Introduction
Aging causes a progressive loss of tissue function
processes (1) marked by numerous
common hallmarks, including genome instability, deregulated
nutrient sensing, molecular damage, telomere attrition, epigenetic
and transcriptional changes, inflammation, cell death and
senescence, and metabolic dysfunction (2,3).
Cellular senescence refers to a process that imposes permanent
proliferative arrest in response to various stressors, emerging as
one of the most important contributors to age-associated disease
and an attractive target for therapy (4,5).
The mechanism underlying senescence remains largely unknown, which
makes it challenging to determine the events involved in aging. An
enduring potential explanation is oxidative stress. Excessive
levels of intracellular reactive oxygen species (ROS) result in
age-associated characteristics, including DNA damage, proteins
oxidation, lipids degradation and increased ROS production,
culminating in significant cellular injury (6). In the nervous system, accumulation
of ROS contributes to the age-associated loss of cognitive, sensory
and motor function (7).
Sirtuins are a conserved family of deacetylase
proteins, which are among the first genes reported to extend
lifespan (8,9). Sirtuin 3 (Sirt3), a mitochondrial
deacetylase (10), has been
reported to regulate major mitochondrial biological processes,
including ATP generation, ROS detoxification, nutrient oxidation,
mitochondrial dynamics and the unfolded protein response (11-15) by removing acetyl modifications
from mitochondrial proteins (10,16) or other acyl modifications and
histone crotonylation (17,18). A number of mitochondrial proteins
involved in aging process are in an acetylated form (19) and are substrates of Sirt3
(10), suggesting that Sirt3 may
mediate a broad spectrum of protection against ROS-induced
aging.
Adjudin, formerly termed AF-2364
[1-(2,4-dichlorobenzyl) 1H-indazole-3-carbo-hydrazide] (20), exhibits reversible
anti-spermatogenic activity through disruption of adherent
junctions of premature sperm to seminiferous epithelium (21,22). We have previously reported that
adjudin reduced ischemia-induced neuroinflammation through the
nuclear factor-κB (NF-κB) pathway (23) and reduced loss of
gentamycin-induced rodent cochlear hair cells through the Sirt3-ROS
axis (24). The current study
aimed to investigate whether adjudin has anti-aging effects and the
underlying mechanisms involved.
Materials and methods
Reagents and animals
Hydroxyurea (HU) and reactive oxygen species (in
dimethyl sulfoxide) were purchased from Sinopharm Chemical Reagent
Co., Ltd. (Beijing, China) and Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany), respectively. Adjudin was provided by Mary M.
Wohlford Laboratory (Population Council, New York, USA). Wild-type
(WT) C57BL/129 mice and Sirt3-knockout (KO) C57BL/129 mice were
acquired from the Jackson Laboratory (Sacramento, CA, USA). Animal
procedures for this research were based on the Institutional Animal
Care and Use Committee of Shanghai Jiao Tong University (Shanghai,
China).
Cell culture and mouse embryo fibroblast
(MEF) isolation
MEFs were isolated from embryonic 13.5 (E13.5) WT or
Sirt3-KO mice. This study was approved by the Bioethics Committee
of School of Biomedical Engineering, Shanghai Jiao Tong University.
The embryos, excluding the head and visceral tissues, were washed
with phosphate-buffered saline (PBS), minced with scissor and then
transferred into 0.1 mM trypsin/1 mM EDTA solution. After
incubation at 37°C for 20 min, twice the amount of medium was added
and cells were cultured in Dulbecco's modified Eagle's medium
(DMEM) containing 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and antibiotics (penicillin,
100 U/ml; 100 µg/ml, streptomycin) at 37°C in a humidified
incubator with 5% CO2.
Cell viability assay
Cell viability was determined using Cell Counting
kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). Briefly, ~10,000 MEFs were seeded into one well of 96-well
plates. Different concentrations of adjudin were tested both with
and without hydroxyurea (24 h, 6 mM: 0, 5, 10, 20 and 40
µM). The cells were pre-treaeted with adjudin for 1 h and
then incubated with/without hydroxyurea for 24 h. Following
treatment, CCK-8 solution was added to each well at the dilution of
1:10. After incubation for ~1.5 h, the absorbance at 450 nm was
measured using a microplate reader (Synergy2; BioTek Instruments,
Inc., Winooski, VT, USA).
Senescence-associated β-galactosidase
(SA-β-gal) staining
Cellular senescence was determined by SA-β-gal
staining with senescence-associated β-galactosidase staining kit
(Beyotime Institute of Biotechnology, Haimen, China). Cells were
fixed with 4% paraformaldehyde for 15 min and washed with PBS three
times. Subsequently, cells were incubated overnight at 37°C in
darkness with the working solution containing 0.05 mg/ml
5-bromo-4-chloro-3-indolyl-b-d-galactopyranoside (X-gal).
Western blot analysis
Western blotting was performed as previously
described (25). Cells were lysed
in radioimmuno-precipitation assay lysis buffer (EMD Millipore,
Billerica, MA, USA) containing Complete Protease Inhibitor Cocktail
(1:100) and 2 mM phenylmethylsulfonyl fluoride. The protein
concentration was quantified by bicinchoninic acid protein assay
kit (Pierce; Thermo Fisher Scientific, Inc.). Total protein (30
µg) was separated by 10% SDS-PAGE and then transferred to a
0.45 µm nitrocellulose membrane (EMD Millipore). Following
the incubation with primary antibodies at 4°C overnight, the
membrane was hybridized with horseradish peroxidase-conjugated
secondary antibody (1:5,000 dilution; 111-035-003; Jackson
Laboratory) at room temperature for 1 h. Protein signals were
visualized by enhanced chemiluminescence detection. The primary
antibodies used were as follows: p16 (1:1,000 dilution; ab51243)
and p21 (1:1,000 dilution; ab109199; both from Abcam, Shanghai,
China); Sirt3 (1:1,000 dilution; 5490S; Cell Signaling Technology,
Inc., Danvers, MA, USA); Sirt6 (1:1,000 dilution; ab191385; Abcam);
MAPK family antibody [extracellular signal-regulated kinase (ERK),
p38, c-Jun N-terminal kinase (JNK); 9926; Cell Signaling
Technology, Inc.]; phospho-MAPK family antibody [phospho-ERK,
phospho-p38, phospho-JNK; 1:1,000 dilution; Cell Signaling
Technology, Inc.)]; manganese superoxide dismutase (SOD2; 1:1,000;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA); Akt (1:1,000;
ab8805), phospho-Akt (1:1,000; ab38449; both from Abcam); forkhead
box O3a (Foxo3a; 1:1,000; 12829; Cell Signaling Technology, Inc.);
and β-tubulin (1:1,000; ab6046; Abcam).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from MEFs with the use of
RNAiso Plus (Takara Biotechnology Co., Ltd., Dalian, China), and
first strand cDNA was synthesized from 1 µg of total RNA
using a PrimeScript RT reagent kit (Takara Biotechnology Co.,
Ltd.). RT-qPCR was performed on an ABI 7900HT (Thermo Fisher
Scientific, Inc.) by using SYBR Premix Ex Taq (Takara Biotechnology
Co., Ltd.) according to the following protocol: 95°C for 30 sec; 40
cycles consisting of 95°C for 5 sec and 60°C for 30 sec, 95°C for
15 sec and 60°C for 1 min; and 95°C for 15 sec. Primers used were
as follows: mSirt1 (sense, 5′-TAG TCC TTC CTA CCC CAA TTTCC-3′ and
antisense, 5′-TTG GTC CTT AGC CAC TCC TTC-3′); mSirt2 (sense,
5′-GCC TGG GTT CCC AAA AGGAG-3′ and antisense, 5′-GAG CGG AAG TCA
GGG ATACC-3′); mSirt3 (sense, 5′-ATC CCG GAC TTC AGA TCCCC-3′ and
antisense, 5′-CAA CAT GAA AAA GGG CTT GGG-3′); mSirt4 (sense,
5′-GTG GAA GAA TAA GAA TGA GCGGA-3′ and antisense, 5′-GGC ACA AAT
AAC CCC GAGG-3′); mSirt5 (sense, 5′-CTC CGG GCC GAT TCA TTTCC-3′
and antisense, 5′-GCG TTC GCA AAA CAC TTCCG-3′); mSirt6 (sense,
5′-ATG TCG GTG AAT TAT GCA GCA-3′ and antisense, 5′-GCT GGA GGA CTG
CCA CATTA-3′); mSirt7 (sense, 5′-AGC ATC ACC CGT TTG CATGA-3′ and
antisense, 5′-GGC AGT ACG CTC AGT CACAT-3′); mSOD2 (sense, 5′-CAG
ACC TGC CTT ACG ACT ATGG-3′ and antisense, 5′-CTC GGT GGC GTT GAG
ATT GTT-3′); m ribosomal protein, large P0 (mRplp0; sense 5′-AGA
TTC GGG ATA TGC TGT TGGC-3′ and antisense, 5′-TCG GGT CCT AGA CCA
GTG TTC-3′). RT-qPCR was performed out in triplicate and the
results are presented as the Cq values. The mean Cq value was
calculated, and the ΔCq value was determined as the mean Cq value
for the target gene minus the mean Cq value for mRplp0. Relative
mRNA expression was calculated using the 2-ΔΔCq
method.
ROS assay
Following treatment, cells were washed with PBS.
Dicholorofluorescein diacetate (DCF-DA; Beyotime Institute of
Biotechnology) was diluted in FBS-free DMEM to 10 µM and
then added to each well. After incubation for 0.5 h, cells were
washed with PBS three times, and the fluorescence was detected
using a FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA) flow
cytometer at an excitation wavelength of 488 nm and an emission
wavelength of 535 nm.
Statistical analysis
Data are presented as the mean ± standard deviation.
Multiple comparisons were analyzed by one-way analysis of variance
followed by Tukey's post hoc test. Statistical analyses were
performed using GraphPad Prism 5 (GraphPad Software, Inc., La
Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Adjudin delays HU-induced cellular
senescence
Cells were treated with HU, which inhibits
ribonucleotide reductase activity (26,27), to produce an in vitro
senescence model. HU-induced senescence has been widely used to
mimic cell aging (28-34). MEFs were pretreated with adjudin
for 1 h, followed by a 24-h incubation period with 6 mM HU. As
presented in Fig. 1A, cell
viability following treatment with the indicated concentrations of
adjudin was not significantly different from the control group in
the absence of HU, while a slight elevation in cell survival was
observed when treated with 20 and 40 µM adjudin in the
presence of HU.
Subsequently, whether adjudin could affect cellular
senescence was determined. The results demonstrated that
pretreatment with adjudin (10, 20 and 40 µM) for 1 h reduced
the percentage of HU-induced SA-β-gal-positive cells at a
dose-dependent manner (Fig. 1B and
C). Furthermore, levels of p16 and p21, two cyclin-dependent
kinase inhibitors considered as senescence markers (35,36), were detected by western blot. HU
increased the expression of p16 and p21, which was notably
decreased by pretreatment with 40 µM adjudin compared with
HU alone (Fig. 1D).
Adjudin elevates Sirt3 expression and
suppresses ROS level
Previous studies have implicated sirtuins as key
mediators of caloric restriction, which is known to inhibit
senescence (37). The effect of
adjudin on sirtuin expression was examined, and adjudin
significantly upregulated mRNA levels of Sirt3 and Sirt6, but there
was no change in Sirt6 protein level between different groups (data
not shown).
Adjudin has been reported to protect cochlear hair
cell via Sirt3-ROS pathway (24)
and Sirt3 mediates antioxidant defense and metabolic adaptation
that greatly influences mammalian lifespan (37), so we speculate whether adjudin
delays senescence through Sirt3-mediated inhibition of ROS
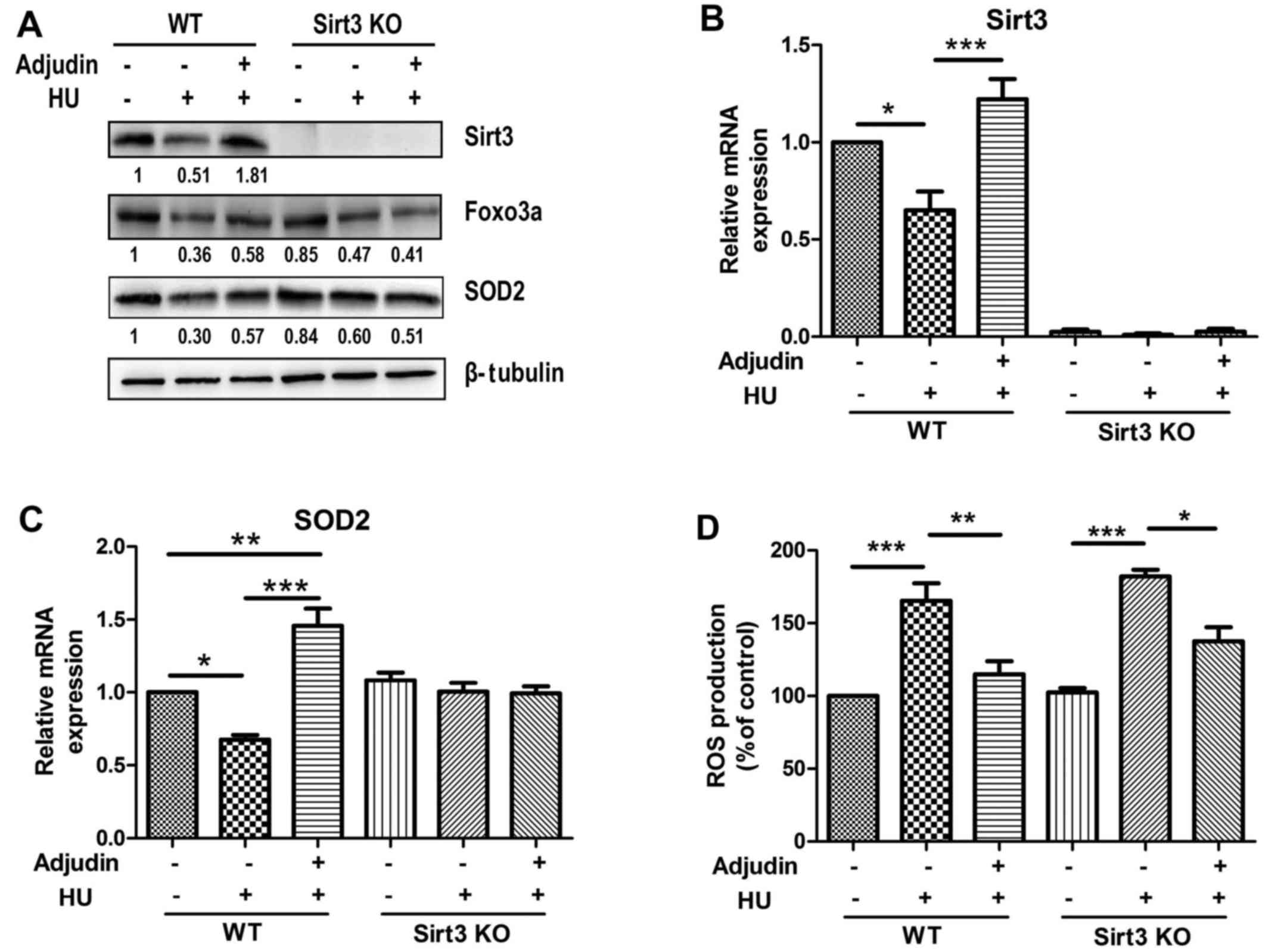
production. Indeed, Sirt3 protein level exhibited a 56% decline
following HU treatment. In addition, Foxo3a and SOD2, two target
proteins of Sirt3 closely associated with ROS, also presented a 73
and 65% decline in senescent cells, relatively. However,
pretreatment with 40 µM adjudin significantly counteracted
those changes in the presence of HU in MEFs (Fig. 2A). Similar results were also
observed at the mRNA level for Sirt3 and SOD2 (Fig. 2B and C).
Subsequently, it was examined whether adjudin could
reduce ROS production by using the DCF-DA method. HU stimulation
resulted in accumulation of intracellular ROS, which was alleviated
by adjudin treatment (Fig.
2D).
Sirt3 mediates anti-senescence effect of
adjudin
To validate the role of Sirt3 in the anti-senescence
effects of adjudin, MEFs from WT and Sirt3-KO mice were isolated.
Both WT and Sirt3-KO MEFs exhibited strong SA-β-gal activity
following exposure to HU. Notably, adjudin co-treatment with HU
caused a 30% reduction in SA-β-gal-positive cells in Sirt3-KO MEFs
compared to a 60% reduction in SA-β-gal-positive cells in WT MEFs,
indicating that Sirt3 may be involved in the anti-aging property of
adjudin (Fig. 3A and B).
In agreement with the SA-β-gal staining results, p16
and p21 levels were also induced by HU in WT and Sirt3-KO MEFs.
Adjudin treatment suppressed the upregulation of p16 and p21
expression by HU in WT MEFs, while the effect was not as potent in
Sirt3-KO MEFs, demonstrating that Sirt3 mediated, at least
partially, the anti-senescence effect of adjudin (Fig. 3C).
Adjudin exerts anti-senescence effects
via Sirt3/ROS
Sirt3 deficiency prevented adjudin from elevating
Foxo3a and SOD2 levels with HU treatment compared with the effects
in WT MEFs (Fig. 4A-C). To
determine whether Sirt3 influences ROS production, experiments were
performed using DCF-DA. Intracellular ROS levels were higher in
Sirt3-KO MEFs than in WT MEFs following treatment with HU and
adjudin, suggesting that Sirt3 is, at least partially, required for
adjudin to perform antioxidant activity (Fig. 4D). This may suggest that other
signaling pathways may be involved in this process.
Adjudin decreases phosphorylation of p38
mitogen-activated protein kinase (MAPK)
ROS generation activates the MAPK pathway, thus, it
was determined whether adjudin affects the MAPK pathway. As
presented in Fig. 5A and B,
stimulation with HU induced phosphorylation of p38 at the early
time point (1 h), which was suppressed by adjudin in WT and
Sirt3-KO MEFs, indicating that Sirt3 and p38 play independent
roles. However, levels of phosphorylated ERK or JNK were not
affected by treatment with adjudin (Fig. 5A). Phosphorylation of Akt was not
affected either (Fig. 5A).
 | Figure 5Adjudin suppress the phosphorylation
of p38. (A) Cells were treated with HU for 1 or 24 h. Cell lysates
were analyzed by immunoblotting with antibodies specific to p-p38
and p38, p-JNK and JNK, p-Akt and Akt, p-ERK1/2 and ERK1/2, and
β-tubulin. (B) WT and Sirt3 KO cells were treated with HU for 24 h.
Western blot analysis of p-p38 and p38 were performed. HU
hydroxy-urea; p-, phospho; t-, total; JNK, c-Jun N-terminal kinase;
ERK, extracellular signal-regulated kinase; WT, wild-type; Sirt3,
sirtuin; KO knockout. |
Discussion
The current study identified a new role of adjudin
in delaying cellular senescence, demonstrated by reduced
SA-β-gal-positive cells, and p16 and p21 levels. The anti-aging
property of adjudin was demonstrated associated with Sirt3-mediated
upregulation of Foxo3a and SOD2 expression and attenuation of ROS
production, which was validated using Sirt3-KO MEFs.
Mammalian sirtuins (Sirt1-7) are a family of highly
conserved NAD+-dependent deacetylases, which are
involved in numerous fundamental cellular processes, including
metabolic regulation, genomic stability maintenance, DNA repair and
stress responses (12). Previous
studies have revealed that Sirt3 KO mice spontaneously develop or
have accelerated progression of multiple age-associated
pathologies, including metabolic syndrome, cancer, cardiovascular
diseases, and neurodegenerative diseases (38). Sirt3 has been reported to block
aging-associated tissue fibrosis (39) and prevent hearing loss during
caloric restriction by stimulating isocitrate dehy-drogenase 2 to
convert NADP to NADPH in mitochondria, leading to decreased ROS
production (40). The results of
the current study illustrated that adjudin may upregulate Sirt3
expression in MEFs and in turn counteract premature senescence
induced by HU, which supports with the role of Sirt3 in aging and
metabolism.
Foxo3a and SOD2 are the two well-established
substrates of Sirt3. Foxo3a forms a physical interaction with Sirt3
in mitochondria, and overexpression of Sirt3 increases DNA-binding
activity and targeted gene expression of Foxo3a (41). Sirt3 deacetylates Foxo3 at K271
and K290, leading to upregulation of a set of genes that are
essential for mitochondrial homeo-stasis. Consequently,
mitochondrial reserve capacity is ensured in response to oxidative
damage (42). In addition, Sirt3
deacetylates two important lysine residues on SOD2, thus promoting
its antioxidant capacity to reduce oxidative stress damage and
extend life span during caloric restriction (43,44). The findings demonstrated that
adjudin increased the expression of Foxo3a and SOD2 in WT MEFs;
however, this effect was not observed in Sirt3-KO MEFs, indicating
Sirt3 is required for the regulation of Foxo3a and SOD2 by
adjudin.
Excessive accumulation of ROS and oxidative stress
damage are the main causes of age-associated diseases (45,46). Mitochondrial ROS is linked to the
elevation of pro-inflammatory mediators and susceptibility to
pathological conditions (47).
Thus, the antioxidant property of adjudin in HU-stimulated MEFs was
examined. DCF-DA signals revealed lower intracellular ROS levels in
adjudin-treated cells, implying contribution to delayed cellular
senescence driven by the antioxidant property of adjudin.
In the current study, adjudin also suppressed
HU-induced phosphorylation of p38, and may have roles independent
of Sirt3, as lack of Sirt3 in the KO cells did not affect
phosphorylation of p38, nor completely abolish the anti-senescence
effect of adjudin. It has been reported that p53 restrains
constitutive activation of p38 MAPK, preventing the
senescence-associated phenotype (48). Another possibility is the
MAPK-nuclear factor-κB (NF-κB) cascade. Our previous work
demonstrated that adjudin inhibited neuroinflammation via
attenuation of NF-κB signaling pathway (23), which functions as a regulator of
redox-sensitive gene expression (49,50).
In conclusion, the results of the current study
demonstrated that adjudin upregulated Sirt3 expression, thus
raising Foxo3a and SOD2 levels and reducing ROS production to delay
cellular senescence. Further investigation on the role of adjudin
in animal models is necessary to confirm that adjudin may be a
promising therapeutic option to age-associated diseases.
Acknowledgements
This study was supported by grants from the Ministry
of Science and Technology (grant no. 2013CB945604), the National
Key Grant (grant no. 2016YFC0906400), the National Natural Science
Foundation, China (grant no. 31270032) and the SJTU funding (grant
no. YG2012ZD05).
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
ROS
|
reactive oxygen species
|
|
HU
|
hydroxyurea
|
|
MEF
|
mouse embryo fibroblast
|
|
SA-β-gal
|
senescence-associated
β-galactosidase
|
|
Foxo3a
|
forkhead box O3a
|
|
SOD2
|
manganese superoxide dismutase
|
|
CCK-8
|
cell counting kit-8
|
References
|
1
|
Flatt T: A new definition of aging? Front
Genet. 3:1482012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liao CY and Kennedy BK: SIRT6, oxidative
stress, and aging. Cell Res. 26:143–144. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
López-Otín C, Blasco MA, Partridge L,
Serrano M and Kroemer G: The hallmarks of aging. Cell.
153:1194–1217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Childs BG, Durik M, Baker DJ and van
Deursen JM: Cellular senescence in aging and age-related disease:
From mechanisms to therapy. Nat Med. 21:1424–1435. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Campisi J: Aging, cellular senescence, and
cancer. Annu Rev Physiol. 75:685–705. 2013. View Article : Google Scholar
|
|
6
|
Harman D: The biologic clock: The
mitochondria? J Am Geriatr Soc. 20:145–147. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin MT and Beal MF: Mitochondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frye RA: Characterization of five human
cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins
(sirtuins) metabolize NAD and may have protein
ADP-ribosyltransferase activity. Biochem Biophys Res Commun.
260:273–279. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaeberlein M, McVey M and Guarente L: The
SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces
cerevisiae by two different mechanisms. Genes Dev. 13:2570–2580.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lombard DB, Alt FW, Cheng HL, Bunkenborg
J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D,
Murphy A, et al: Mammalian Sir2 homolog SIRT3 regulates global
mitochondrial lysine acetylation. Mol Cell Biol. 27:8807–8814.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ahn BH, Kim HS, Song S, Lee IH, Liu J,
Vassilopoulos A, Deng CX and Finkel T: A role for the mitochondrial
deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad
Sci USA. 105:14447–14452. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Samant SA, Zhang HJ, Hong Z, Pillai VB,
Sundaresan NR, Wolfgeher D, Archer SL, Chan DC and Gupta MP: SIRT3
deacetylates and activates OPA1 to regulate mitochondrial dynamics
during stress. Mol Cell Biol. 34:807–819. 2014. View Article : Google Scholar :
|
|
13
|
Hirschey MD, Shimazu T, Goetzman E, Jing
E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S,
Ilkayeva OR, et al: SIRT3 regulates mitochondrial fatty-acid
oxidation by reversible enzyme deacetylation. Nature. 464:121–125.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jing E, O'Neill BT, Rardin MJ,
Kleinridders A, Ilkeyeva OR, Ussar S, Bain JR, Lee KY, Verdin EM,
Newgard CB, et al: Sirt3 regulates metabolic flexibility of
skeletal muscle through reversible enzymatic deacetylation.
Diabetes. 62:3404–3417. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Papa L and Germain D: SirT3 regulates the
mitochondrial unfolded protein response. Mol Cell Biol. 34:699–710.
2014. View Article : Google Scholar :
|
|
16
|
Onyango P, Celic I, McCaffery JM, Boeke JD
and Feinberg AP: SIRT3, a human SIR2 homologue, is an NAD-dependent
deacetylase localized to mitochondria. Proc Natl Acad Sci USA.
99:13653–13658. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Feldman JL, Baeza J and Denu JM:
Activation of the protein deacetylase SIRT6 by long-chain fatty
acids and widespread deacylation by mammalian sirtuins. J Biol
Chem. 288:31350–31356. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bao X, Wang Y, Li X, Li XM, Liu Z, Yang T,
Wong CF, Zhang J, Hao Q and Li XD: Identification of ‘erasers’ for
lysine crotonylated histone marks using a chemical proteomics
approach. eLife. Nov 4–2014. View Article : Google Scholar
|
|
19
|
Kim SC, Sprung R, Chen Y, Xu Y, Ball H,
Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al: Substrate and
functional diversity of lysine acetylation revealed by a proteomics
survey. Mol Cell. 23:607–618. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Floridi A, Paggi MG, D'Atri S, De Martino
C, Marcante ML, Silvestrini B and Caputo A: Effect of lonidamine on
the energy metabolism of Ehrlich ascites tumor cells. Cancer Res.
41:4661–4666. 1981.PubMed/NCBI
|
|
21
|
Cheng CY, Silvestrini B, Grima J, Mo MY,
Zhu LJ, Johansson E, Saso L, Leone MG, Palmery M and Mruk D: Two
new male contraceptives exert their effects by depleting germ cells
prematurely from the testis. Biol Reprod. 65:449–461. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xia W and Geng K: A sirtuin activator and
an anti-inflammatory molecule-multifaceted roles of adjudin and its
potential applications for aging-related diseases. Semin Cell Dev
Biol. 59:71–78. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shao J, Liu T, Xie QR, Zhang T, Yu H, Wang
B, Ying W, Mruk DD, Silvestrini B, Cheng CY, et al: Adjudin
attenuates lipopolysaccharide (LPS)- and ischemia-induced
microglial activation. J Neuroimmunol. 254:83–90. 2013. View Article : Google Scholar
|
|
24
|
Quan Y, Xia L, Shao J, Yin S, Cheng CY,
Xia W and Gao WQ: Adjudin protects rodent cochlear hair cells
against gentamicin ototoxicity via the SIRT3-ROS pathway. Sci Rep.
5:81812015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xia W, Mruk DD and Cheng CY: C-type
natriuretic peptide regulates blood-testis barrier dynamics in
adult rat testes. Proc Natl Acad Sci USA. 104:3841–3846. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Krakoff IH, Brown NC and Reichard P:
Inhibition of ribonu-cleoside diphosphate reductase by hydroxyurea.
Cancer Res. 28:1559–1565. 1968.PubMed/NCBI
|
|
27
|
Moore EC and Hurlbert RB: The inhibition
of ribonucleoside diphosphate reductase by hydroxyurea, guanazole
and pyrazolo-imidazole (IMPY). Pharmacol Ther. 27:167–196. 1985.
View Article : Google Scholar
|
|
28
|
Yeo EJ, Hwang YC, Kang CM, Kim IH, Kim DI,
Parka JS, Choy HE, Park WY and Park SC: Senescence-like changes
induced by hydroxyurea in human diploid fibroblasts. Exp Gerontol.
35:553–571. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park MS, Choi JS, Lee W, Yang YJ, Kim J,
Lee GJ, Kim SS, Park SH, Kim SC, et al: Pharmacogenomic analysis
indicates potential of 1,5-isoquinolinediol as a universal
anti-aging agent for different tissues. Oncotarget. 6:17251–17260.
2015.PubMed/NCBI
|
|
30
|
Dong CM, Wang XL, Wang GM, Zhang WJ, Zhu
L, Gao S, Yang DJ, Qin Y, Liang QJ, et al: A stress-induced
cellular aging model with postnatal neural stem cells. Cell Death
Dis. 5:e11162014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Min JN, Tian Y, Xiao Y, Wu L, Li L and
Chang S: The mINO80 chromatin remodeling complex is required for
efficient telomere replication and maintenance of genome stability.
Cell Res. 23:1396–1413. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moiseeva O, Mallette FA, Mukhopadhyay UK,
Moores A and Ferbeyre G: DNA damage signaling and p53-dependent
senescence after prolonged beta-interferon stimulation. Mol Biol
Cell. 17:1583–1592. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stewart SA, Ben-Porath I, Carey VJ,
O'Connor BF, Hahn WC and Weinberg RA: Erosion of the telomeric
single-strand overhang at replicative senescence. Nat Genet.
33:492–496. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Di Micco R, Sulli G, Dobreva M, Liontos M,
Botrugno OA, Gargiulo G, dal Zuffo R, Matti V, d'Ario G, et al:
Interplay between oncogene-induced DNA damage response and
heterochromatin in senescence and cancer. Nat Cell Biol.
13:292–302. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim WY and Sharpless NE: The regulation of
INK4/ARF in cancer and aging. Cell. 127:265–275. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Herbig U, Jobling WA, Chen BP, Chen DJ and
Sedivy JM: Telomere shortening triggers senescence of human cells
through a pathway involving ATM, p53, and p21(CIP1), but not
p16(INK4a). Mol Cell. 14:501–513. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Baur JA, Ungvari Z, Minor RK, Le Couteur
DG and de Cabo R: Are sirtuins viable targets for improving
healthspan and lifespan? Nat Rev Drug Discov. 11:443–461. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
McDonnell E, Peterson BS, Bomze HM and
Hirschey MD: SIRT3 regulates progression and development of
diseases of aging. Trends Endocrinol Metab. 26:486–492. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sundaresan NR, Bindu S, Pillai VB, Samant
S, Pan Y, Huang JY, Gupta M, Nagalingam RS, Wolfgeher D, Verdin E,
et al: SIRT3 blocks aging-associated tissue fibrosis in mice by
deacetylating and activating glycogen synthase kinase 3β. Mol Cell
Biol. 36:678–692. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Someya S, Yu W, Hallows WC, Xu J, Vann JM,
Leeuwenburgh C, Tanokura M, Denu JM and Prolla TA: Sirt3 mediates
reduction of oxidative damage and prevention of age-related hearing
loss under caloric restriction. Cell. 143:802–812. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jacobs KM, Pennington JD, Bisht KS,
Aykin-Burns N, Kim HS, Mishra M, Sun L, Nguyen P, Ahn BH, Leclerc
J, et al: SIRT3 interacts with the daf-16 homolog FOXO3a in the
mitochondria, as well as increases FOXO3a dependent gene
expression. Int J Biol Sci. 4:291–299. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tseng AH, Shieh SS and Wang DL: SIRT3
deacetylates FOXO3 to protect mitochondria against oxidative
damage. Free Radic Biol Med. 63:222–234. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qiu X, Brown K, Hirschey MD, Verdin E and
Chen D: Calorie restriction reduces oxidative stress by
SIRT3-mediated SOD2 activation. Cell Metab. 12:662–667. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tao R, Vassilopoulos A, Parisiadou L, Yan
Y and Gius D: Regulation of MnSOD enzymatic activity by Sirt3
connects the mitochondrial acetylome signaling networks to aging
and carcinogenesis. Antioxid Redox Signal. 20:1646–1654. 2014.
View Article : Google Scholar :
|
|
45
|
Salminen A, Ojala J, Kaarniranta K and
Kauppinen A: Mitochondrial dysfunction and oxidative stress
activate inflam-masomes: Impact on the aging process and
age-related diseases. Cell Mol Life Sci. 69:2999–3013. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sena LA and Chandel NS: Physiological
roles of mitochondrial reactive oxygen species. Mol Cell.
48:158–167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li X, Fang P, Mai J, Choi ET, Wang H and
Yang XF: Targeting mitochondrial reactive oxygen species as novel
therapy for inflammatory diseases and cancers. J Hematol Oncol.
6:192013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Freund A, Patil CK and Campisi J: p38MAPK
is a novel DNA damage response-independent regulator of the
senescence-associated secretory phenotype. EMBO J. 30:1536–1548.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bakunina N, Pariante CM and Zunszain PA:
Immune mechanisms linked to depression via oxidative stress and
neuroprogression. Immunology. 144:365–373. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hawkes HJ, Karlenius TC and Tonissen KF:
Regulation of the human thioredoxin gene promoter and its key
substrates: A study of functional and putative regulatory elements.
Biochim Biophys Acta. 1840:303–314. 2014. View Article : Google Scholar
|