Introduction
Pulmonary arterial hypertension (PAH) comprises a
group of disorders that involve pulmonary vasoconstriction and
vascular remodeling, as well as right ventricular (RV) hypertrophy
(1,2). The response of the RV to the
increased afterload, including electrical, mechanical and
structural changes, may ultimately induce left ventricular (LV)
atrophic remodeling and dysfunction (3,4).
RV failure (RVF) in particular is the immediate cause of death in
the majority of PAH patients (1,2).
Clinically updated techniques and approved PAH-specific therapeutic
agents have been effectively applied to identify RV dysfunction and
improved the quality of life of the respective patients (5,6).
However, PAH is associated with a poor outcome with an estimated
5-year survival rate of <60% (7).
The key role of RV remodeling in PAH has received
increasing attention, as larger volumes, pathological hypertrophy,
diffuse inflammatory infiltration and interstitial fibrosis
adversely were reported to affect the ejection fraction and
ultimately lead to ventricular failure (1,2,8-12).
Myocardial apoptosis, a highly regulated process of cell death, has
also been reported to occur in a rat model of monocrotaline
(MCT)-induced PAH and may have an important role in cardiac
remodeling after PAH (10,11).
Therefore, remodeling as well as apoptosis have key roles in the
pathogenesis of RV maladaptation and failure.
β-adrenergic receptor (β-AR) signaling is a primary
event in the regulation of myocardial contractility under normal
physiological conditions, whereas its sustained activation also
contributes to myocardial dysfunction during the progression of
heart failure (HF). It has been confirmed that overexpression of
human β1-AR leads to early hypertrophy and interstitial
fibrosis, followed by marked cardiac dysfunction in transgenic mice
(13). Several studies reported
that Ca2+/calmodulin-dependent kinase II (CaMKII)
mediates β-AR-induced myocyte death through either a protein kinase
A (PKA)-dependent or a PKA-independent process (14,15). Of note, β1-AR
activation enhances cardiomyocyte apoptosis, whereas
β2-AR exerts anti-apoptotic effects on the heart. This
crucial difference between the two receptor subtypes is associated
with the signaling of β2-AR through the inhibitory G
protein (Gi), which improves cardiac function and
decreases apoptosis (16,17). In contrast to the current dogma,
recent studies demonstrated that β2-AR signaling may
also have cardiotoxic effects (17-21). In certain types of HF,
β2-AR signaling may lose its normally cardioprotective
properties and change to β1-AR-like global signaling,
thus contributing to the HF phenotype (22). In addition, accumulating evidence
demonstrated that β2-AR activation increases the risk of
sudden cardiac death in HF patients, and that non-selective β-AR
blockade provides a greater survival benefit compared with
selective β1-AR blockade in chronic HF patients
(23,24). Thus, a state of toxic and
protective effects coexists with respect to the β2-AR
status in HF. However, the role of β2-AR signaling in
the progression of PAH has not been fully elucidated.
To investigate this issue, the present study used a
rat model of MCT-induced PAH, since it is an informative animal
model for identifying the progression of cardiopulmonary
alterations associated with a profound remodeling of the heart with
RV hypertrophy and dysfunction (25-28). In the present study, the
time-course of the alterations of cardiopulmonary function and
structure was investigated during the progression of PAH. In
addition, the potential roles of G protein-coupled receptor (GPCR)
signaling and further β2-AR signaling in the progression
of the pathophysiology of MCT-induced PAH were determined in a
stage-dependent manner.
Methods
MCT-induced PAH
The present study conformed to the Guide for the
Care and Use of Laboratory Animals published by the National
Institutes of Health (NIH) and all of the procedures were approved
by the Animal Ethics Committee of Tianjin University of Traditional
Chinese Medicine (Tianjin, China; no. TCM-LAEC2015034). A total of
160 Male Wistar rats (age, 7 weeks; weight, 180-200 g; certificate
no. SCXK 2012-0001) were obtained from Beijing Vital River
Laboratory Animal Technology Co., Ltd. (Beijing, China). The rats
were maintained at a room temperature of 22±2°C and humidity of
50±5%, with a 12 h light/dark cycle. Food and water were available
ad libitum. The PAH rat model was induced by a single
intraperitoneal injection of MCT (60 mg/kg body weight; GuangRun
Bio Technology, Co., Ltd., Nanjing, China) as described in a
previous study by our group (29). Saline-injected rats served as the
control (CON) group. At week 0 (prior to the MCT injection), and at
weeks 1-6, cardiopulmonary function and pathology, as well as the
molecular mechanisms underlying RV remodeling and apoptosis were
compared between the CON and MCT-treated rats (n=10/group) at a
total of seven sequential time-points.
Lung respiratory function
Pulmonary respiratory function, including
respiratory frequency (RF), tidal volume (TV) and minute volume
(MV), were measured using whole-body barometric plethysmography
(WBP PLT-UNR-RT-2; EMKA Technologies, Paris, France). The rats were
placed in a whole-body plethysmograph and were allowed to
acclimatize for at least 10 min prior to analysis. The box pressure
wave was recorded via a transducer and computer system (iOX2
software; version 2.5; EMKA Technologies) and the measurements were
recorded.
Pulmonary artery pressure, RV and LV
haemodynamics
Baseline hemodynamic data were obtained during the
routine pulmonary artery, right and left heart catheterization, as
previously described (30). In
brief, the rats were anesthetized with pentobarbital sodium (50
mg/kg) and a cervical midline skin incision was performed. The
right carotid artery was cannulated with a polyethylene catheter
[PE-50; 0.58 mm inner diameter (ID); 0.99 mm outer diameter (OD);
American Health & Medical Supply International Corp. Co., Ltd.,
Scarsdale, NY, USA]. LV systolic pressure (LVSP), LV end-diastolic
pressure (LVEDP) and heart rate (HR) were measured via this
catheter, which was advanced into the LV cavity, and the parameters
were recorded with a PowerLab System (ML870 PowerLab; AD
Instruments Pty Ltd., Sydney, Australia) to obtain the maximum
rates of LV pressure increase and decrease as a measure of systolic
and diastolic function, respectively (LV +dP/dtmax and
−dP/dtmin). The right jugular vein was cannulated with a
micro-urethane catheter (BB520-40, 0.635 mm ID, 1.02 mm OD;
American Health & Medical Supply International Corp. Co., Ltd.)
and, after recording the RV hemodynamic data [RV systolic pressure
(RVSP), RV end-diastolic pressure (RVEDP), and maximal rate of RV
pressure increase and decrease (RV +dP/dtmax and
−dP/dtmin, respectively)], the measuring catheter was
advanced across the pulmonary valve into the pulmonary artery to
measure the pulmonary artery systolic pressure (PASP) and pulmonary
arterial diastolic pressure (PADP). All these data were entered
into a computer using PowerLab 8 channel physiological recorder and
analyzed using LabChart 7.3.7 software (AD Instruments Pty Ltd.,
Sydney, Australia). Following hemodynamic measurements, the animals
were euthanized for further processing.
Serum brain natriuretic peptide (BNP)
level assessment
Blood samples were collected and centrifuged at
1,000 × g at 4°C for 15 min to separate the supernatant. Serum BNP
was detected with an ELISA kit (cat. no. CSB-E07972r; Cusabio
Biotech Co., Ltd., Wuhan, China) according to the manufacturer's
protocols.
Gross evaluation
The lungs and heart were rapidly dissected and
weighed. The lung index was assessed as follows: Lung index =
[total lung wet weight (g)/body weight (g)] ×100. The heart was
divided into RV and LV including the intraventricular septum
(defined as LVS) to calculate the RV/LVS weight ratio, which
resembled the RV hypertrophy index (RVHI). The RV wall was
snap-frozen and stored at -80°C for further analysis.
Histological analysis
Heart and lung tissues were fixed with 10%
formaldehyde, embedded in paraffin, sectioned at 3.5-4 μm
intervals, stained with hematoxylin and eosin staining (H&E,
hematoxylin for 10 min, followed by counterstaining with eosin for
4 min at room temperature) and visualized under a Leica DM4000B LED
light microscope equipped with a Leica DFC450C camera (Leica
Microsystems, Wetzlar, Germany). Interstitial fibrosis of the RV
was determined by using a Masson's trichrome-staining kit (cat. no.
G1340; Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) according to the manufacturer's protocol. Briefly,
deparaffinization and rehydration were accomplished as follows:
immersion in xylene, twice (5 min each); 100% ethanol, twice (5 min
each); immersion in 95% ethanol, twice (5 min each); distilled
water (5 min). These sections were stained with
hematoxylin-iodineferric chloride solution (1:1) for 10 min at room
temperature, differentiated in hydrochloride-alcohol solution for 6
sec and then quickly rinsed under running tap water (15 min).
Sections were stained in ponceau staining solution (8 min, room
temperature) and then quickly rinsed in phosphomolybdic acid
solution (1%, room temperature). The sections were stained with
aniline blue solution and followed by dehydration through a series
of alcohols (95%, three times; 100%, twice) to xylene (3 times) and
then coverslipped. The collagen fibers appeared blue. Micrographs
were captured from each sample at ×400 magnification and quantified
using ImageJ software (version 1.42q; NIH, Bethesda, MD, USA).
Terminal deoxynucleotidyl transferase
(TdT) deoxyuridine triphosphate nick end labeling (TUNEL)
assay
The TUNEL assay was performed using the In
Situ Cell Death Detection kit (POD; cat. no. 11684817910; Roche
Applied Science, Mannheim, Germany), according to the
manufacturer's instructions. Following deparaffinization and
rehydration, the sections were treated with 3%
H2O2 in methanol for 10 min, and then
incubated with proteinase K (20 μg/ml in 10 mM Tris/HCl, pH
7.4) for 30 min at 37°C. The slides were immersed in TUNEL reaction
mixture for 60 min at 37°C in a humidified atmosphere in the dark.
A converter peroxidase was added, followed by incubation of the
slides for 30 min. The reaction was developed with diaminobenzidine
substrate (brown) and counterstained with hematoxylin (blue).
Negative (omission of TdT) and positive controls (treatment of the
sections with DNase I for 10 min at room temperature prior to
labeling procedures) were prepared for contrast. TUNEL-positive
cells were defined as cells with clear brown or brown-blue nuclear
labeling. To assess the TUNEL index of the RV, 10 micrographs per
tissue section were randomly selected at a magnification of ×400.
The TUNEL index (%) was calculated as the ratio of the number of
TUNEL-positive cells divided by the total number of cells.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) GPCR
arrays
The expression levels of 84 GPCR-associated genes
were quantified using a G-Protein-Coupled Receptor Signaling
Pathway Finder™ PCR Array from SA Biosciences (Qiagen, Hilden,
Germany) at weeks 1, 3, 5 and 6 (corresponding to groups 1, 3, 5
and 6). Total RNA from RVs was isolated using the Qiagen RNA
extraction kits (cat. no. 74704; Qiagen) in accordance with the
manufacturer's protocols. Complementary (c)DNA was synthesized from
1 μg RNA using the SABiosciences RT2 First Strand
kit (cat. no. 330401; Qiagen). Of the 84 genes analyzed by the cDNA
array, 95% were amplified with Ct values of <35 cycles by qPCR
and were considered to be suitable for relative expression
analysis. RT-qPCR was performed by using the RT2
Profiler PCR Array Rat GPCR kit (cat. no. 330231; Qiagen). The
reaction conditions comprised 1 cycle of initiation at 95°C for 10
min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min
using the ABI 7500 Fast Real-Time PCR System (Applied Biosystems;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). Each 96-well
plate included primers for five housekeeping genes, as well as
positive and negative controls. The ΔCt method was used for PCR
array data analysis (31,32). Only expression values with a
difference of >2- or <0.5-fold with respect to the controls
were considered statistically significant. Three repeated tests
were performed for each set of measurements and the resulting data
were averaged.
Protein analysis of β-AR signaling
Standard western blot analysis was performed using
RV lysates, as previously described (33). The proteins were probed with the
following primary antibodies: Rabbit anti-tubulin (1:500 dilution;
cat. no. SC-91C4; Santa Cruz Biotechnology, Inc., Danvers, MA,
USA), rabbit anti-β1-AR (1:1,000 dilution; cat. no.
ab3442) rabbit anti-β2-AR (1:1,000 dilution; cat. no.
ab137494) and rabbit anti-G protein-coupled receptor kinase 2
(GRK2; 1:500 dilution; cat. no. ab32558; all from Abcam, Cambridge,
MA, USA), rabbit anti-phospho (p)-β2-AR (pSer346;
1:1,000 dilution; cat. no. SAB4504272) and rabbit
anti-p-β2-AR [pSer(355,356); 1:500 dilution; cat. no.
SAB4504074; both from Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany], rabbit anti-PKA (1:1,000 dilution; cat. no. 06-903; EMD
Millipore, Billerica, MA, USA) and rabbit anti-CaMKII (1:1,000
dilution; cat. no. 3362; Cell Signaling Technologies, Inc.,
Danvers, MA, USA). Horseradish peroxidase-conjugated goat
anti-rabbit immunoglobulin G (1:3,000 dilution; cat. no. ZB-2301;
Zhongshan Goldenbridge Bio, Beijing, China) was used as a secondary
antibody. Enhanced chemiluminescence (WBKLS0500; Merck KGaA) was
visualized using a Vilber Fusion FX7 RT-ECL scanner (Vilber
Lourmat, Marne-la-Vallée, France). Band signals were quantified by
densitometry with Bio1D software (version 15.06a; Vilber Lourmat).
The protein expression data of β1-AR, β2-AR,
PKA, GRK2 and CaMKII were normalized to tubulin, whereas data on
the phosphorylation status of β2-AR at Ser346 and
Ser(355,356) were normalized to total β2-AR protein
levels.
Statistical analysis
Values are expressed as the mean ± standard
deviation (SD). SPSS 13 software (SPSS Inc., Chicago, IL, USA) was
used to perform statistical analysis. All data were normally
distributed. The unpaired Student's t-test was used to determine
the significance between two groups. One-way analysis of variance
was used for multiple comparisons, followed by post-hoc analysis
[Dunett's test or least significant differences test]. P<0.05
was considered to indicate a statistically significant
difference.
Results
Progressive deterioration of lung
function and histopathology
PASP and PADP were increased from week 2 onwards
(Fig. 1A). As an indication of
respiratory dysfunction, the MV exhibited a declining trend from
the early stages of PAH, becoming significantly lower compared with
the baseline value at week 6, which was concurrent with a
significantly increased RF and a reduced TV during PAH development
(Fig. 1B). Concomitant with the
results of pulmonary dysfunction, MCT-induced rats displayed an
increasingly higher lung weight and lung index compared with that
of CON-rats over the experimental period (Fig. 1C). The gradually aggravating
histopathological changes of the lungs were characterized by
interstitial edema, congestion, thickening of the alveolar septum,
thickening of the tunica media of the pulmonary arteries,
inflammatory cell infiltration into the thickened area and
pulmonary hemosiderosis (Fig.
1D).
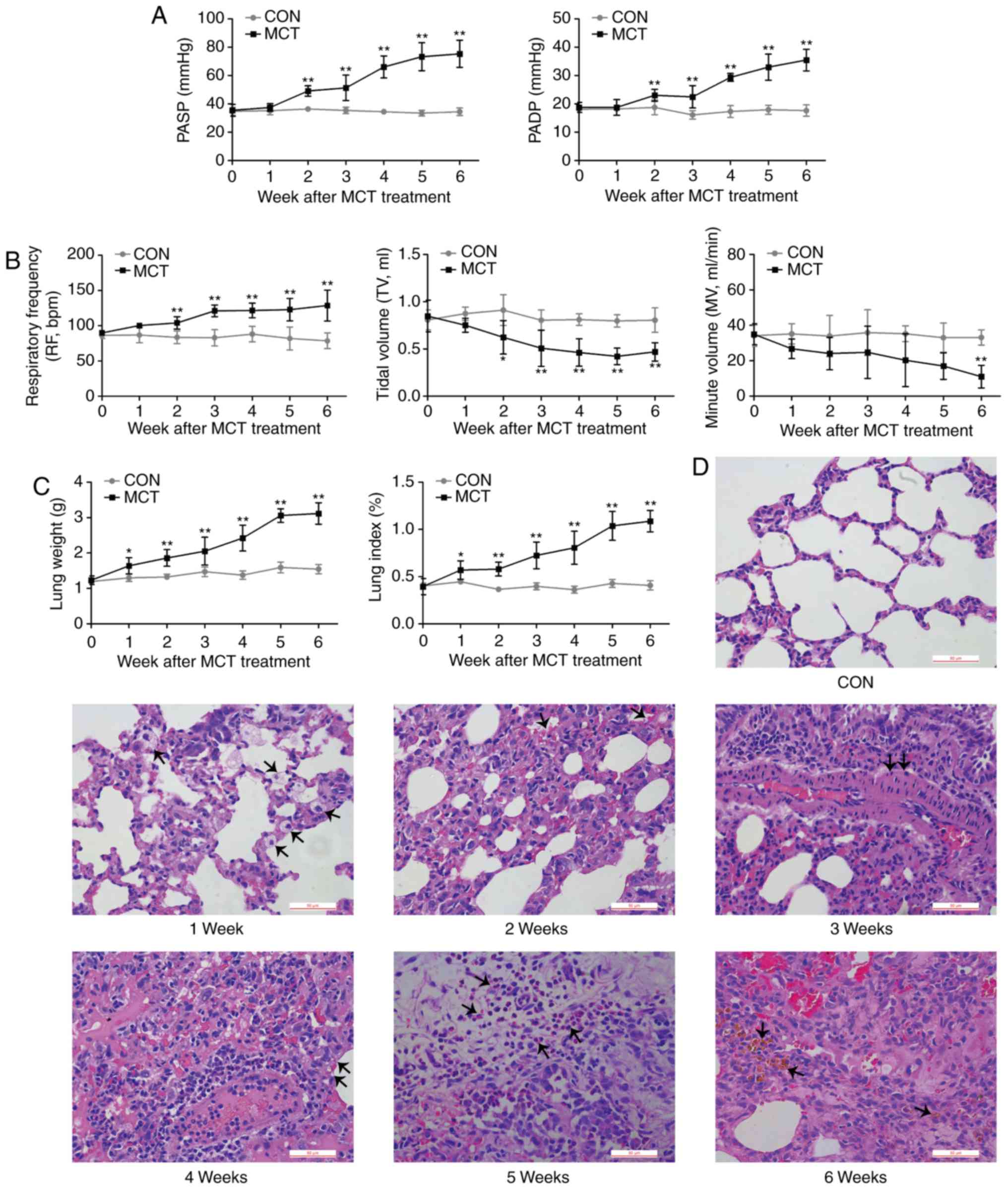 | Figure 1In vivo effects of MCT on lung
function and structure. (A) PASP and PADP in the MCT group
displayed a significant increase at weeks 2-6. (B) During disease
progression, respiratory parameters, including respiratory
frequency, TV and MV we recorded. A decreasing MV trend mirrored
the significant decline in TV, while the RF remained significantly
accelerated from week 2 to week 6 in MCT-treated rats compared with
that in the control rats. (C and D) Histopathological changes in
rats with MCT-induced pulmonary arterial hypertension were
progressively aggravated from W1-6 (scale bar, 50 μm). W1,
edema; W2, congestion; W3, thickening of arterial wall; W4,
thickening of alveolar wall; W5, inflammatory cell infiltration;
W6, pulmonary hemosiderosis (respective changes were indicated by
arrows). Values are expressed as the mean ± standard deviation
(n=10). *P<0.05 and **P<0.01 vs. the
CON group. MCT, monocrotaline; CON, control; PASP, pulmonary artery
systolic pressure; PADP, pulmonary arterial diastolic pressure; MV,
minute volume; TV, tidal volume; RF, respiratory frequency; W,
week. |
Dynamic changes of the heart
hemodynamics
In MCT-induced rats, RVSP and RVEDP exhibited a
continuous elevation from week 2 onwards, reaching relative
increases of ~1.8- and ~2.7-fold, respectively, at week 6 (Fig. 2A and B). Of note, improved RV
contraction and relaxation were evidenced by +dP/dtmax
increasing from week 3 onwards and −dP/dtmin
significantly declining during weeks 5-6 (Fig. 2C and D). By contrast, LV function
appeared to be initially compensated during weeks 1-2, as indicated
by the elevated LVSP and +dP/dtmax, as well as the
lowered LVEDP, and was eventually decompensated by weeks 5-6, as
confirmed by a significantly decreased LVSP and
+dP/dtmax, as well as an increased LVEDP and
−dP/dtmin (Fig. 2E–H).
Similarly, the chronotropic curve of the HR was increased at weeks
1-2 and was significantly decreased by weeks 5-6 (Fig. 2I). In agreement with previous
hemodynamic results, the ventricular response to persistent high
pressure caused a time-dependent increase of the BNP in MCT-exposed
rats by weeks 3-6, which was already notably increased by 65.39% at
week 6 compared with the CON group (Fig. 2J).
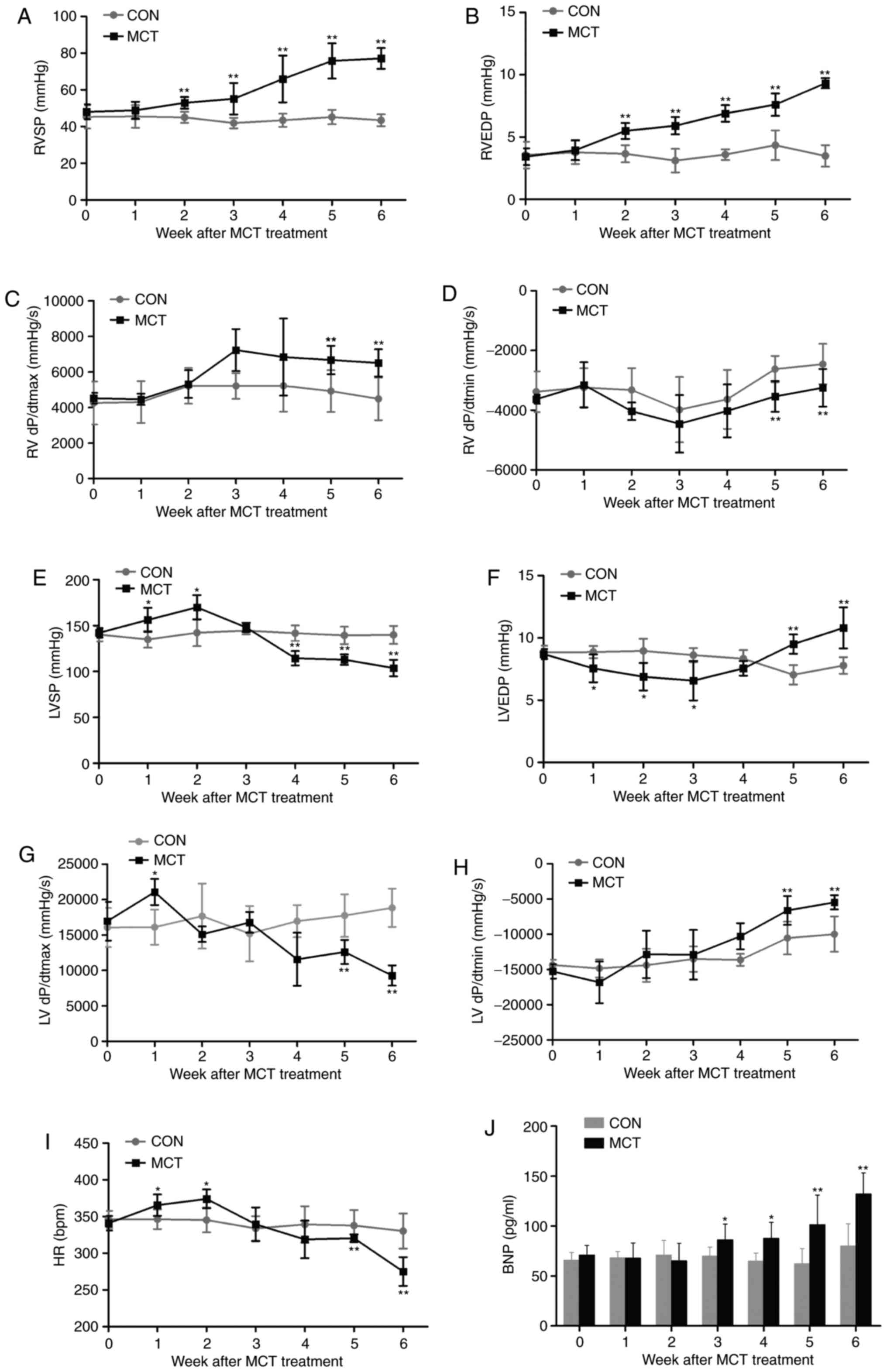 | Figure 2Hemodynamic characteristics of the
heart and BNP augmentation during 6 weeks of MCT treatment. (A) The
RVSP and (B) RVEDP significantly increased in rats with MCT-induced
pulmonary arterial hypertension. (C and D) RV function was
enhanced, as confirmed by the increase in the +dP/dtmax
and decrease in the −dP/dtmin. (E) The cardiac inotropic
LVSP, (F) lusitropic LVEDP, (G) +dP/dtmax in the LV, (H)
−dP/dtmin in the LV and (I) chronotropic HR in rats in
the presence of MCT were evaluated by catheterization. (J) A
significant increase in the level of plasma BNP due to the
persistent pressure overload was revealed in MCT-treated rats.
Values are expressed as the mean ± standard deviation (n=10).
*P<0.05 and **P<0.01 vs. the CON group.
MCT, monocrotaline; CON, control; RVSP/LVSP, right/left ventricular
systolic pressure; RVEDP, right ventricular end-diastolic pressure;
+dP/dtmax, maximal rate of ventricular pressure;
−dP/dtmin, minimal rate of ventricular pressure; HR,
heart rate; BNP, brain natriuretic peptide. |
Progressive maladaptive RV remodeling in
the MCT-induced PAH model
The degree of RV hypertrophy was determined by the
RV weight and the RVHI, and was identified to reach a significant
level from week 3 onwards, whereas the LVS weight exhibited no
significant change over time (Fig.
3A–C). In addition, on gross anatomical examination, RV
dilatation was evident as a gradually progressive enlargement of
the right atrial and RV volumes (Fig.
3D and E). These changes were accompanied by diffuse
inflammatory cell infiltration and widening of the interstitial
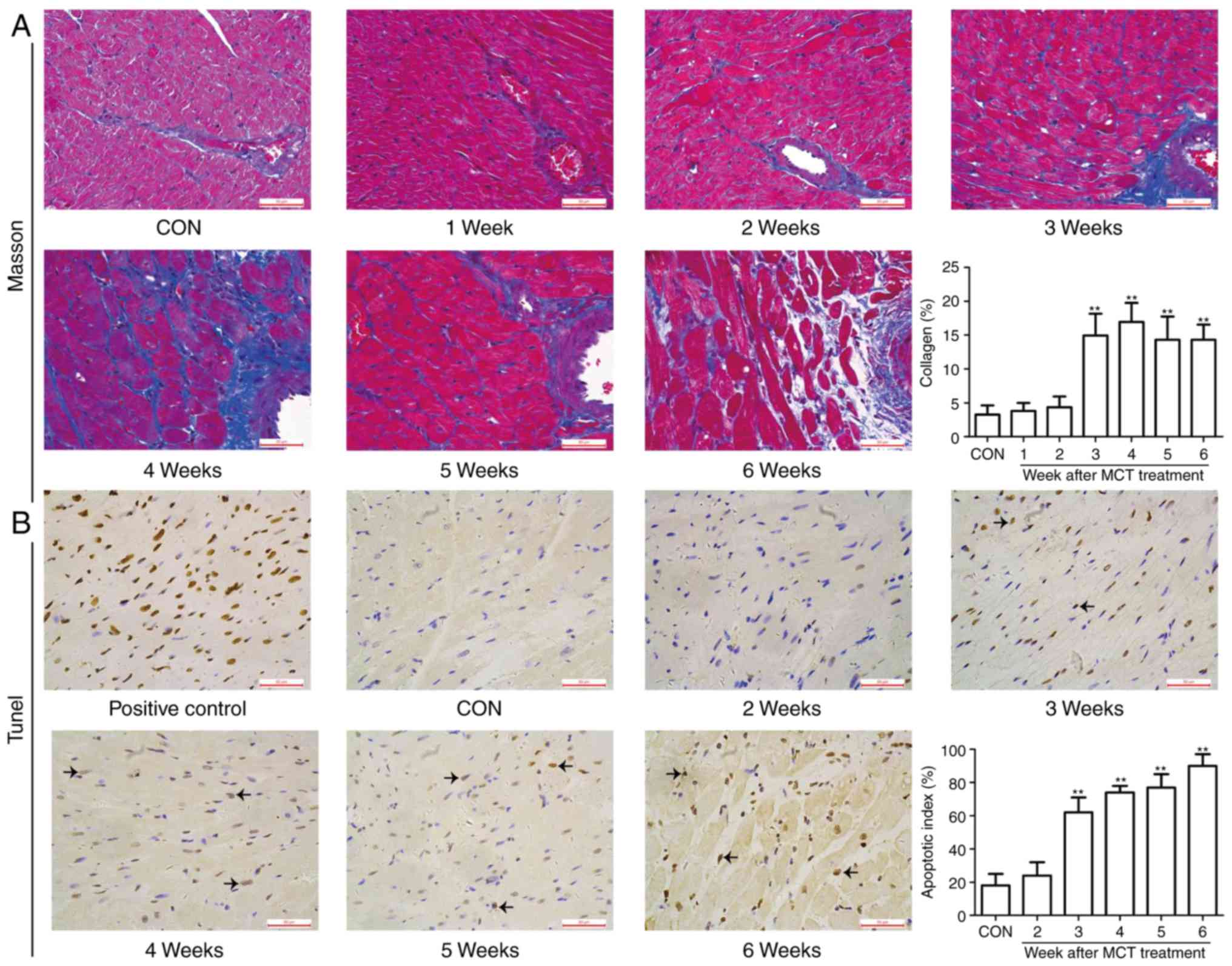
space from weeks 4-6, as assessed by H&E staining (Fig. 3F). In addition, as the disease
progressed, the RV muscle exhibited typical characteristics of
fibrosis, including the appearance of extensive interstitial and
perivascular collagen deposition or fibrosis. Similar results were
obtained by apoptosis detection using the TUNEL assay. As presented
in Fig. 4, no marked myocardial
fibrosis and apoptosis were observed at weeks 1-2, whereas they
were significantly higher in the MCT group compared with those in
the CON group from week 3 onwards. Taken together, these
observations suggest that progressive maladaptive remodeling
occurred in the RV during the MCT-induced development of PAH.
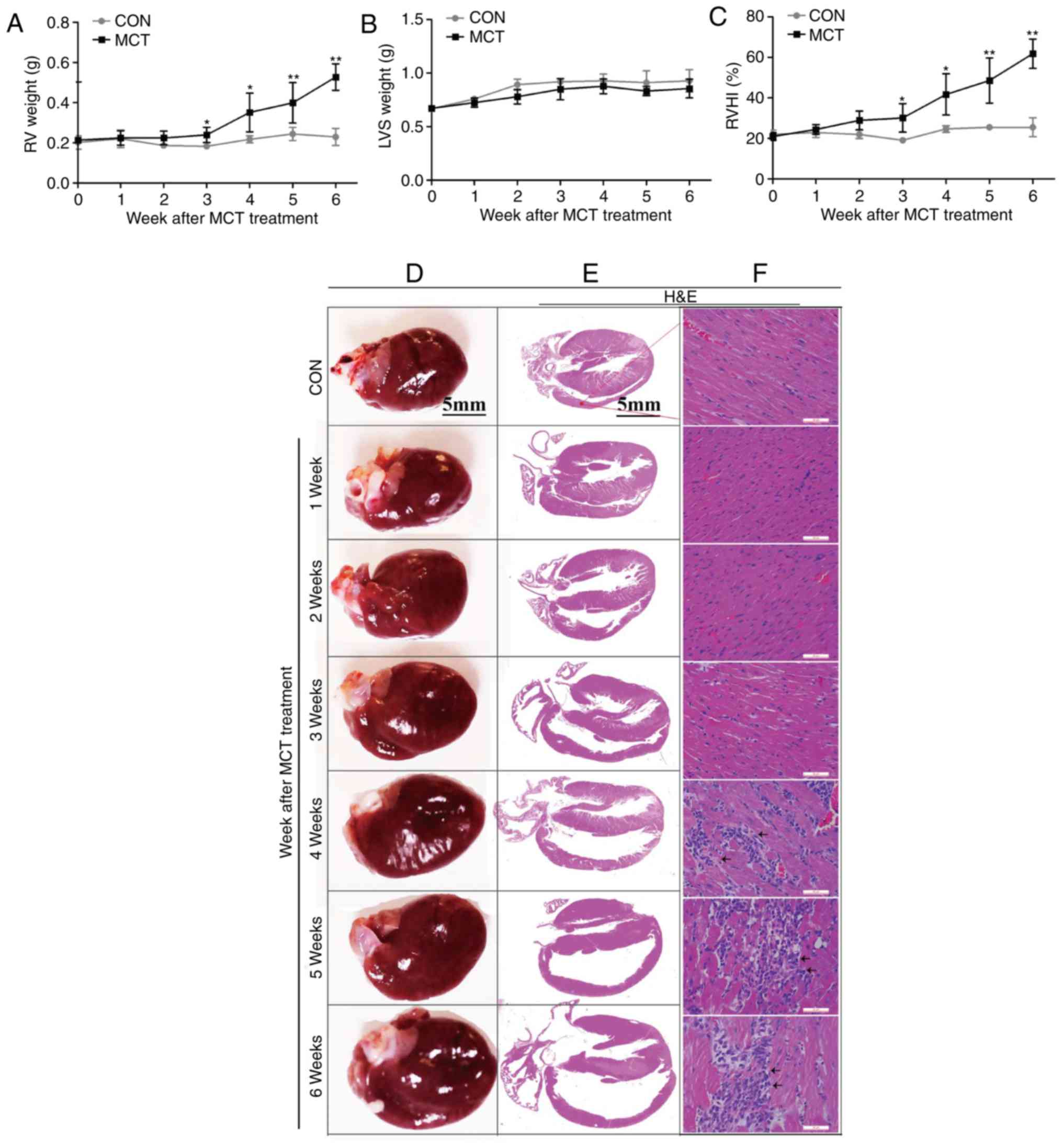 | Figure 3RV hypertrophy, dilatation and
inflammation occurred concurrently during the development of
pulmonary arterial hypertension. (A) The RV weight sharply
increased at weeks 3-6. (B) The left ventricular weight, including
the LVS, exhibited no significant change throughout the course of
the experiment in MCT-induced rats. (C) The RVHI increased at weeks
3-6. (D) Gradually enlarged heart size and progressive dilatation
of the right atrium and RV were observed on gross pathological
examination (scale bar, 5 mm). (E and F) Histomorphometric analysis
revealed a significant increase in diffuse inflammatory cell
infiltration and widening of the interstitial space associated with
progressive degeneration of cardiomyocytes from weeks 4-6
[indicated by arrows; scale bar, 5 mm in (E) and 50 μm in
(F)]. Values are expressed as the mean ± standard deviation (n=10).
*P<0.05 and **P<0.01 vs. the CON group.
MCT, monocrotaline; CON, control; RV, right ventricular; RVHI, RV
hypertrophy index; LVS, left ventricular and intraventricular
septum. |
GPCR signaling contributes to RV
remodeling
PCR array analysis demonstrated that pathological
changes of RV remodeling were accompanied with an upregulation of
genes associated with inflammation, hypertrophy, apoptosis and
fibrosis, including interleukin (IL)1b, IL-1 receptor type 2
(IL1R2), endothelin 1 (EDN1), connective tissue growth factor
(CTGF), matrix metalloproteinase 9 (MMP9) and serpin family E
member 1 (Serpine1) (P<0.05; Fig.
5). In addition, in MCT-induced rats, angiotensin receptor
(Agtr)-associated genes (Agt, Agtr1b and Agtr2) were upregulated at
weeks 1-3 and then downregulated compared with the CON group by
weeks 5 and/or 6 (Fig. 5). In
addition, the expression of β1-AR was downregulated by
weeks 5-6 (Fig. 5).
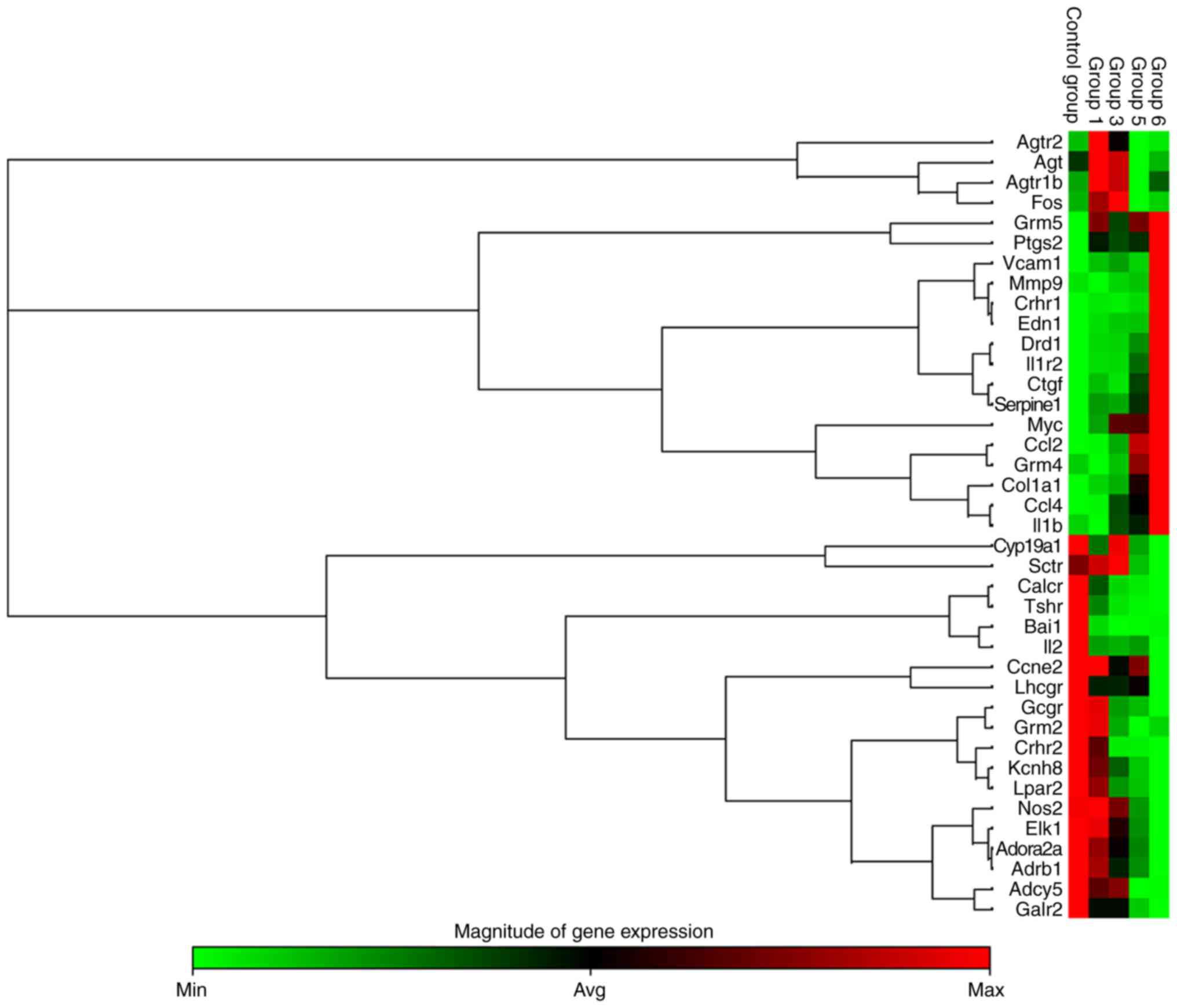 | Figure 5Heat map cluster representing 39
differentially expressed G protein-coupled receptor mRNAs from RV
tissue determined by reverse transcription-quantitative polymerase
chain reaction analysis. Differentially expressed mRNAs were
selected among those that displayed fold changes of >2 or
<0.5 compared with the control group (n=3 per group). Groups 1,
3, 5 and 6 correspond to the monocrotaline groups at weeks 1, 3, 5
and 6, respectively. Agtr2, angiotensin II receptor, type 2; Agt,
angiotensinogen; Agtr1b, angiotensin II receptor, type 1b; Fos, fos
proto-oncogene; Grm5, glutamate metabotropic receptor 5; Ptgs2,
prostaglandin-endoperoxide synthase 2; Vcam1, vascular cell
adhesion molecule 1; Mmp9, matrix metalloproteinase 9; Crhr1,
corticotropin releasing hormone receptor1; EDN1, endothelin 1;
Drd1, dopamine receptor D1; Il1r2, interleukin 1 receptor, type 2;
Ctgf, connective tissue growth factor; Serpine1, serpin family E
member 1; Myc, myc proto-oncogene; Ccl2, C-C motif chemokine ligand
2; Grm4, glutamate metabotropic receptor 4; Col1a1, collagen type I
α1 chain; Ccl4, C-C motif chemokine ligand 4; Il1b, interleukin 1β;
Cyp19a1, cytochrome P450, family 19, subfamily a, polypeptide 1;
Sctr, secretin receptor; Calcr, calcitonin receptor; Tshr, thyroid
stimulating hormone receptor; Bai1, brain-specific angiogenesis
inhibitor 1; Il2, interleukin 2; Ccne2, Cyclin E2; Lhcgr,
luteinizing hormone/choriogonadotropin receptor; Gcgr, glucagon
receptor; Grm2, glutamate metabotropic receptor 2; Crhr2,
corticotropin releasing hormone receptor 2; Kcnh8, potassium
voltage-gated channel subfamily H member 8; Lpar2, lysophosphatidic
acid receptor 2; Nos2, nitric oxide synthase 2; EIK-1, Ets-like
transcription factor-1; Adora2a, adenosine A2a receptor; Adrb1,
adrenoceptor β1; Adcy5, adenylate cyclase 5; Galr2, galanin
receptor 2; RV, right ventricular; min, minimum; max, maximum; avg,
average. |
β2-AR signaling is involved in
RV remodeling
To further investigate the molecular mechanisms of
RV remodeling induced by PAH, the expression of β1-AR
and β2-AR, as well as the phosphorylation of
β2-AR at two sites [pSer346 and pSer(355,356)] was
measured, as was the expression of PKA, CaMKII and GRK2 (Fig. 6A). The results demonstrated that
the expression of β1-AR exhibited a slight increasing
trend by weeks 2-3 and then rapidly declined by weeks 4-6 compared
with the value at week 3. The expression of β2-AR
exhibited a slight decreasing trend over time (Fig. 6B). The PKA-dependent level of
p-β2-AR (Ser346) initially remained unchanged and then
sharply increased by weeks 5-6, with a concomitant increase in the
expression of PKA and CaMKII (Fig. 6C
and D). However, the GRK-dependent level of p-β2-AR
[Ser(355,356)] was reduced by week 3 in comparison with that in the
CON group, and was increased by weeks 5-6, while the levels of GRK2
remained unchanged (Fig. 6C and
D).
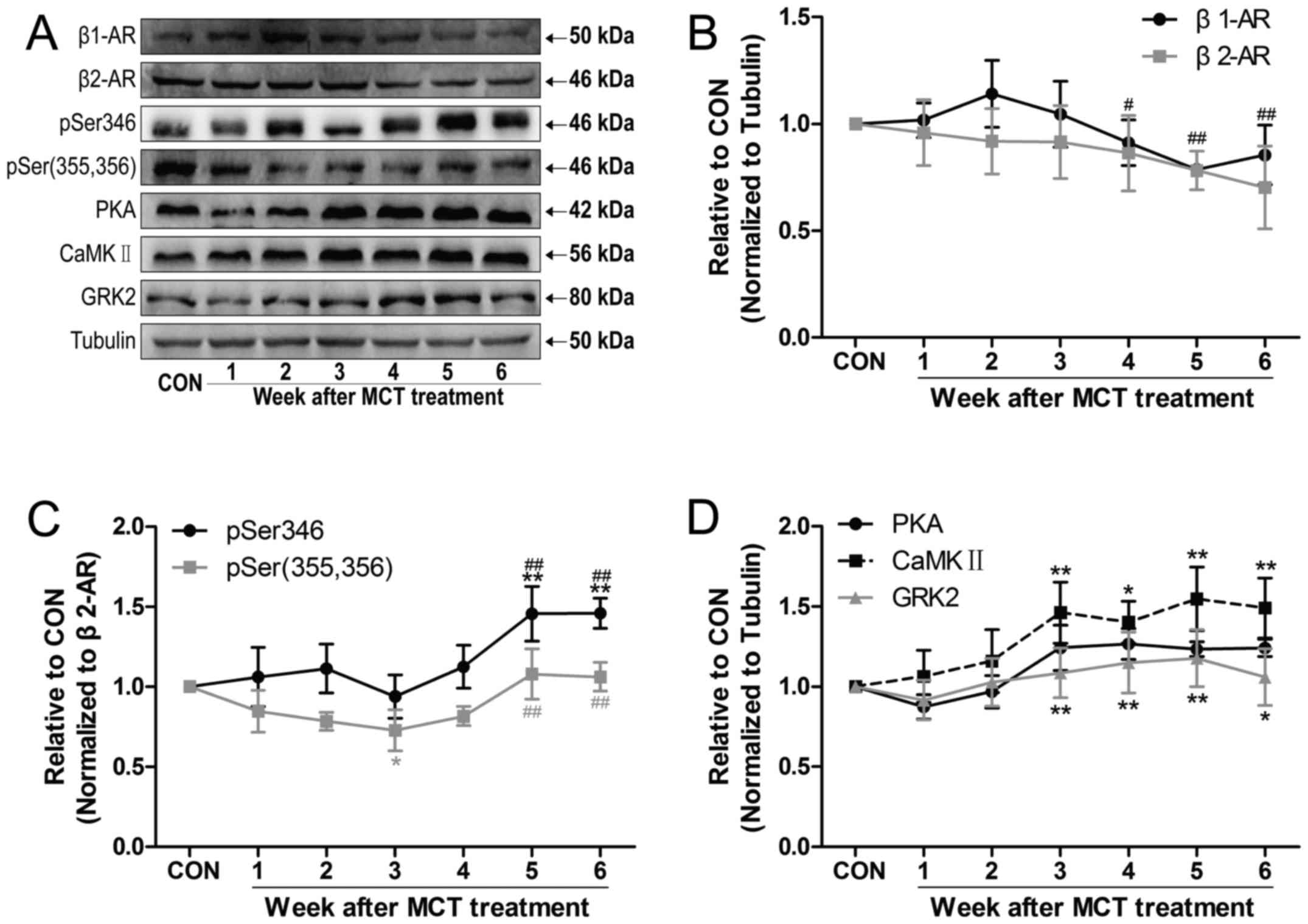 | Figure 6MCT treatment caused changes in the
relative protein levels of β-AR in the RV free wall. (A)
Representative western blot image of β-AR signaling-associated
proteins in different groups. (B-D) Time-dependent changes in (B)
β1-AR and β2-AR subtypes, (C) phosphorylation
of the β2-AR at the Ser346 and Ser(355,356) sites, and
(D) PKA and CaMKII (a PKA-mediated downstream target of β-AR). A
slight increasing trend was observed in the density of GRK2
expression. Values are expressed as the mean ± standard deviation
(n=5 per group). *P<0.05 and **P<0.01
vs. the CON group; #P<0.05 and ##P<0.01 vs. the
week 3 group. MCT, monocrotaline; CON, control; RV, right
ventricular; β-AR, β-adrenergic receptor; PKA, protein kinase A;
CaMKII, Ca2+/calmodulin-dependent kinase II; GRK2, G
protein-coupled receptor kinase 2. |
Discussion
In the present study, the RV transitioned from
initially adaptive remodeling caused by persistent PAH, to
hyperfunction and maladaptive remodeling, as evidenced by
hypertrophy, ventricular dilatation, inflammation, fibrosis and
apoptosis, during the course of the disease. MCT injection
initially resulted in functional and structural lung damage, which
was accompanied by LV hyperfunction and subsequent adaptive
hypertrophy of the RV. Consistently, in the early stages of PAH,
the expression of the pSer(355,356) site of β2-AR was
significantly decreased in rats with MCT-induced PAH. As the
disease progressed, concomitant with late maladaptive RV remodeling
and apoptosis, upregulation of GPCR signaling and further
β2-AR-stimulatory G protein (Gs)-PKA/CaMKII
signaling activation were observed in the late stages of PAH. The
key finding of the present study was that the adaptive and
maladaptive RV remodeling in the MCT-induced PAH model was
accompanied by a distinct evolution in β2-AR signaling,
which may facilitate the treatment of progressive RV failure during
PAH with appropriately timed drug intervention.
Following MCT treatment, structural damage and
functional impairment were initially observed in the lungs, thereby
producing irreversible PAH (increased PASP and PADP), further
leading to RV hypertrophy and failure, which manifested with a
marked elevation in RV weight and RVHI, hemodynamically by an
increase in RVSP and RVEDP, and histopathologically by evidenced
dilatation and inflammation. Of note, the present results, taken
together with those of associated studies (11,34,35), revealed enhanced RV performance
(increased +dP/dtmax and decreased −dP/dtmin)
in the late stages of PAH. However, despite this result, the RV
pumping function was reduced (36), likely due to the markedly elevated
afterload (increased PASP, PADP and RVSP) and preload (increased
RVEDP). Hyperfunction leads to increased energy consumption and
adverse cardiac remodeling in the failing heart. In the present
study, Masson and TUNEL staining further confirmed the exacerbation
of adverse RV remodeling, as evidenced by increased myocardial
fibrosis and more apparent myocyte apoptosis. Mechanically,
upregulation of GPCR-associated genes, including IL1b, IL1R2, EDN1,
CTGF, MMP9 and Serpine1, were partly responsible for aggravated
pathological alterations of the RV in the late stages of PAH.
Assessment of the physiology revealed a noteworthy yet differential
functional response of the heart to MCT. Although the RV systolic
and diastolic performance increased following remodeling, the
effects on the LV appeared to be different, with the initial
increase being followed by reduced and compromised ventricular
function (28). Overall, LV
failure may be attributed to global neurohormonal adaptations, as
defined by hyperactivity of the sympathetic nervous system (SNS)
and the renin-angiotensin-aldosterone system (RAAS) (37,38). Hyperactivity of the SNS and RAAS
were further demonstrated by upregulated angiotensin
receptor-associated genes (Agt, Agtr1b and Agtr2) in the RV, and
accelerated HR in the early stages of PAH. In advanced PAH, rats
that were injected with MCT developed resistance to the sustained
activation of SNS and RAAS, as validated by downregulated
angiotensin receptor-associated genes and β1-AR, as well
as slow HR in the late stages of PAH. In addition, an elevation in
BNP, which is considered a hallmark of HF (39,40), further confirmed the presence of
overt HF. Taken together, these results support that the
advancement of MCT-induced pulmonary inflammation and consolidation
resulted in PAH, and further led to adverse RV remodeling and
apoptosis and, eventually, overt HF.
A recent study suggested that nebivolol, a
β1-antagonist and β2,3-agonist, induced
partial relaxation of the pulmonary artery, altered PAH-associated
endothelial dysfunction in vitro and improved experimental
PAH in rats (26). In fact, the
benefits of nebivolol in rats administered MCT may be restricted in
the early stages (days 14-21) of PAH (7). Routinely, the prevalence of PAH
increases as chronic obstructive pulmonary disease (COPD) worsens
(5). β2-agonists
generally have bronchodilatory action and exert beneficial effects
on pulmonary circulatory hemodynamics and RV performance in
patients with COPD, hence delaying the occurrence and development
of PAH. Furthermore, the present study documented that the
GRK-dependent phosphorylation of β2-AR at Ser(355,356)
in the RV was significantly reduced, without changes in the
PKA-dependent expression of pSer346 of β2-AR in the
early stages of PAH. In addition, it is known that β2-AR
activation by catecholamines has a key role in promoting the M2
regulatory macrophage (anti-inflammatory) phenotype during
endotoxemia and acute lung injury through the phosphoinositide
3-kinase pathway, but not the canonical cyclic adenosine
monophosphate (cAMP)/PKA signaling pathway (41). Additionally, circulating
monocyte/macrophage lineage cells contribute significantly to the
pulmonary artery (PA) remodeling process in experimental PAH
(42). On the basis of these
findings, a preliminarily hypothesis may be that β2-AR
activation could promote the M2 regulatory macrophage phenotype to
improve pulmonary vascular remodeling in PAH. Thus, there is a
rationale for a potential benefit of β2-agonist therapy
in this stage.
As the disease progressed, however, cardiotoxic
effects develop with respect to β2-AR activation in the
failing heart (17-22,43,44). Under normal physiological
conditions, the predominant β1-AR subtype is the major
mediator of cardiac contractility. However, under pathological
conditions (failing heart, advanced age), the β1-AR
response to sustained high circulating catecholamine levels may
result in β1-AR downregulation and mediate myocyte
apoptosis through PKA-dependent or PKA-independent CaMKII
activation (1,14,15,37). By contrast, the contribution of
the more minor β2-AR subtype to cardiac contractility is
more prominent (45). This is an
important hypothesis, as β2-AR activation may be
associated with enhanced RV contractility mediated by
Gs-biased β2-AR (increased phosphorylation of
the pSer346 site)/PKA signaling activation in the late stages of
PAH. Moreover, Gs-biased β2-AR activation may
also change to a cell-wide cyclic adenosine monophosphate (cAMP)
signal propagation pattern and acquire the characteristics of the
β1-AR response in the failing heart, as evidenced by
PKA-dependent stimulation of CaMKII signaling in the present study,
thus promoting the HF phenotype (22).
In support of this result, compelling evidence has
demonstrated that β-blockade treatment prevented pathological RV
remodeling and improved RV function in MCT-induced PAH models
(25,27,28). Furthermore, several clinical
studies have reported the beneficial effect of β-blockade therapy
on PAH-RVF (46-48). In addition, β-blockade also
improved LV function (28,38,49).
Finally, a series of studies by our group have demonstrated that
the combination of the traditional Chinese medicines Fuzi
(Aconiti Lateralis radix praeparata, which may activate
β2-AR) and Beimu (Fritillariae Thunbergii bulbus)
significantly improved lung function and reduced pulmonary
histopathological changes in the early stages of PAH (29,50). However, as the disease progressed,
the combination of Fuzi and Beimu increased the risk of developing
severe cardiac adverse effects by synergistically activating the
cAMP-PKA-CaMKII signaling pathway in the late stages of the same
PAH model. These results suggest that the enhanced RV contractility
and concomitant activation of β2-AR signaling occur in
the late stages of PAH, which subsequently accelerates the adverse
RV remodeling and apoptosis during the progression of PAH.
Of note, the present study had certain limitations.
First, since β1-AR and β2-AR are associated
with cardiotoxic effects and acceleration of HF, further study is
required to demonstrate the causality of whether changes in
β1-AR or β2-AR signaling may be associated
with RV remodeling and apoptosis during the progression of PAH,
e.g. by using β-AR inhibitors in the same model or β-AR agonists in
similar experiments. The results of the present study indicated
changes in the expression of PKA, GRK2 and β-ARs proteins, which
may provide a novel theoretical approach according to which β-ARs
are the key factors leading to RV remodeling induced by PAH.
However, this role remains to be determined by gain- and
loss-of-function strategies (including overexpression and silencing
of these genes) in further studies. In addition, it is required to
further investigate whether the variability in the expression of
GRKs, including GRK-5 and -6, has any significance in MCT-treated
rat hearts. Furthermore it should be examined whether the dynamic
changes observed in RV are associated with concomitant changes in
the LV. Finally, the significance of inflammatory cells in the
pulmonary artery remodeling process is currently attracting
increasing attention (42). It is
also known that macrophage activation and β2-AR have an
important role in inflammation (41). The hypoxic rat model is
particularly useful in the evaluation of anti-inflammatory
therapies, since circulating monocytic cell populations
significantly contributing to the pulmonary artery remodeling
process is a key feature in this PAH model (42). However, the present study mainly
focuses on the association of β2-AR with the progression
of RV remodeling. Further investigation is required to confirm the
role of β2-AR through the anti-inflammatory M2
regulatory macrophage phenotype in PA remodeling, particularly in
an animal model of hypoxic PAH.
In conclusion, the present study observed initial
lung injury following MCT injection, which was accompanied by
unchanged PKA-mediated phosphorylation of β2-AR and
decreased GRK-mediated phosphorylation of β2-AR in the
early stages of PAH. As the disease progressed, a marked increase
in maladaptive RV remodeling and concomitant cardiomyocyte
apoptosis were observed in the late stages of PAH, which was
accompanied by GPCR signaling alterations and
β2-AR-Gs-PKA/CaMKII signaling activation.
Having established that the development of PAH in rats is
characterized by distinct evolutionary β2-AR signaling
changes, this knowledge must be applied in the clinical setting by
appropriately timed administration of either β2-agonists
or non-selective β-AR blockade therapy, to avoid the development of
PAH.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81773920), the
National Basic Research Program of China (973 program; grant nos.
2011CB505300 and 2011CB505302) and the Program for Changjiang
Scholars and Innovative Research Team in University (grant no.
IRT_14R41).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Ryan JJ, Huston J, Kutty S, Hatton ND,
Bowman L, Tian L, Herr JE, Johri AM and Archer SL: Right
ventricular adaptation and failure in pulmonary arterial
hypertension. Can J Cardiol. 31:391–406. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vonk-Noordegraaf A, Haddad F, Chin KM,
Forfia PR, Kawut SM, Lumens J, Naeije R, Newman J, Oudiz RJ,
Provencher S, et al: Right heart adaptation to pulmonary arterial
hypertension: Physiology and pathobiology. J Am Coll Cardiol.
62(Suppl 25): D22–D33. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hardziyenka M, Campian ME, Reesink HJ,
Surie S, Bouma BJ, Groenink M, Klemens CA, Beekman L, Remme CA,
Bresser P and Tan HL: Right ventricular failure following chronic
pressure overload is associated with reduction in left ventricular
mass: Evidence for atrophic remodeling. J Am Coll Cardiol.
57:921–928. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hsia HH and Haddad F: Pulmonary
hypertension: A stage for ventricular interdependence? J Am Coll
Cardiol. 59:2203–2205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee-Chiong TL Jr and Matthay RA: Pulmonary
hypertension and cor pulmonale in COPD. Semin Respir Crit Care Med.
24:263–272. 2003. View Article : Google Scholar
|
|
6
|
Montani D, Gunther S, Dorfmuller P, Perros
F, Girerd B, Garcia G, Jaïs X, Savale L, Artaud-Macari E, Price LC,
et al: Pulmonary arterial hypertension. Orphanet J Rare Dis.
8:972013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rubin LJ: The beta-adrenergic receptor in
pulmonary arterial hypertension: A novel therapeutic target. J Am
Coll Cardiol. 65:681–683. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mendes-Ferreira P, Santos-Ribeiro D, Adão
R, Maia-Rocha C, Mendes-Ferreira M, Sousa-Mendes C, Leite-Moreira
AF and Brás-Silva C: Distinct right ventricle remodeling in
response to pressure overload in the rat. Am J Physiol Hear Circ
Physiol. 311:H85–H95. 2016. View Article : Google Scholar
|
|
9
|
Rain S, Handoko ML, Trip P, Gan CT,
Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CA, Marcus JT,
Dorfmüller P, et al: Right ventricular diastolic impairment in
patients with pulmonary arterial hypertension. Circulation.
128:2025. 2013. View Article : Google Scholar
|
|
10
|
Campian ME, Verberne HJ, Hardziyenka M, de
Bruin K, Selwaness M, van den Hoff MJ, Ruijter JM, van Eck-Smit BL,
de Bakker JM and Tan HL: Serial noninvasive assessment of apoptosis
during right ventricular disease progression in rats. J Nucl Med.
50:1371–1377. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Paffett ML, Hesterman J, Candelaria G,
Lucas S, Anderson T, Irwin D, Hoppin J, Norenberg J and Campen MJ:
Longitudinal in vivo SPECT/CT imaging reveals morphological changes
and cardiopulmonary apoptosis in a rodent model of pulmonary
arterial hypertension. PLoS One. 7:e409102012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Ouyang M, Wang Q and Jian Z:
MicroRNA-142-3p inhibits hypoxia/reoxygenation-induced apoptosis
and fibrosis of cardiomyocytes by targeting high mobility group box
1. Int J Mol Med. 38:1377–1386. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bisognano JD, Weinberger HD, Bohlmeyer TJ,
Pende A, Raynolds MV, Sastravaha A, Roden R, Asano K, Blaxall BC,
Wu SC, et al: Myocardial-directed overexpression of the human
beta(1)-adrenergic receptor in transgenic mice. J Mol Cell Cardiol.
32:817–830. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Iwai-Kanai E, Hasegawa K, Araki M, Kakita
T, Morimoto T and Sasayama S: alpha- and beta-adrenergic pathways
differentially regulate cell type-specific apoptosis in rat cardiac
myocytes. Circulation. 100:305–311. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu
Q, Makarewich C, Ai X, Li Y, Tang A, et al: Cardiotoxic and
cardioprotective features of chronic β-adrenergic signaling. Circ
Res. 112:498–509. 2013. View Article : Google Scholar
|
|
16
|
Lymperopoulos A, Rengo G and Koch WJ:
Adrenergic nervous system in heart failure: Pathophysiology and
therapy. Circ Res. 113:739–753. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Woo AY, Song Y, Xiao RP and Zhu W: Biased
β2-adrenoceptor signalling in heart failure: Pathophysiology and
drug discovery. Br J Pharmacol. 172:5444–5456. 2015. View Article : Google Scholar
|
|
18
|
Daaka Y, Luttrell LM and Lefkowitz RJ:
Switching of the coupling of the beta2-adrenergic receptor to
different G proteins by protein kinase A. Nature. 390:88–91. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rengo G, Lymperopoulos A, Leosco D and
Koch WJ: GRK2 as a novel gene therapy target in heart failure. J
Mol Cell Cardiol. 50:785–792. 2011. View Article : Google Scholar
|
|
20
|
Salazar NC, Vallejos X, Siryk A, Rengo G,
Cannavo A, Liccardo D, De Lucia C, Gao E, Leosco D, Koch WJ and
Lymperopoulos A: GRK2 blockade with βARKct is essential for cardiac
β2-adrenergic receptor signaling towards increased contractility.
Cell Commun Signal. 11:642013. View Article : Google Scholar
|
|
21
|
Zhu W, Petrashevskaya N, Ren S, Zhao A,
Chakir K, Gao E, Chuprun JK, Wang Y, Talan M, Dorn GW II, et al:
Gi-biased β2AR signaling links GRK2 upregulation to heart failure.
Circ Res. 110:265–274. 2012. View Article : Google Scholar
|
|
22
|
Nikolaev VO, Moshkov A, Lyon AR, Miragoli
M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE and Gorelik J:
Beta2-adrenergic receptor redistribution in heart failure changes
cAMP compartmentation. Science. 327:1653–1657. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lang D, Holzem K, Kang C, Xiao M, Hwang
HJ, Ewald GA, Yamada KA and Efimov IR: Arrhythmogenic remodeling of
β2 versus β1 adrenergic signaling in the human failing heart. Circ
Arrhythmia Electrophysiol. 8:409–419. 2015. View Article : Google Scholar
|
|
24
|
Wang Y, Yuan J, Qian Z, Zhang X, Chen Y,
Hou X and Zou J: β2 adrenergic receptor activation governs cardiac
repolarization and arrhythmogenesis in a guinea pig model of heart
failure. Sci Rep. 5:76812015. View Article : Google Scholar
|
|
25
|
Bogaard HJ, Natarajan R, Mizuno S, Abbate
A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN
and Voelkel NF: Adrenergic receptor blockade reverses right heart
remodeling and dysfunction in pulmonary hypertensive rats. Am J
Respir Crit Care Med. 182:652–660. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Perros F, Ranchoux B, Izikki M, Bentebbal
S, Happé C, Antigny F, Jourdon P, Dorfmüller P, Lecerf F, Fadel E,
et al: Nebivolol for improving endothelial dysfunction, pulmonary
vascular remodeling, and right heart function in pulmonary
hypertension. J Am Coll Cardiol. 65:668–680. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ishikawa M, Sato N, Asai K, Takano T and
Mizuno K: Effects of a pure alpha/beta-adrenergic receptor blocker
on monocrotaline-induced pulmonary arterial hypertension with right
ventricular hypertrophy in rats. Circ J. 73:2337–2341. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Okumura K, Kato H, Honjo O, Breitling S,
Kuebler WM, Sun M and Friedberg MK: Carvedilol improves
biventricular fibrosis and function in experimental pulmonary
hypertension. J Mol Med. 93:663–674. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhuang P, Huang Y, Lu Z, Yang Z, Xu L, Sun
F, Zhang Y and Duan J: cAMP-PKA-CaMKII signaling pathway is
involved in aggravated cardiotoxicity during fuzi and beimu
combination treatment of experimental pulmonary hypertension. Sci
Rep. 6:349032016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Deten A, Millar H and Zimmer HG:
Catheterization of pulmonary artery in rats with an ultraminiature
catheter pressure transducer. Am J Physiol Circ Physiol.
285:H2212–H2217. 2003. View Article : Google Scholar
|
|
31
|
Schmittgen TD, Lee EJ, Jiang J, Sarkar A,
Yang L, Elton TS and Chen C: Real-time PCR quantification of
precursor and mature microRNA. Methods. 44:31–38. 2008. View Article : Google Scholar
|
|
32
|
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee
DH, Nguyen JT, Barbisin M, Xu L, Mahuvakar VR, Andersen MR, et al:
Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic
Acids Res. 33:e1792005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhuang P, Zhang Y, Cui G, Bian Y, Zhang M,
Zhang J, Liu Y, Yang X, Isaiah AO, Lin Y and Jiang Y: Direct
stimulation of adult neural stem/progenitor cells in vitro and
neurogenesis in vivo by salvianolic acid B. PLoS One. 7:e356362012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hessel MH, Steendijk P, den Adel B,
Schutte CI and van der Laarse A: Characterization of right
ventricular function after monocrotaline-induced pulmonary
hypertension in the intact rat. Am J Physiol Hear Circ Physiol.
291:H2424–H2430. 2006. View Article : Google Scholar
|
|
35
|
Werchan PM, Summer WR, Gerdes AM and
McDonough KH: Right ventricular performance after
monocrotaline-induced pulmonary hypertension. Am J Physiol Circ
Physiol. 256:H1328–H1336. 1989. View Article : Google Scholar
|
|
36
|
Kuehne T, Yilmaz S, Steendijk P, Moore P,
Groenink M, Saaed M, Weber O, Higgins CB, Ewert P, Fleck E, et al:
Magnetic resonance imaging analysis of right ventricular
pressure-volume loops in vivo validation and clinical application
in patients with pulmonary hypertension. Circulation.
110:2010–2016. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ameri P, Bertero E, Meliota G, Cheli M,
Canepa M, Brunelli C and Balbi M: Neurohormonal activation and
pharmacological inhibition in pulmonary arterial hypertension and
related right ventricular failure. Hear Fail Rev. 21:539–547. 2016.
View Article : Google Scholar
|
|
38
|
Usui S, Yao A, Hatano M, Kohmoto O,
Takahashi T, Nagai R and Kinugawa K: Upregulated neurohumoral
factors are associated with left ventricular remodeling and poor
prognosis in rats with monocrotaline-induced pulmonary arterial
hypertension. Circ J. 70:1208–1215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Giannakoulas G, Mouratoglou SA, Gatzoulis
MA and Karvounis H: Blood biomarkers and their potential role in
pulmonary arterial hypertension associated with congenital heart
disease. A systematic review. Int J Cardiol. 174:618–623. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Luchner A, Hengstenberg C, Löwel H,
Riegger GA, Schunkert H and Holmer S: Effect of compensated renal
dysfunction on approved heart failure markers direct comparison of
brain natriuretic peptide (BNP) and N-terminal pro-BNP.
Hypertension. 46:118–123. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Grailer JJ, Haggadone MD, Sarma JV,
Zetoune FS and Ward PA: Induction of M2 regulatory macrophages
through the β2-adrenergic receptor with protection during
endotoxemia and acute lung injury. J Innate Immun. 6:607–618. 2014.
View Article : Google Scholar :
|
|
42
|
Burke DL, Frid MG, Kunrath CL, Karoor V,
Anwar A, Wagner BD, Strassheim D and Stenmark KR: Sustained hypoxia
promotes the development of a pulmonary artery-specific chronic
inflammatory microenvironment. Am J Physiol Lung Cell Mol Physiol.
297:L238–L250. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Baker AJ: Adrenergic signaling in heart
failure: A balance of toxic and protective effects. Pflugers Arch.
466:1139–1150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fajardo G, Zhao M, Urashima T, Farahani S,
Hu DQ, Reddy S and Bernstein D: Deletion of the β2-adrenergic
receptor prevents the development of cardiomyopathy in mice. J Mol
Cell Cardiol. 63:155–164. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rybin VO, Pak E, Alcott S and Steinberg
SF: Developmental changes in beta2-adrenergic receptor signaling in
ventricular myocytes: The role of Gi proteins and caveolae
microdomains. Mol Pharmacol. 63:1338–1348. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
So PP, Davies RA, Chandy G, Stewart D,
Beanlands RS, Haddad H, Pugliese C and Mielniczuk LM: Usefulness of
beta-blocker therapy and outcomes in patients with pulmonary
arterial hypertension. Am J Cardiol. 109:1504–1509. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Thenappan T, Roy SS, Duval S,
Glassner-Kolmin C and Gomberg-Maitland M: β-blocker therapy is not
associated with adverse outcomes in patients with pulmonary
arterial hypertension: A propensity score analysis. Circ Hear Fail.
7:903–910. 2014. View Article : Google Scholar
|
|
48
|
Bandyopadhyay D, Bajaj NS, Zein J, Minai
OA and Dweik RA: Outcomes of β-blocker use in pulmonary arterial
hypertension: A propensity-matched analysis. Eur Respir J.
46:750–760. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chatterjee S, Udell JA, Sardar P,
Lichstein E and Ryan JJ: Comparable benefit of β-blocker therapy in
heart failure across regions of the world: Meta-analysis of
randomized clinical trials. Can J Cardiol. 30:898–903. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang Z, Lu ZQ, Zhang YJ, Li YB, Wang ZY,
Zhang YL, Zhuang PW and Bai G: Looking for agonists of β2
adrenergic receptor from Fuzi and Chuanwu by virtual screening and
dual-luciferase reporter assay. J Asian Nat Prod Res. 18:550–561.
2016. View Article : Google Scholar
|