Introduction
Non-Hodgkin lymphoma (NHL) is a common hematological
cancer with multiple subtypes, derived from various differentiation
stages of the B cell lineage. Burkitt lymphoma (BL) is the most
common NHL subtype (69%), followed by lymphoblastic lymphoma,
diffuse large B cell lymphoma (DLBCL) and anaplastic large-cell
lymphoma, accounting for 18.3, 10.6 and 2.1% of the cases,
respectively (1). Although
high-dose multiagent chemotherapy and targeted agents induce high
remission rates in patients with previously untreated NHL, relapse
and drug resistance within a few years is common. Therefore,
discovering new therapeutic agents for NHL is required (2).
Cancer cells develop acquiring a set of functional
capabilities for malignant growth, such as self-sufficiency in
growth signals, insensitivity to growth-inhibitory signals and
evasion from apoptosis (3,4).
These essential alterations in cell physiology are achieved by the
constitutive activation of oncogenes and the loss of function of
the tumor-suppressor genes (5).
Genetic and epigenetic mechanisms can all contribute to the
inactivation of tumor-suppressor genes (6). Methylation of cytosine residues by
DNA methyltransferases (DNMTs) at CpG dinucleotides in the promoter
region of genes is a major epigenetic modification in mammalian
genomes that can have profound effects on gene expression (7,8).
One study has shown that DNMTs, including DNMT1, DNMT3A and DNMT3B
are overexpressed in 48, 13 and 45% of de novo DLBCLs,
respectively, which correlates with advanced clinical stage
(9). In addition, the therapeutic
efficacy of the demethylating agents, such as decitabine and
5-azacytidine (5-Aza), can induce significant clinical responses
and even prolong the survival of patients with higher-risk
myelodysplastic syndrome (10).
PTPL1 maps to the human chromosomal locus
4q21, and encodes a cytoplasmic tyrosine phosphatase with a
molecular mass of 270 kDa with roles in numerous physiological and
pathological processes. Among the potential roles in
carcinogenesis, the PTPL1 gene product can impact cancer
development through its capacity to counteract the activity of
oncogenic tyrosine kinases or its inhibitory interaction with the
death receptor Fas (11). Several
studies have shown that hypermethylation of the PTPL1 gene
promoter is involved in various types of cancers, such as non-small
cell lung cancer (11),
esophageal cancer, gastric and hepatocellular tumors (2,12).
The aim of the present study was to analyze
PTPL1 methylation patterns in a broad spectrum of
NHL-derived cell lines and de novo DLBCL samples. Epigenetic
regulation of PTPL1 was confirmed in experiments with a DNA
demethylating agent. The results obtained from the study
significantly contribute towards an improved understanding of the
role of PTPL1 as a tumor-suppressor gene in NHL, and 5-Aza
may offer a potential new therapeutic approach to improve the poor
outcomes associated with NHL.
Materials and methods
Human cell lines
Cell culture
The study included 7 cell lines, Hut78 (cutaneous T
cell lymphoma cell line), Maver, Z138 (mantle cell lymphoma cell
lines), CA46, Raji (Burkitt lymphoma cell lines), Jurkat (acute T
cell lymphoma cell line) and DB (DLBCL cell line). All the cell
lines, except CA46, were maintained in RPMI-1640 supplemented with
10% fetal bovine serum (FBS) (HyClone, Logan, UT, USA) and 1%
antibiotics (Gibco-Invitrogen, Carlsbad, CA, USA). CA46 was
maintained with RPMI-1640 supplemented with 20% FBS (HyClone) and
1% antibiotics (Gibco-Invitrogen). Cells were incubated at 37°C in
a humid atmosphere at 5% CO2 and split every 2–3 days
depending on cell density.
In vitro cytotoxicity assays
Raji and Jurkat cells in the logarithmic growth
phase were inoculated in a 96-well plate, with 100 µl/well
and a cell suspension density of 2.5×105/ml. The cells
were randomly divided into the control and test groups medially
with 4 duplicates/group. They were subsequently treated with 5-Aza
at 0.1, 0.5, 1, 2, 5, 10, 20 and 50 µmol/l for 24, 48 and 72
h, respectively. CCK-8 (10 µl; Dojindo, Kumamoto, Japan)
accompanied every sampling in each well. After 2 h of incubation,
the absorption value (A) of each well was detected at the
wavelength of 450 nm in a Quant spectrophotometer. Drug-free wells
were used as a control and the no-cell wells with the same amounts
of 5-Aza were used as blank controls. Cell inhibition rate (I%) was
calculated using the following equation: I% = [A(control) −
A(treated)/A(control) − A(blank)] × 100%
Treatment with 5-Aza
5-Aza dissolved in normal saline was used to verify
the effect on PTPL1 expression. Three cells lines (CA46,
Raji and Jurkat) were seeded at a density of 2.5×105
cells/ml and 5-Aza was added at a final concentration of 20
µmol/l for CA46, 15 µmol/l for Raji and 3.5
µmol/l for Jurkat. Cells were randomly assigned into 3
groups: Negative control group (added in normal saline), 5-Aza-24 h
group (treated with 5-Aza for 24 h) and 5-Aza-48 h group (treated
with 5-Aza-Cdr for 48 h). Cells were harvested, respectively, to
prepare DNA and RNA.
DNA extraction and bisulfite
conversion
Genomic DNA was extracted by the E.Z.N.A®
Tissue DNA kit (Omega Bio-Tek, Lilburn, GA, USA). DNA (200 ng) in a
volume of 1–5 µl and was subjected to treatment with sodium
bisulfite using a CpGenome DNA modification kit (Epigentek,
Farmingdale, NY, USA), according to the manufacturer's
instructions. Modified DNA was stored at −80°C until use.
Methylation-specific polymerase chain
reaction (MSP)
Modified DNA was subjected to two separate PCRs. MSP
primers were designed to amplify the methylated or unmethylated
alleles, and the Methylamp Universal Methylated DNA kit (Epigentek)
was used as a positive control. Promoter meth-ylation status was
analyzed by MSP using methylated and unmethylated gene-specific
primers for PTPL1 (12).
Primers for PTPL1 were 5′-CGAGTAGTTTTA GCGGTTAC-3′ (sense)
and 5′-AAAACCTTCTAACGCGAA CGA-3′ (antisense) for the methylated
reaction and 5′-TGTGAGTAGTTTTAGTGGTTAT-3′ (sense) and 5′-CAAAACCTT
CTAACACAAACAA-3′ (antisense) for the unmethylated reaction. These
primer sets were designed to amplify 160 and 163 bp, respectively.
The methylation promoter was: 95°C for 5 min; 40 cycles of 95°C for
50 sec, 58°C for 1 min and 72°C for 1 min; and a final extension at
72°C for 10 min; the unmethylation promoter was: 95°C for 5 min; 40
cycles of 95°C for 50 sec, 60°C for 1 min and 72°C for 1 min; and a
final extension at 72°C for 10 min. Amplified products were
visualized under an ultraviolet gel imaging system using the
GeneSnap System (Multi Genius; Syngene, Cambridge, UK) following
electrophoresis in 2% agarose gels containing the GelRed Nucleic
Acid Gel Stain. For each case, MSP results were scored when a
clearly visible band on the electrophoresis gel with the methylated
and/or the unmethylatated primers were observed. Results from
triplicate experiments were used to determine methylation
status.
RNA isolation and reverse
transcription-polymerase chain reaction (PCR)
RNA was isolated using TRIzol (Gibco-Invitrogen),
according to the manufacturer's instructions. Total cellular RNA (1
µg) was reverse transcribed using the GoScript™ Reverse
Transcription system (Promega, Madison, WI, USA). Primers were:
PTPL1 forward, 5′-GCG CTCCAGTAGCAGGAC-3′ and reverse,
5′-TCATCTGTA AATGACACACTAC-3′; and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH; as a control) forward, 5′-GGAGCG
AGATCCCTCCAAAAT-3′ and reverse, 5′-GGCTGTTGT CATACTTCTCATGG-3′.
Amplified products were visualized under an ultraviolet gel imaging
system using the GeneSnap System (Multi Genius; Syngene) following
electrophoresis in 2% agarose gels containing a GelRed Nucleic Acid
Gel Stain.
Western blot analysis
Protein was extracted from Hut78, Maver, Z138, CA46,
Raji, Jurkat and DB cell lines. Protein concentrations of cells
were determined using a bicinchoninic acid protein assay kit
(Applygen Technologies Inc., Beijing, China). Western blot analyses
were performed using the following primary antibodies: Anti-PTPL1
(1:200; sc-15356) and anti-β-actin (1:1,000; sc-130656) (both from
Santa Cruz Biotechnology, Inc., Dallas, TX, USA). Lysates (60
µg) were resolved on sodium dodecyl sulfate-polyacrylamide
gel electrophoresis gels (PTPL1 8% and β-actin 10%) and transferred
to NC membranes. Membranes were blocked with 5% bovine serum
albumin in Tris-buffered saline and Tween 20 and primary antibodies
(Abs) were added overnight. Fluorescently labeled secondary
antibodies (1:10,000) were used and the membranes were scanned
using the Odyssey Infrared Imaging system (both from LI-COR
Biosciences, Lincoln, NE, USA).
Patients
Patient selection
The formalin-fixed paraffin-embedded (FFPE) tissues
of 47 DLBCL patients and 16 reactive lymph nodes (as control) were
evaluated. The archived FFPE tissues were obtained from the
Department of Pathology, Peking University Third Hospital (Beijing,
China). The patients were diagnosed according to the criteria of
the 2008 World Health Organization classification and were
clinically staged according to the Ann Arbor classification.
Clinical outcomes were evaluated according to the standard
international criteria.
DNA extraction, bisulfite conversion
and MSP
Genomic DNA of 47 patient samples and 16 reactive
lymph nodes were extracted using the E.Z.N.A® FFPE DNA
kit (Omega Bio-Tek). DNA (200 ng) in a volume of 1–5 µl was
subjected to treatment with sodium bisulfite using a CpGenome DNA
modification kit (Epigentek). The reaction system and reaction
conditions of MSP were the same as the experimental cell lines.
Statistical analysis
Statistical analyses were carried out with Social
Sciences software (SPSS, version 20.0; IBM Corp., Armonk, NY, USA).
Pairwise correlations between the methylation status of DLBCL and
control patients, and the germinal center phenotype (GCB) and
non-GCB patients were investigated by χ 2 test or
Fisher's exact test where appropriate. Statistical significance was
set at the two-sided 5% comparison wise. P<0.05 was considered
to indicate a statistically significant difference.
Results
Human cell lines
Analysis of PTPL1 gene methylation in
lymphoma cell lines
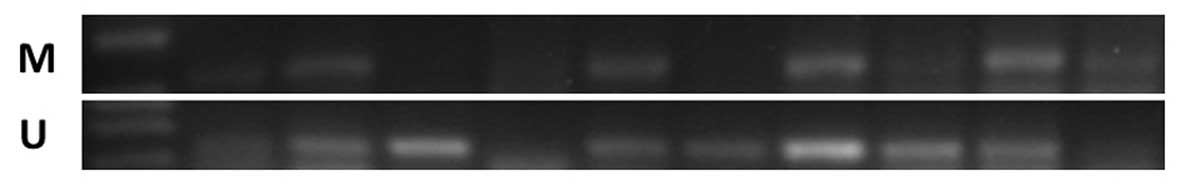
The PTPL1 methylation pattern was analyzed by
MSP. Following bisulfite conversion of DNA, the methylation status
of PTPL1 was determined with MSP in the lymphoma cell lines.
According to MSP, PTPL1 was methylated in the CA46, Raji,
Jurkat and DB cell lines and unmethylated in the Hut78, Maver and
Z138 cell lines (Fig. 1).
PTPL1 mRNA expression in lymphoma cell
lines
To evaluate the correlation between methylation of
the PTPL1 and PTPL1 transcription, reverse
transcription PCR was performed with cDNA from the lymphoma cell
lines. The expression of PTPL1 mRNA was ubiquitously
expressed at different levels in Hut78, Maver and Z138 cells, but
silenced in CA46, Raji, Jurkat and DB cells (Fig. 2).
PTPL1 protein expression in lymphoma
cell lines
Further examination was performed on the PTPL1
protein. The expression of the PTPL1 protein was ubiquitously
expressed at different levels in Hut78, Maver and Z138 cells, but
silenced in CA46, Raji, Jurkat and DB cells (Fig. 3). In the majority of the lymphoma
cell lines, PTPL1 gene expression was inversely correlated
with PTPL1 hypermethylation. This suggests that PTPL1
is regulated by DNA methylation in lymphoma cells.
 | Figure 3Western blott analysis of the PTPL1
protein in Hut78, Maver, Z138, CA46, Raji, Jurkat and DB cell
lines, with β-actin as a control. The PTPL1 protein was
ubiquitously expressed at different levels in Hut78, Maver and Z138
cells, but silenced in CA46, Raji, Jurkat and DB cells. |
5-Aza induces growth inhibition of
Raji and Jurkat cells lines
Cell proliferation was detected using the CCK8 kit
after 12, 24, 48 and 72 h treatment (Fig. 4). 5-Aza inhibited the
proliferation of Raji and Jurkat cells in a concentration-dependent
manner. Patterns of the inhibition efficiency differ in different
cell lines.
Restoration of PTPL1 gene expression
by 5-Aza, a DNMTs inhibitor
PTPL1 re-expression was investigated
following treatment of CA46, Raji and Jurkat cells lines with the
DNMTs inhibitor 5-Aza. 5-Aza treatment increased PTPL1 mRNA
expression compared to the untreated control in the cell lines. In
CA46, Raji and Jurkat cells, treatment with 5-Aza lead to
re-expression of PTPL1 at 48 h (Fig. 5). The final half inhibitory
concentrations were 20 µM for CA46, 15 µM for Raji
and 3.5 µM for Jurkat, respectively.
Patients
Patient characteristics
Forty-seven samples were screened and 23 samples
were followed up. Among the 23 follow-up patients, there were 11
males and 12 females, with a median age of 63 years (range, 26–81
years). Of the 23 patients, 5 (21.7%) were stage I, 6 (26.1%) were
stage II, 2 (8.7%) were stage III, and 10 (43.5%) were stage IV.
Using the Hans classification model, 9 cases were GCB and 14 were
non-GCB, with a GCB:non-GCB ratio of 1:1.5 (Table I).
 | Table IClinical characteristics of 23
patients with DLBCL. |
Table I
Clinical characteristics of 23
patients with DLBCL.
| Clinical
characteristic | Patients, n
(%) |
|---|
| Gender | |
| Male | 11 (47.8) |
| Female | 12 (52.2) |
| Age, years | |
| <65 | 15 (65.2) |
| ≥65 | 8 (34.8) |
| Stage | |
| I | 5 (21.7) |
| II | 6 (26.1) |
| III | 2 (8.7) |
| IV | 10 (43.5) |
| Type | |
| GCB | 9 (39.1) |
| Non-GCB | 14 (61.9) |
Promoter methylation status of DLBCL
and reactive lymph node patients
Among the 47 DLBCL cases, the promoter of gene
PTPL1 was methylated in 59.6% (28/47) (Fig. 6), and unmethylated in 40.4%
(19/47) (Table II).
| Table IIPTPL1 methylation pattern in
DLBCL patients. |
Table II
PTPL1 methylation pattern in
DLBCL patients.
| Patients | Methylated, n
(%) | Unmethylated, n
(%) |
|---|
| DLBCL, n=47 | 28 (59.6) | 19 (40.4) |
| Reactive
lymphnodes, n=16 | 1 (6.3) | 15 (93.7) |
In 9 GCB patients, the promoter of the PTPL1
gene was methylated in 22.2% (2/9) and unmethylated in 77.8% (7/9).
In the 14 non-GCB patients, the promoter of PTPL1 was
methylated in 64.3% (9/14) and unmethylated in 35.7% (5/14)
(Table III). In the 16 reaction
lymph node cases, the frequency of methylation was 6.3% (1/16), and
the frequency of unmethylation was 93.8% (15/16).
| Table IIIPTPL1 methylation pattern in
GCB and non-GCB patients. |
Table III
PTPL1 methylation pattern in
GCB and non-GCB patients.
| Patients | Methylated, n
(%) | Unmethylated, n
(%) |
|---|
| GCB, n=9 | 2 (22.2) | 7 (77.8) |
| Non-GCB, n=14 | 9 (64.3) | 5 (35.7) |
Statistical analysis
The Fisher exact probability method was used to
evaluate the difference of the number of methylated PTPL1
promoters between DLBCL patients and reactive lymph node cases, GCB
group and non-GCB group. The difference of the number of methylated
PTPL1 promoters between DLBCL patients and reactive lymph
node proliferation cases was statistically significant
(P<0.001). The difference of the number of methylated
PTPL1 promoters between the GCB and non-GCB group was not
statistically significant (P=0.089).
Discussion
The aim of the present study was to identify novel
methylated biomarkers in lymphoma and to explore potential new
therapeutic targets. The methylation pattern of the PTPL1
gene was investigated in certain lymphoma-derived cell lines and 47
DLBCL cases. PTPL1 was methylated in two Burkitt lymphoma
cell lines (CA46 and Raji), one acute T cell lymphoma cell line
(Jurkat) and one DLBCL cell line (DB); and unmethylated in the
cutaneous T cell lymphoma cell line (Hut78), and in two mantle cell
lymphoma cell lines (Maver and Z138). The methylated frequency of
PTPL1 in DLBCL patients was significantly higher compared to
the non-malignant lymphoid control. Shi et al (13) reported that there were significant
differences in DNA methylation between pre-germinal and germinal
center-derived NHL. In general, germinal center-related lymphomas
(follicular lymphoma and DLBCL) have more methylation compared to
non-germinal center lymphoma (mantle cell lymphoma and chronic
lymphocytic leukemia/lymphoma) (14). The present study shows that the
PTPL1 methylation frequency of non-GCB was higher compared
with GCB. Clinically, the malignancy of non-GCB is higher compared
with GCB, and prior to the appearance of rituximab, the prognosis
of non-GCB was worse compared with GCB (15). Hypermethylation of the
PTPL1 promoter was also identified in a small number of
carcinomas, including gastric and hepatocellular tumors, with 8/12
hepatocellular tumors presenting with significant methylation
patterns (16). In addition, the
methylation pattern of several genes were identified in lymphoma,
such as SHP1, CD44, DAPK, GSTP1, MGMT, P14, P15, P16, P33, RB1,
hMLH1, CDH1, APC, RASSFA1, TIMP3, VHL and BLU (17–20). Epigenetic abnormalities affecting
histone-modifying enzymes and regulators, such as histone
deacetylases (HDACs), have also been described in lymphoma
(21). The methylation of lysine
9 and lysine 27 of histone H3 (H3K9me and H3K27me) can lead to
transcriptional repression of the target gene; however, the
methylation of lysine 4 and lysine 36 of histone H3 (H3K4me and
H3K36me) can lead to transcriptional activation of the target gene
(22,23). These all indicate that epigenetic
alterations of gene expression are important in the development of
tumorigenesis. The present study also confirmed this by showing
that methylation of the promoter region of PTPL1 correlates
with lymphoma.
In addition, the present study has detected
PTPL1 mRNA in cell lines. To compare this finding with the
methylation patterns of the previously described cell lines, the
expression of PTPL1 mRNA was ubiquitously expressed at
different levels in the unmethylated cell lines (Hut78, Maver and
Z138) and silenced in the total methylated cell lines (CA46, Raji,
Jurkat and DB). Methylation of cytosine residues at CpG
dinucleotides in the promoter region of genes is a major epigenetic
modification in mammalian genomes and can lead to the silencing of
gene expression (24,25). Epigenetic regulation of
PTPL1 expression was also documented in other cancers. In a
study using a total of 82 tumor cell lines, Ying et al
(26) showed that the expression
of PTPL1 was frequently downregulated or silenced in NHL
(94%, 15/16), Hodgkin lymphoma (50%, 3/6), breast (30%, 3/10),
gastric (60%, 6/10) and hepatocellular (67%, 8/12) carcinoma cell
lines. In another study, Lee et al (27) identified that PTPL1 can be
detected in 80% of hepatocellular carcinoma with a significant
variation of the protein expression level by immunohistochemistry
staining. The present findings indicate that this epigenetic
alteration of PTPL1 is a common phenomenon in lymphoma and
may be an important approach to inactivate cancer-related genes in
this disease. However, these results also show that DNA methylation
is not the only reason for PTPL1 silencing.
The PTPL1 re-expression pattern was also
investigated following treatment with the DNMTs inhibitor 5-Aza to
further confirm the role of DNA methylation in PTPL1
regulation. Re-expression of PTPL1 mRNA emerged at 48h after
treated with 5-Aza. 5-Aza exerts its action by inhibiting DNA
methylation (via its incorporation into DNA at cytosine positions)
during DNA replication. In general, their transport is mediated by
the human concentrative nucleoside transporter 1 (hCNT1) followed
by their phosphorylation and conversion into their active
tri-phosphate forms, namely 5-Aza-CTP (28). In this way, 5-Aza is able to
interact with DNMTs, inhibit their activity and decrease overall
DNA methylation levels. Therefore, the effect of 5-Aza on cell
lines may be associated with the activity or expression of DNMT1,
DNMT3A and DNMT3B (29). Overall,
these data suggest that the DNA methyltransferase inhibitor 5-Aza
was able to successfully lead to re-expression of PTPL1
mRNA. The results confirmed that hypermethylation of PTPL1
was responsible for gene silencing, as DNA demethylation resulted
in reactivation of PTPL1 transcription in the PTPL1
hypermethylated cell lines. This may also support a
tumor-suppressor role for PTPL1 in lymphoma.
By contrast, the relative increase of PTPL1
level in tumor tissues supports the role in tumor promotion. A high
level of PTPL1 mRNA expression in Kaposi's sarcoma,
hepatocellular carcinomas, pancreatic adenocarcinomas, as well as
with higher expression in T helper cells type 1 (which are
resistant to apoptosis) versus T helper cells type 2 (which are
sensitive to Fas ligand), also shows a correlation between tumor
cell survival in the presence of PTPL1 expression (11,30–32). In addition, investigators have
shown relatively higher levels of PTPL1 expression in
multiple carcinomas compared to the normal adjacent tissue as
detected by immunohistochemistry (33). Another previous study showed that
in the process of dimethyl sulfoxide- and all-trans retinoic
acid-induced differentiation in HL-60 cells, the increased
resistance to death receptor-mediated apoptosis coincided with an
increase in PTPL1 (34).
In CML, the resistance to death receptor-mediated apoptosis and the
existence of leukemic stem cells were associated with an increase
in PTPL1 (35). A positive
correlation between PTPL1 expression and resistance to
Fas-induced apoptosis has been shown in human T lymphotrophic virus
(HTLV-I) infected T cell lines, ovarian cancer cell lines, human
pancreatic cancer cell lines and squamous cell carcinomas of the
head and neck cell lines (36).
The presence of high PTPL1 levels in tumor tissues may
oppose PTPL1 as a tumor suppressor. This may indicate that
PTPL1 has a role as a tumor promoter. The induction of
PTPL1 by an oncogene and relative increase of PTPL1
levels in tumor tissues supports a role in tumor promotion. By
contrast, epigenetic studies are more consistent with a role for
PTPL1 as a tumor suppressor. The impact of PTPL1 on
cancer is divided between its capacity to counteract the activity
of oncogenic tyrosine kinases and its inhibitory interaction with
the death receptor, Fas. The ability of PTPL1 to inhibit
signaling from growth factor receptors or oncogenes with tyrosine
kinase activity can suppress tumor occurrence (37,38). By contrast, the ability of
PTPL1 to interact with the Fas receptor can promote tumor
occurrence (39). Therefore,
according to the tissue type and the cellular environment,
different proportions of these two signaling pathways can lead to
different biological effects. A complete understanding of
epigenetic modifications of PTPL1 and various PTPL1
domains in mediating protein-lipid and protein-protein interactions
will be critical in resolving the functional role of PTPL1
in cancer. Establishing the precise function of PTPL1 in NHL
and understanding its mode of action will aid in our understanding
of the use of PTPL1 as a therapeutic target in NHL.
In the present study, the number of DLBCL cases was
less, and that of subjects lost to follow-up was greater. More
cases and future molecular studies are required to determine the
role of PTPL1 methylation in the development and progression
of NHL.
In conclusion, the study showed that PTPL1
expression is regulated by DNA methylation, not only in lymphoma
cell lines, but also in the DLBCL patients. The loss of
PTPL1 mRNA is the consequence of PTPL1 methylation
and can be reversed by 5-Aza. Thus, 5-Aza may be further
investigated as a novel therapeutic agent for NHL.
Acknowledgments
The authors would like to thank Professor Junmin Li
(Shanghai Ruijin Hospital, China) for the provision of the DB cell
lines.
References
|
1
|
Sherief AM, Elsafy UR, Abdelkhalek ER,
Kamal NM, Youssef DM and Elbehedy R: Disease patterns of pediatric
non-Hodgkin lymphoma: A study from a developing area in Egypt. Mol
Clin Oncol. 3:139–144. 2015.
|
|
2
|
Bradley WD, Arora S, Busby J,
Balasubramanian S, Gehling VS, Nasveschuk CG, Vaswani RG, Yuan CC,
Hatton C, Zhao F, et al: EZH2 inhibitor efficacy in non-Hodgkin's
lymphoma does not require suppression of H3K27 monomethylation.
Chem Biol. 21:1463–1475. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miyazaki T, Atarashi Y, Yasumura S,
Minatoya I, Ogawa K, Iwamoto M, Minemura M, Shimizu Y, Sato TA,
Watanabe A, et al: Fas-associated phosphatase-1 promotes
Fas-mediated apoptosis in human colon cancer cells: Novel function
of FAP-1. J Gastroenterol Hepatol. 21:84–91. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yeh SH, Wu DC, Tsai CY, Kuo TJ, Yu WC,
Chang YS, Chen CL, Chang CF, Chen DS and Chen PJ: Genetic
characterization of fas-associated phosphatase-1 as a putative
tumor suppressor gene on chromosome 4q21.3 in hepatocellular
carcinoma. Clin Cancer Res. 12:1097–1108. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eberth S, Schneider B, Rosenwald A,
Hartmann EM, Romani J, Zaborski M, Siebert R, Drexler HG and
Quentmeier H: Epigenetic regulation of CD44 in Hodgkin and
non-Hodgkin lymphoma. BMC Cancer. 10:5172010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abaan OD and Toretsky JA: PTPL1: A large
phosphatase with a split personality. Cancer Metastasis Rev.
27:205–214. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y,
Yan J, Ren X, Lin S, Li J, et al: The DNA methylation landscape of
human early embryos. Nature. 511:606–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lübbert M: DNA methylation inhibitors in
the treatment of leukemias, myelodysplastic syndromes and
hemoglobinopathies: Clinical results and possible mechanisms of
action. Curr Top Microbiol Immunol. 249:135–164. 2000.PubMed/NCBI
|
|
9
|
Amara K, Ziadi S, Hachana M, Soltani N,
Korbi S and Trimeche M: DNA methyltransferase DNMT3b protein
over-expression as a prognostic factor in patients with diffuse
large B-cell lymphomas. Cancer Sci. 101:1722–1730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bejar R and Steensma DP: Recent
developments in myelodys-plastic syndromes. Blood. 124:2793–2803.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Freiss G and Chalbos D: PTPN13/PTPL1: An
important regulator of tumor aggressiveness. Anticancer Agents Med
Chem. 11:78–88. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Herman JG, Graff JR, Myöhänen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: A novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi H, Guo J, Duff DJ, Rahmatpanah F,
Chitima-Matsiga R, Al-Kuhlani M, Taylor KH, Sjahputera O, Andreski
M, Wooldridge JE, et al: Discovery of novel epigenetic markers in
non-Hodgkin's lymphoma. Carcinogenesis. 28:60–70. 2007. View Article : Google Scholar
|
|
14
|
Lossos IS: The DNA methylome: A novel
biomarker. Blood. 123:1627–1628. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bittenbring JT, Neumann F, Altmann B,
Achenbach M, Reichrath J, Ziepert M, Geisel J, Regitz E, Held G and
Pfreundschuh M: Vitamin D deficiency impairs rituximab-mediated
cellular cytotoxicity and outcome of patients with diffuse large
B-cell lymphoma treated with but not without rituximab. J Clin
Oncol. 32:3242–3248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hunter T: The role of tyrosine
phosphorylation in cell growth and disease. Harvey Lect. 94:81–119.
1998–1999.PubMed/NCBI
|
|
17
|
Paz MF, Fraga MF, Avila S, Guo M, Pollan
M, Herman JG and Esteller M: A systematic profile of DNA
methylation in human cancer cell lines. Cancer Res. 63:1114–1121.
2003.PubMed/NCBI
|
|
18
|
Bodoor K, Haddad Y, Alkhateeb A, Al-Abbadi
A, Dowairi M, Magableh A, Bsoul N and Ghabkari A: DNA
hypermethylation of cell cycle (p15 and p16) and apoptotic (p14,
p53, DAPK and TMS1) genes in peripheral blood of leukemia patients.
Asian Pac J Cancer Prev. 15:75–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kupčinskaitė-Noreikienė R, Skiecevičienė
J, Jonaitis L, Ugenskienė R, Kupčinskas J, Markelis R, Baltrėnas V,
Sakavičius L, Semakina I, Grižas S, et al: CpG island methylation
of the MLH1, MGMT, DAPK, and CASP8 genes in cancerous and adjacent
noncancerous stomach tissues. Medicina (Kaunas). 49:361–366.
2013.
|
|
20
|
Ng HY, Wan TS, So CC and Chim CS:
Epigenetic inactivation of DAPK1, p14ARF, mir-34a and -34b/c in
acute promyelocytic leukaemia. J Clin Pathol. 67:626–631. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hutt DM, Roth DM, Vignaud H, Cullin C and
Bouchecareilh M: The histone deacetylase inhibitor, Vorinostat,
represses hypoxia inducible factor 1 alpha expression through
translational inhibition. PLoS One. 9:e1062242014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Witzig TE, Hu G, Offer SM, Wellik LE, Han
JJ, Stenson MJ, Dogan A, Diasio RB and Gupta M: Epigenetic
mechanisms of protein tyrosine phosphatase 6 suppression in diffuse
large B-cell lymphoma: Implications for epigenetic therapy.
Leukemia. 28:147–154. 2014. View Article : Google Scholar :
|
|
23
|
Kroesen M, Gielen P, Brok IC, Armandari I,
Hoogerbrugge PM and Adema GJ: HDAC inhibitors and immunotherapy; a
double edged sword? Oncotarget. 5:6558–6572. 2014.PubMed/NCBI
|
|
24
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
25
|
Jones PA: Overview of cancer epigenetics.
Semin Hematol. 42(Suppl 2): S3–S8. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ying J, Li H, Cui Y, Wong AH, Langford C
and Tao Q: Epigenetic disruption of two proapoptotic genes
MAPK10/JNK3 and PTPN13/FAP-1 in multiple lymphomas and carcinomas
through hypermethylation of a common bidirectional promoter.
Leukemia. 20:1173–1175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee SH, Shin MS, Lee HS, Bae JH, Lee HK,
Kim HS, Kim SY, Jang JJ, Joo M, Kang YK, et al: Expression of Fas
and Fas-related molecules in human hepatocellular carcinoma. Hum
Pathol. 32:250–256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sripayap P, Nagai T, Uesawa M, Kobayashi
H, Tsukahara T, Ohmine K, Muroi K and Ozawa K: Mechanisms of
resistance to azacitidine in human leukemia cell lines. Exp
Hematol. 42:294–306. 2014. View Article : Google Scholar
|
|
29
|
Zhou Y and Hu Z: Genome-wide demethylation
by 5-aza-2′-de-oxycytidine alters the cell fate of stem/progenitor
cells. Stem Cell Rev. 11:87–95. 2015. View Article : Google Scholar
|
|
30
|
Chaudhry P, Srinivasan R and Patel FD:
Differential expression of Fas family members and Bcl-2 family
members in benign versus malignant epithelial ovarian cancer (EOC)
in North Indian population. Mol Cell Biochem. 368:119–126. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mori S, Murakami-Mori K, Jewett A,
Nakamura S and Bonavida B: Resistance of AIDS-associated Kaposi's
sarcoma cells to Fas-mediated apoptosis. Cancer Res. 56:1874–1879.
1996.PubMed/NCBI
|
|
32
|
Zhang X, Brunner T, Carter L, Dutton RW,
Rogers P, Bradley L, Sato T, Reed JC, Green D and Swain SL: Unequal
death in T helper cell (Th)1 and Th2 effectors: Th1, but not Th2,
effectors undergo rapid Fas/FasL-mediated apoptosis. J Exp Med.
185:1837–1849. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nariai Y, Mishima K, Yoshimura Y and
Sekine J: FAP-1 and NF- κB expressions in oral squamous cell
carcinoma as potential markers for chemo-radio sensitivity and
prognosis. Int J Oral Maxillofac Surg. 40:419–426. 2011. View Article : Google Scholar
|
|
34
|
Vondrácek J, Sheard MA, Krejcí P, Minksová
K, Hofmanová J and Kozubík A: Modulation of death receptor-mediated
apoptosis in differentiating human myeloid leukemia HL-60 cells. J
Leukoc Biol. 69:794–802. 2001.PubMed/NCBI
|
|
35
|
Michor F, Hughes TP, Iwasa Y, Branford S,
Shah NP, Sawyers CL and Nowak MA: Dynamics of chronic myeloid
leukaemia. Nature. 435:1267–1270. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Arai M, Kannagi M, Matsuoka M, Sato T,
Yamamoto N and Fujii M: Expression of FAP-1 (Fas-associated
phosphatase) and resistance to Fas-mediated apoptosis in T cell
lines derived from human T cell leukemia virus type 1-associated
myelopathy/tropical spastic paraparesis patients. AIDS Res Hum
Retroviruses. 14:261–267. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Glondu-Lassis M, Dromard M, Lacroix-Triki
M, Nirdé P, Puech C, Knani D, Chalbos D and Freiss G: PTPL1/PTPN13
regulates breast cancer cell aggressiveness through direct
inactivation of Src kinase. Cancer Res. 70:5116–5126. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
He RJ, Yu ZH, Zhang RY and Zhang ZY:
Protein tyrosine phosphatases as potential therapeutic targets.
Acta Pharmacol Sin. 35:1227–1246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kamihira S, Yamada Y, Hirakata Y, Tomonaga
M, Sugahara K, Hayashi T, Dateki N, Harasawa H and Nakayama K:
Aberrant expression of caspase cascade regulatory genes in adult
T-cell leukaemia: Survivin is an important determinant for
prognosis. Br J Haematol. 114:63–69. 2001. View Article : Google Scholar : PubMed/NCBI
|