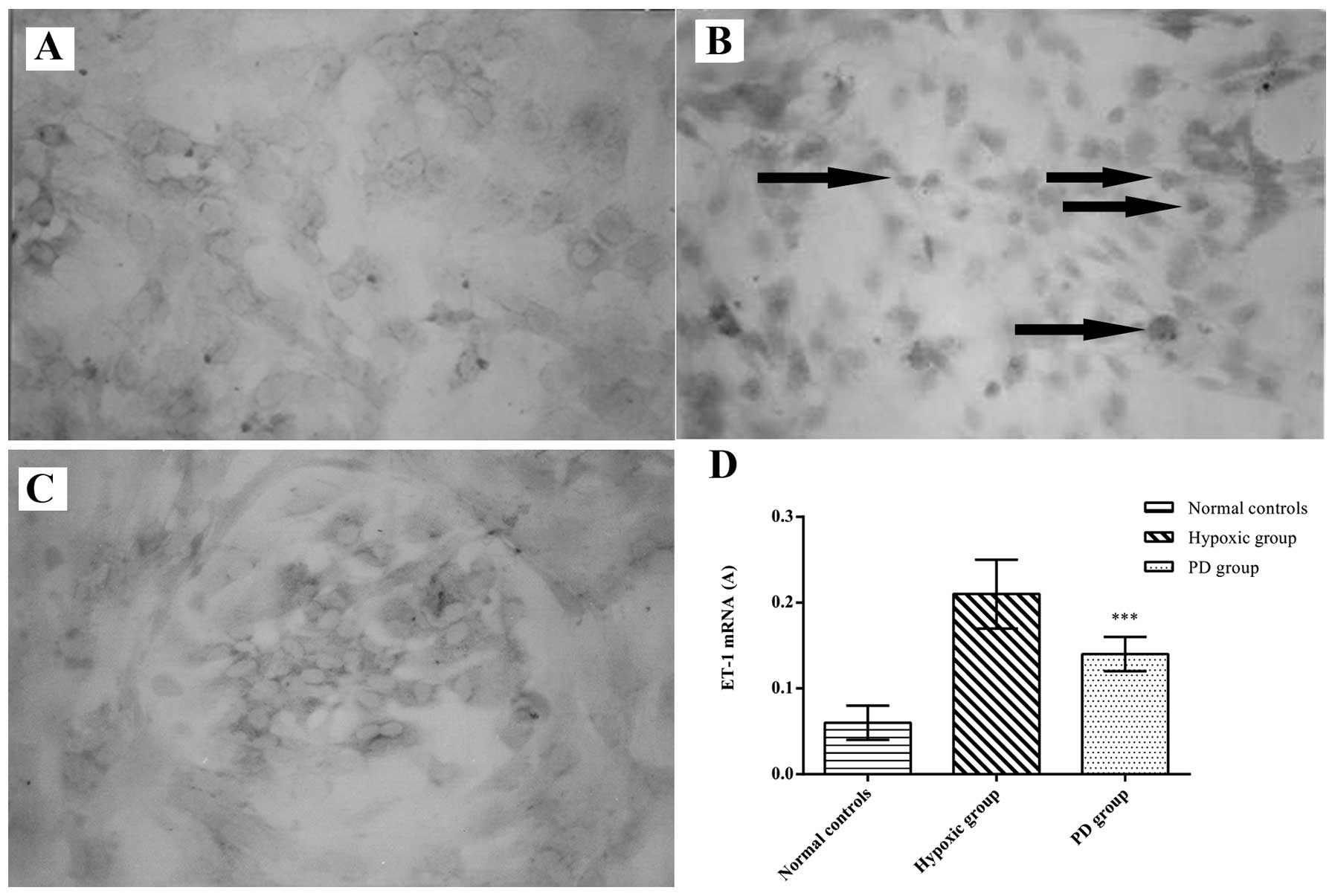
Effects of PD155080 on the ET-1 mRNA
level in BMECs
To further determine the effects of PD155080 on
hypoxia-induced ET-1 gene expression in the BMECs, we measured the
ET-1 mRNA expression in the BMECs by in situ hybridization
and RT-PCR. In the image analysis of in situ hybridization,
the ET-1 mRNA level was significantly higher in the hypoxic group
compared with the normal controls (p<0.01) (Fig. 7). However, treatment with PD155080
reduced the increased ET-1 mRNA expression induced by hypoxia
(p<0.01) (normal controls, 0.06±0.02; hypoxic group, 0.21±0.04;
PD155080 group, 0.14±0.02) (Fig.
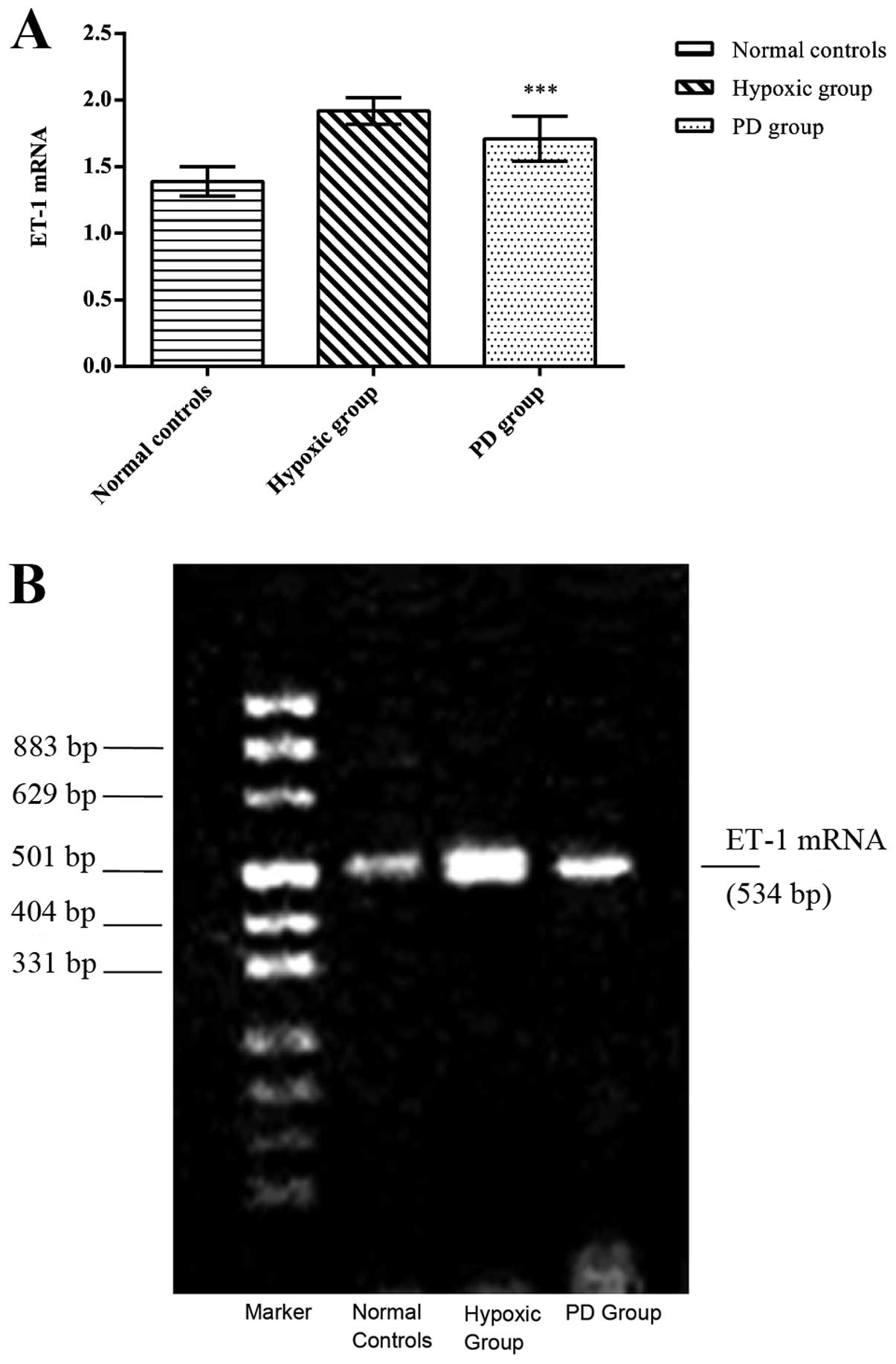
7). Using RT-PCR, we also observed that the ET-1 mRNA level was
significantly higher in the hypoxic group compared to the normal
controls (p<0.01) (Fig. 8).
PD155080 administration also reduced the hypoxia-induced ET-1 mRNA
expression (p<0.05) (normal controls, 1.39±0.11; hypoxic group,
1.92±0.10; PD155080 group, 1.71±0.17) (Fig. 8).
Discussion
Ischemic stroke is one of the major causes of
disability and death; however, the therapeutic strategies for
ischemic stroke are limited. Cerebral vascular endothelial cells,
which regulate the vasomotion and metabolism of cerebral vessels by
releasing a series of vasoactive substances, play an important role
in the structural and functional abnormality of vessels in cerebral
vascular diseases. It has been reported that artery segments with
an intact endothelium show vasodilation during pressure decrease
and vasoconstriction during pressure increase, while artery
segments without an intact endothelium do not respond to pressure
changes (12). In the present
study, we investigated the effects of PD155080 on hypoxia-induced
brain microvascular endothelial cell injury using an in
vitro rat BMEC model. Our results demonstrated that hypoxia
leads to the ultrastructural impairment of BMECs, characterized by
pyknosis, condensation of nuclear chromatin, edema and
vacuolization of the cells. Importantly, following pre-treatment of
the BMECs with PD155080, the ultrastructure of the BMECs only
showed mild mitochondrial swelling and the expansion of a rough
surfaced endoplasmic reticulum following exposure to hypoxia.
Compared with the hypoxic group, the viability of the BMECs in the
PD155080 group was significantly higher and the mortality rate was
reduced. The release of LDH, which is in proportion to the membrane
permeability changes, is a sensitive and convenient marker for cell
injury and death (13). In our
study, we found that the LDH level in the PD155080 group was lower
compared to the hypoxic group. These results suggested that the
inhibition of the ETA receptor using PD155080 increased the
survival rate and reduced the release of LDH from the BMECs;
therefore, PD155080 protects the BMECs from hypoxia-induced injury
and may have a membrane-stabilizing effect on these cells. As
previously demonstrated, PD155080 dilates vessels in ischemic
stroke. An experiment on feline pial arterioles showed that the
perivascular microapplication of PD155080 (30 μM) around pial
vessels within the territory of occluded middle cerebral artery
elicited an increase in the caliber of both dilated and constricted
pial arterioles following middle cerebral artery occlusion (MCAO)
(14). A similar effect of
PD155080 has also been reported in studies on coronary arteries
using isolated perfused rat hearts, in which the addition of
PD155080 resulted in a significant increase in the recovery of
coronary flow after 30 min of reperfusion (15). More importantly, it has also been
reported that ‘increasing concentrations of PD155080 caused a
progressive, parallel rightward shift of ET-1
concentration-response curve without detrimental effect on the
maximal response to ET-1’ (8).
One of the most important vasoactive substances
released by cerebral endothelial cells is ET-1, the most powerful
vasoconstrictor involved in the regulation of cerebral capillary
microcirculation and vascular remodeling. The overproduction of
ET-1 is related to the damage of cerebrovascular endothelial cells,
and plays an important role in the pathogenesis of atherosclerosis
and ischemic cerebral vascular diseases (16–19). Hypoxia results in the deficiency
of energy metabolism, intracellular acidosis and increased membrane
permeability, all of which lead to the release of intracellular
ET-1 (2). Similarly, our study
revealed a higher level of ET-1 secretion in the BMECs in the
hypoxic group, indicating that hypoxia can induce ET-1 secretion by
BMECs. Of note, in the PD155080 group, ET-1 secretion was markedly
reduced compared to the hypoxic group, suggesting that PD155080
inhibited hypoxia-induced ET-1 secretion from the BMECs.
The decrease in intracellular ET-1 and hypoxia
activates the mRNA expression of ET-1, resulting in the increased
synthesis of ET-1 (20). In our
study, ET-1 mRNA levels in the hypoxic group were higher than those
in the normal controls, and the ET-1 mRNA level in the PD155080
group was significantly reduced compared to that in the hypoxic
group, indicating that PD155080 inhibits hypoxia-induced ET-1 gene
overexpression. However, the underlying mechanisms remain unclear.
A previous study demonstrated that LU135252 (an ETAR antagonist)
reduced the tissue concentration of ET-1, but increased the plasma
levels of ET-1 in angiotensin II-treated animals, and suggested
that this may be related to the displacement of ET-1 from tissues
to the circulation or may be related to the direct interference of
LU135252 with the production of ET in vascular tissue through its
ETAR interaction (21). Another
study using LU135252 suggested that ET-1 may act as an autocrine
modulator of its own production in vivo through the
activation of ETAR; thus, the blockade of ETAR prevented the
increase in vascular ET-l (22).
It has also been reported that the binding of ET-1 to endothelin B
receptor (ETBR) inhibits endothelin-converting enzyme-1 expression
in endothelial cells and ETBR mediates the reuptake of ET-1 by
endothelial cells (23).
Therefore, the blockade of ETAR may also increase the binding of
ET-1 to ETBR, which results in a reduced ET-1 level and its
production. In this study, a significant decrease in ET-1 mRNA
expression in the BMECs was detected in the PD155080 group compared
to the hypoxic group. It is possible that PD155080 directly
interferes with the production of ET-1, but it remains unclear
whether this interference is related to the blockade of ETAR or to
the activation of ETBR. Further studies are required to determine
whether the decrease in ET-1 expression in the PD155080 group was
primarily due to the effects of PD155080 directly, or due to the
reduced release of intracellular ET-1.
ET-1 leads to vasoconstriction, by activating ETAR,
which is expressed in endothelial cells (6,12,24), indicating that the effects of ET-1
on BMECs are mainly mediated by ETAR. ETAR antagonists can compete
against ET-1 to bind specifically to ETAR and block the biological
interaction between ET-1 and ETAR. A recent study on the effects of
selective ETAR antagonists in an ischemic animal model reported
that ETAR antagonists appear to offer multiple neuroprotective
mechanisms, including the prevention of the blood-brain barrier
disruption and leukocyte infiltration (25). BQ-485, an ETAR antagonist, has
been shown to prevented the oxygen saturation decrease in ischemic
cerebral tissue, and increase the cerebral oxygen utilization
coefficiency (26). ABT-627 and
A-147627 are both ETAR antagonists that partially normalize the
neurological deficits and infarct volume of MCAO mice, whilst the
ETBR antagonist has no such effect (4,27).
In addition, rats treated with Clazosentan® (an ETAR
antagonist) have shown a reduction of encephalic edema at 72 h and
at day 7, and a decreased serum ET-1 level at 72 h and at day 7
(7). SB 234551 (an ETAR
antagonist) protected MCAO rats by enhancing collateral blood flow
and the salvage of penumbra (28). However, other studies have
reported that ABT-627 had no effect on cerebral edema, infarct
volume and neural function of ischemic rats (29). There are only limited in
vitro studies that have reported that the effects of selective
ETAR antagonists, such as BQ-123, may improve the survival of human
fetal astroglial and neuronal cells upon hypoxic injury (30). To date, there are no studies on
their effects on ischemic cerebral vascular endothelial cells.
PD155080 is a non-peptide specific ETAR antagonist,
its oral bioavailability is 87%, and it has up to 1,000-fold
selectivity for the human ETA receptor than for ETBR (8,31).
PD155080 exerts a variety of effects on ET-1-related physiological
and pathophysiological processes by blocking ETA receptors.
Long-term ETAR antagonism can normalize myocardial cytosolic
Ca2 + modulation, which may contribute to the
anti-hypertrophic and cardioprotective effects of ETAR therapy
(32). In a previous study on
isolated rabbit atrial cardiomyocytes, PD155080 (1 μM) was found to
prevent the ET-1-induced inhibition of IK (ACh) (33). A combination of A-192621 (an ETBR
antagonist) and PD155080 abolished endotoxin-induced pulmonary
hypertension, enhanced cardiac performance and improved systemic
oxygen delivery and acid-base balance (34). In experimental chronic renal
failure rats, PD155080 reduced their high blood pressure without
affecting renal function (35).
Our study demonstrated that PD155080 significantly inhibited the
hypoxia-induced release of ET-1 and ET-1 mRNA expression and
protected BMECs from hypoxic injury. Therefore, PD155080 and other
ETAR antagonists may provide therapeutic strategies for many
ET-1-related diseases. Further studies on the anti-hypoxic
mechanisms of PD155080 are required in order to develop novel drugs
for the treatment and prevention of ischemic vascular endothelial
injury.
Acknowledgements
PD155080 in this study was kindly provided by Mr.
Donnie W. Ovens from Pfizer Pharmaceuticals Ltd. (New York, NY,
USA). We sincerely thank Mr. Weixue Tang from the Department of
Pathophysiology of Chongqing Medical University for his assistance
with our study.
References
|
1
|
Shostak HDC, Lemasters JJ, Edgell CJ, et
al: Role of ICE-like proteases in endothelial cell hypoxic and
reperfusion injury. Biochem Biophys Res Commun. 231:844–847. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Luo J, Martinez J, Yin X, et al: Hypoxia
induces angiogenic factors in brain microvascular endothelial
cells. Microvasc Res. 83:138–145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Faller DV: Endothelial cell responses to
hypoxic stress. Clin Exp Pharmacol Physiol. 26:74–84. 1999.
View Article : Google Scholar
|
|
4
|
Lo AC, Chen AY, Hung VK, et al:
Endothelin-1 overexpression leads to further water accumulation and
brain edema after middle cerebral artery occlusion via aquaporin 4
expression in astrocytic end-feet. J Cereb Blood Flow Metab.
25:998–1011. 2005. View Article : Google Scholar
|
|
5
|
Kourembanas S, Marsden PA, Mcquillan LP,
et al: Hypoxia induced endothelin gene expression and secretion in
cultured human endothelium. J Clin Invest. 88:1054–1060. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fernandez N, Monge L, Garcia-Villalon AL,
et al: Endothelin-1-induced in vitro cerebral venoconstriction is
mediated by endothelin ETA receptors. Eur-J-Pharmacol. 294:483–490.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moldes O, Sobrino T, Blanco M, et al:
Neuroprotection afforded by antagonists of endothelin-1 receptors
in experimental stroke. Neuropharmacology. 63:1279–1285. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maguire JJ, Kuc RE, Doherty AM, et al:
Potency of 155080, an orally active ETA receptor antagonist,
determined for human endothelin receptors. J Cardiovasc Pharmacol.
26(suppl 3): S362–S364. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nagy Z, Vastag M, Kolev K, et al: Human
cerebral microvessel endothelial cell culture as a model system to
study the blood-brain interface in ischemic/hypoxic conditions.
Cell Mol Neurobiol. 25:201–210. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nagy Z, Vastag M, Skopal J, et al: Human
brain microvessel endothelial cell culture as a model system to
study vascular factors of ischemic brain. Keio J Med. 45:200–206.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wenbin Wu, Changlin HU and Weixue TANG:
Microvascular endothelial cell culture of Wistar rat cerebral
cortex. Journal of Chongqing Medical University. 27:151–152.
2002.
|
|
12
|
Martinez-Orgado J, Gonzalez R, Alonso MJ,
et al: Endothelial factors and autoregulation during pressure
changes in isolated newborn piglet cerebral arteries. Pediatr-Res.
44:161–167. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Korzeniewski C and Callewaert DM: An
enzyme-release assay for natural cytotoxicity. J Immunol Methods.
64:313–320. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Patel TR, Galbraith S, McAuley MA and
McCulloch J: Endothelin-mediated vascular tone following focal
cerebral ischaemia in the cat. J Cereb Blood Flow Metab.
16:679–687. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goodwin AT, Smolenski RT, Gray CC,
Jayakumar J, Amrani M and Yacoub MH: Role of endogenous endothelin
on coronary reflow after cardioplegic arrest. J Thorac Cardiovasc
Surg. 122:1167–1173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen Y, McCarron RM, Golech S, et al:
ET-1- and NO-mediated signal transduction pathway in human brain
capillary endothelial cells. Am J Physiol Cell Physiol.
284:C243–C249. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang CZ, Winardi D, Lin CL, et al:
Attenuation of hemolysate-induced cerebrovascular endothelial cell
injury and of production of endothelin-1 and big endothelin-1 by an
endothelin-converting enzyme inhibitor. Surg Neurol. 58:181–187.
2002. View Article : Google Scholar
|
|
18
|
Schaller BJ: The role of endothelin in
stroke: experimental data and underlying pathophysiology. Arch Med
Sci. 2:1462006.
|
|
19
|
Ergul A: Endothelin-1 and diabetic
complications: focus on the vasculature. Pharmacol Res. 63:477–482.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamashita K, Discher DJ, Hu J, Bishopric
NH and Webster KA: Molecular regulation of the endothelin-1 gene by
hypoxia. Contributions of hypoxia-inducible factor-1, activator
protein-1, GATA-2, AND p300/CBP. J Biol Chem. 276:12645–12653.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moreau P, d’Uscio LV, Shaw S, Takase H,
Barton M and Lüscher TF: Angiotensin II increases tissue endothelin
and induces vascular hypertrophy: reversal by ET(A)-receptor
antagonist. Circulation. 96:1593–1597. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barton M, d’Uscio LV, Shaw S, Meyer P,
Moreau P and Lüscher TF: ET(A) receptor blockade prevents increased
tissue endothelin-1, vascular hypertrophy, and endothelial
dysfunction in salt-sensitive hypertension. Hypertension.
31:499–504. 1998. View Article : Google Scholar
|
|
23
|
Lüscher Thomas F and Barton Matthias:
Endothelins and endothelin receptor antagonists: therapeutic
considerations for a novel class of cardiovascular drugs.
Circulation. 102:2434–2440. 2000.PubMed/NCBI
|
|
24
|
Terese PR and Nilsson GE: Endothelin
induced cerebral vasoconstriction in rainbow trout, detected in a
novel in vitro preparation. Neurosci Lett. 325:195–198. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaundal RK, Deshpande TA, Gulati A, et al:
Targeting endothelin receptors for pharmacotherapy of ischemic
stroke: current scenario and future perspectives. Drug Discov
Today. 17:793–804. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takasu A, Matsushima S, Takino M, et al:
Effect of an endothelin-1 antagonist, BQ-485, on cerebral oxygen
metabolism after complete global cerebral ischemia in dogs.
Resuscitation. 34:65–69. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leung JW, Chung SS and Chung SK:
Endothelial endothelin-1 over-expression using receptor tyrosine
kinase tie-1 promoter leads to more severe vascular permeability
and blood brain barrier breakdown after transient middle cerebral
artery occlusion. Brain Res. 1266:121–129. 2009. View Article : Google Scholar
|
|
28
|
Legos JJ, Lenhard SC, Haimbach RE, et al:
selective ET(A) receptor antagonism: perfusion/diffusion MRI used
to define treatable stroke model, time to treatment and mechanism
of protection. Exp Neurol. 212:53–62. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Khatibi NH, Lee LK, Zhou Y, et al:
Endothelin receptor-A (ETa) inhibition fails to improve neonatal
hypoxic-ischemic brain injury in rats. Acta Neurochir Suppl.
111:207–212. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Danielyan L, Mueller L, Proksch B, et al:
Similar protective effects of BQ-123 and erythropoietin on survival
of neural cells and generation of neurons upon hypoxic injury. Eur
J Cell Biol. 84:907–913. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Doherty AM, Patt WC, Repine J, et al:
Structure-activity relationships of a novel series of orally active
nonpeptide ETA and ETA/B endothelin receptor-selective antagonists.
J Cardiovasc Pharmacol. 26(suppl 3): S358–S361. 1995. View Article : Google Scholar
|
|
32
|
Friedrich B, Gerald W and Stephen H:
Defective intracellular calcium handling in monocrotaline-induced
right ventricular hypertrophy: protective effect of long-term
endothelin-A receptor blockade with
2-benzo[1,3]dioxol-5-yl-3-benzyl-4-(4-methoxy-phenyl-)-4-oxobut-2-enoate-sodium
(PD 155080). J Pharmacol Exp Ther. 300:442–449. 2002.PubMed/NCBI
|
|
33
|
Spiers JP, Kelso EJ, McDermott BJ,
Scholfield CN and Silke B: Endothelin-1 mediated inhibition of the
acetylcholine-activated potassium current from rabbit isolated
atrial cardiomyocytes. Br J Pharmacol. 119:1427–1437. 1996.
View Article : Google Scholar
|
|
34
|
Wanecek M, Oldner A, Sundin P, Alving K,
Weitzberg E and Rudehill A: Effects on haemodynamics by selective
endothelin ET(B) receptor and combined endothelin ET(A)/ET(B)
receptor antagonism during endotoxin shock. Eur J Pharmacol.
386:235–245. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Potter GS, Johnson RJ and Fink GD: Role of
endothelin in hypertension of experimental chronic renal failure.
Hypertension. 30:1578–1584. 1997. View Article : Google Scholar : PubMed/NCBI
|