Introduction
Ischemic stroke (IS) is a severe neurological
disease and a major cause of death and disability throughout the
world (1). Although rates of IS
mortality and financial burden vary greatly among countries,
low-income and middle-income countries are the most affected
(2). Thrombotic stroke, embolic
stroke, systemic hypoperfusion and venous thrombosis may lead to
IS. IS has great heterogeneity since various pathophysiological
mechanisms are usually involved (3). The application of unbiased
genome-wide approaches has contributed to the understanding of IS.
Quarta et al (4)
investigated the relationship between cholesteryl ester transfer
protein gene variants and the risk of IS. O'Connell et al
(5) identified a pattern of gene
expression in the peripheral blood of IS patients. However, the
etiology of and mechanism underlying IS are unknown in most
patients and identifying novel signatures or biomarkers that
enhance clinical decisions in the treatment of IS are
essential.
In addition to the construction of globally altered
mRNA expression profiles in IS, the expression profiles of long
non-coding (lnc)RNAs have also contributed to IS research (6). lncRNAs are defined as transcripts of
more than 200 nucleotides that do not code for proteins and are
pervasive across the genome (7).
A previous study have demonstrated that the lncRNA expression
profiles are altered in patient blood after IS and several lncRNAs
have been shown to play roles in animal models of stroke and in
vitro models of oxygen-glucose deprivation (8). Zhu et al (9) suggested that the lncRNA H19 rs217727
gene polymorphism contributes to IS susceptibility and may serve as
a potential indicator for IS susceptibility. lncRNA
rhabdomyosarcoma 2 associated transcript silencing has been shown
to protect against middle cerebral artery occlusion-induced IS
(10). However, studies into
lncRNAs in IS are just beginning and little is known about their
roles in IS. Numerous lncRNAs remain undiscovered in human IS and
the functions of the majority of lncRNAs have not yet been
elucidated.
Transcription factors (TFs) determine the level of
gene expression by recognizing specific DNA sequences in diverse
cell types (11). TFs could also
regulate the expression of lncRNAs in a number of diseases
(12,13). Regulatory relationships also exist
between genes and lncRNAs (14,15). LncRNAs, TFs and genes can form
lncRNA-mediated regulatory triplets (LncMRTs), which have been
widely observed in human diseases (16). However, the variety of LncMRT
roles in IS has not been studied in a systematic manner.
In the present study, a global LncMRT network was
constructed using experimentally verified TF-lncRNA, TF-gene and
gene-lncRNA interactions. A dysregulated LncMRT network for IS was
also constructed using a comprehensive computational approach based
on lncRNA, TF and gene expression profiles of IS patients. In
LncMRT networks, lncRNAs showed specific topological
characteristics similar to coding genes in IS. The dysregulated
LncMRT network exhibited a closer network structure than the global
LncMRT network. Several patterns in these dysregulated LncMRTs were
found, including the absence and presence of regulatory
relationships. Moreover, several core clusters were identified and
these core clusters could distinguish between matched control and
IS patient samples. Functional analyses revealed that dysregulated
LncMRTs in IS participate in the regulation of gene expression and
cell proliferation. The phosphatidylinositol 3-kinase
(PI3K)/protein kinase B (Akt) signaling pathway was identified as
an IS-associated pathway. In conclusion, the present study
highlighted the effect of dysregulated LncMRTs in IS, which
revealed their possibility as novel biomarkers and treatment
targets in IS.
Materials and methods
Construction of an experimentally
validated global LncMRT network
TF-lncRNA interaction data were download from
SNP@lincTFBS, which identified the TF binding sites of lncRNAs
using genome-wide chromatin immunoprecipitation sequencing data
(17). TF-gene interaction data
were obtained from TRANScription FACtor database (18). Gene-lncRNA interaction data were
obtained from RNA Association Interaction Database, which
integrates experimental gene-lncRNA interactions from manually
reading the literature and other database resources (19). Then the global LncMRT network was
constructed following the aforementioned experimentally validated
interactions.
Expression profiles of TFs, genes and
lncRNAs for IS
LncRNA, TF and gene expression profiles of IS
patients and matched controls were downloaded from the Gene
Expression Omnibus database (www.ncbi.nlm.nih.gov/geo). The study selected
contained peripheral blood mononuclear cells of 20 IS patients and
20 sex- and age-matched controls (GSE22255) (20). The IS patients in the
aforementioned study suffered only one stroke episode ≥6 months
before the blood collection and controls could not have a family
history of stroke. The authors also excluded participants with
severe anemia or active allergies (Table SI).
Identification of dysregulated LncMRTs in
IS
A comprehensive computational approach was developed
to identify significantly dysregulated LncMRTs in IS based on the
global LncMRT network and expression data. Initially, a t-test was
used to identify the differential level of expression of the
lncRNA, TF and gene in each LncMRT; the t-test compared the
expression levels between IS patients and matched controls,
resulting in P-values. Second, Pearson Correlation Coefficients
(PCCs) were calculated for each interacting pair (TF-lncRNA,
TF-gene and lncRNA-gene) in a LncMRT based on the expression
profile of IS patients. Different PCCs were represented using the
absolute difference of PCCs between the IS patients and matched
controls. Third, an integrated approach was performed based on two
comprehensive risk scores, including differential expression
P-values (RSdif) and PCCs (RSPCC) for each
LncMRT as follows:
RSdif=PTx PLx PGRSPCC=[(ISTL−CONTL)x(ISTG−CONTG)x(ISGL−CONGL)]
where PT, PL, PG represented the
P-values of the differentially expressed TF, lncRNA and gene,
respectively, in each LncMRT. The integral differential expression
level between IS patient and matched control samples of a LncMRT
was indicated by RSdif. ISTL, ISTG
and ISGL represented the PCCs of interactions between
the TF and lncRNA, the TF and gene, and the lncRNA and gene,
respectively in IS patients. CONTL, CONTG and
CONGL represented the PCCs of interactions between the
TF and lncRNA, the TF and gene, and the lncRNA and gene,
respectively in matched controls. The difference in the
co-expression level of a LncMRT between IS patients and matched
controls was indicated by RSPCC.
Fourth, an equal-weighted multiple ranking approach
was performed to rank all LncMRTs based on their RSdif
and RSPCC. After this step, each LncMRT received a final
risk score and was ranked by these final risk scores. Finally,
1,000 sample labels of the expression profiles were randomly
permuted to compare the final risk score with the permutation risk
score and obtain significant results. A permutation result of
P<0.05 was selected as the threshold value to generate
significantly dysregulated LncMRTs for IS.
Topological features of the global LncMRT
network and the dysregulated LncMRT network for IS
Topological features including degree, clustering
coefficient and network density analyses were performed for all the
nodes in the two networks using Cytoscape 3.0 (http://www.cytoscape.org/).
Identification of core clusters from the
dysregulated LncMRT network for IS
Core clusters were extracted from the dysregulated
LncMRT network for IS using the Clustering with Overlapping
Neighborhood Expansion (ClusterONE) package in Cytoscape, with
default parameters (http://apps.cytoscape.org/apps/ClusterONE). ClusterONE
is a package that clusters a given network based on topology to
identify densely connected regions. Finally, four core clusters
were extracted based on the number of nodes and the cluster
scores.
Classification power of the core clusters
in IS
A consensus clustering approach was used to classify
20 IS patients and 20 matched controls based on expression data of
TFs, genes and lncRNAs (21). The
ConsensusClusterPlus package in R (https://www.r-project.org/) was used to perform this
process. The smallest increase in the area under the cumulative
distribution function (CDF) curve was defined as the best category
number. A Chi-square test was applied to evaluate whether IS
patients and matched controls could be classified using this method
(P<0.05).
Functional enrichment analysis for
dysregulated LncMRTs in IS
TFs and genes were selected for functional
enrichment analyses to assess the functionality of the LncMRTs.
Online Enrichr tool was applied with default parameters to perform
the functional enrichment analyses (22). Enriched Gene Ontology (GO) terms
and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were
identified using P<0.01 and P<0.05, respectively, as the
thresholds.
Results
The construction of the global LncMRT
network and topological analysis
A global LncMRT network was constructed using
experimentally verified interactions (Fig. 1A). Details of the interactions are
listed in Table SII. The global
LncMRT network contained 4,554 LncMRTs, 1,153 nodes (including 74
TFs and genes, 501 genes, 524 lncRNAs, and 54 TFs) and 5,383 edges
(Fig. 1B). The lncRNAs occupied a
large proportion in all nodes and may play essential roles in the
LncMRT network. The global LncMRT network exhibited scale-free
distribution (R2=0.728), which is a specific topological
feature of transcriptional regulatory networks (Fig. 1C). Next, the authors found that
the degrees of TFs, genes and lncRNAs approximated the scale-free
network (Fig. 1D-F).
Transcription factor E2F (E2F)1, E2F6 and E2F4 were the TFs with
the highest degrees and are all members of the E2F family. The E2F
family plays crucial roles in the control of the cell cycle and was
also a target of transforming proteins of small DNA tumor viruses
(23). Metastasis associated lung
adenocarcinoma transcript 1, taurine upregulated gene 1 and nuclear
enriched abundant transcript 1 (NEAT1) were the lncRNAs with the
highest degrees and were previously associated with IS (24,25). Proto-oncogene c-Fos, myc
proto-oncogene protein (MYC) and transcription factor Sp1 (SP1)
were the genes with the highest degrees. These results indicated
that the global LncMRT network could be useful background
information to help design IS studies.
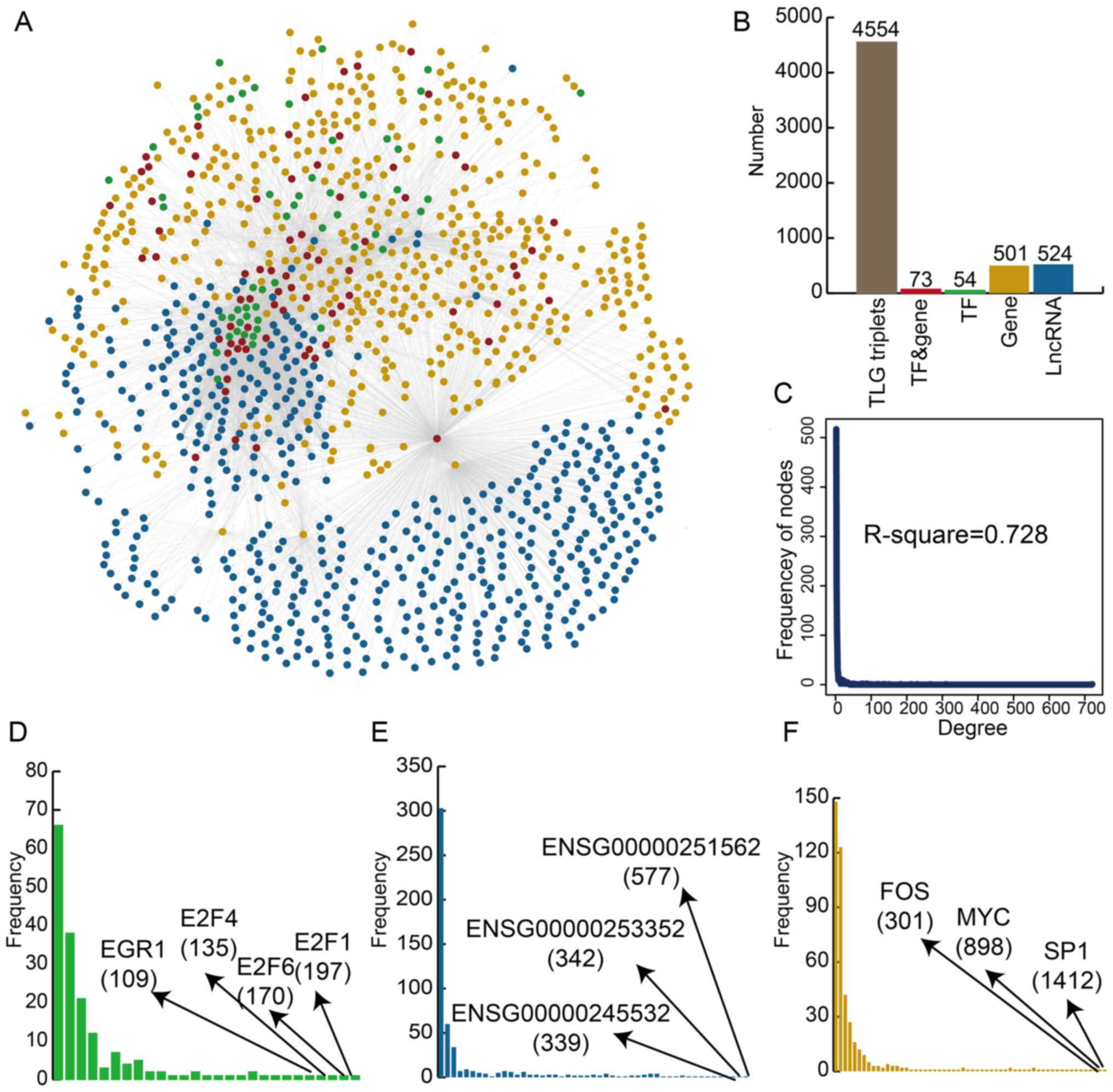 | Figure 1Construction and global
characteristics of the global LncMRT network. (A) TFs and genes,
TFs, genes, and lncRNAs are colored by red, green, orange and blue,
respectively. (B) The number of molecules in the global LncMRT
network. (C) The degree distribution of the global LncMRT network.
The degree distribution of (D) TFs, (E) genes and (F) lncRNAs. TF,
transcription factor; lncRNA, long non-coding RNA; LncMRT,
lncRNA-mediated regulatory triplet; MYC, myc proto-oncogene
protein; SP1, transcription factor Sp1. |
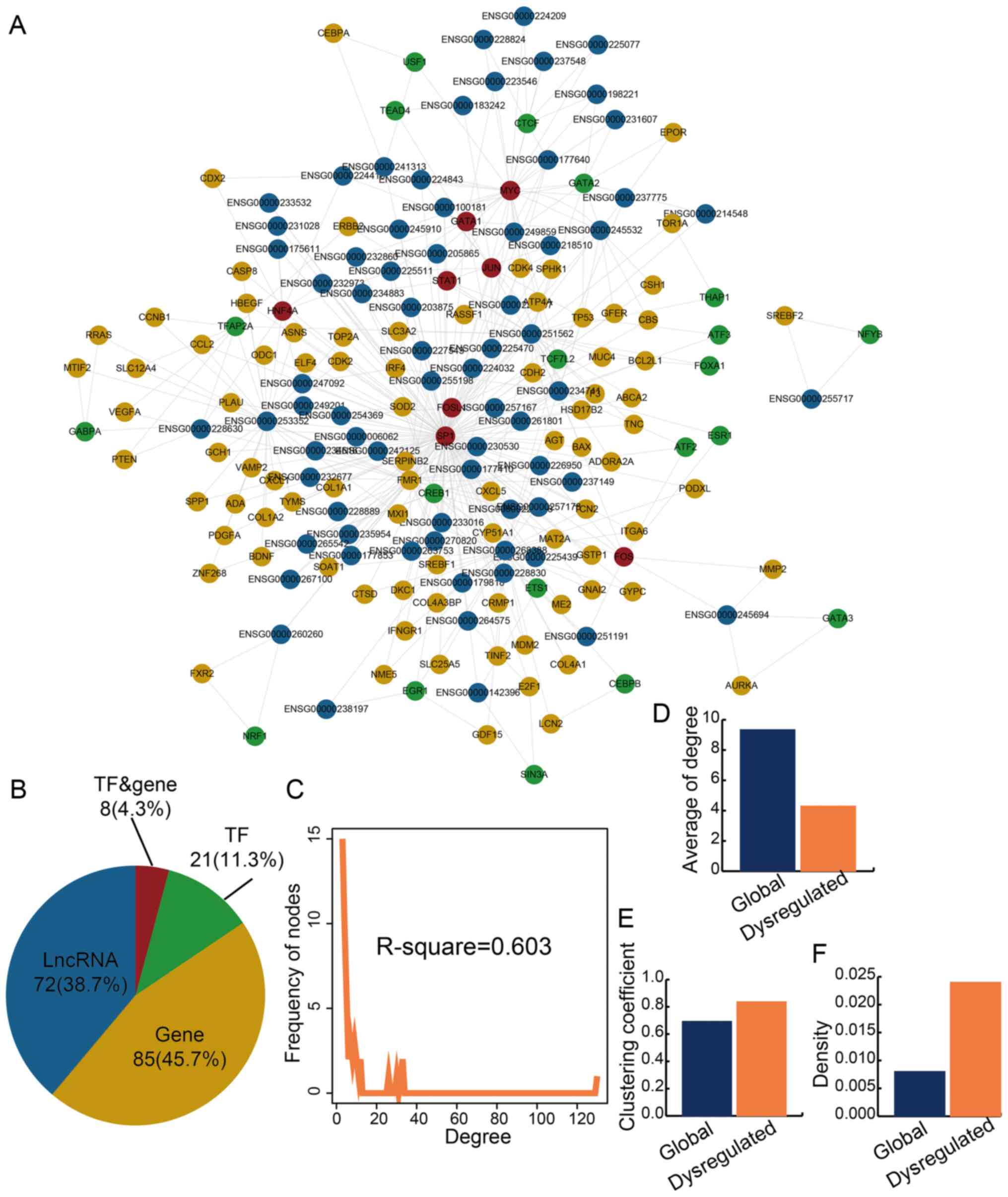
Specific LncMRTs are dysregulated in IS
patients
An integrated computational pipeline was used to
dissect the functional significance of LncMRTs in IS and 196
significantly dysregulated LncMRTs that were identified in IS
(P<0.05). A significantly dysregulated LncMRT network was
constructed and the network contained 183 nodes and 396 edges
(Fig. 2A). There were eight TFs
or genes, 21 TFs, 85 genes and 71 lncRNAs in the significantly
dysregulated LncMRT network (Fig.
2B; Table SIII). The result
indicated that lncRNAs occupied a large proportion in all nodes and
may play essential roles in the LncMRT network; this phenomenon was
similar to the global LncMRT network. Some of the lncRNAs in the
dysregulated LncMRT network had been identified by other in
vitro or mouse model studies (Table SIV). The degree of all nodes
still approximated a scale-free network (R2=0.603;
Fig. 2C). The average degree of
the significantly dysregulated LncMRT network was smaller than the
global LncMRT network and indicated the specific features of the
LncMRTs (Fig. 2D). The
dysregulated LncMRT network showed a higher clustering coefficient
and network density compared with the global LncMRT network
(Fig. 2E and F). The result
indicated that the dysregulated LncMRT network had closer network
structure features than the global LncMRT network and may be a
functional network for IS.
Risk score profiles and multiple
dysregulated patterns of LncMRTs in IS
Risk score profiles were created to characterize the
compactness of each dysregulated LncMRT in IS (Fig. 3A). The two key risk score
(RSdif and RSPCC) profiles were constructed
and they showed similar unimodal distribution. The results
indicated that the trends of the two risk scores were consistent.
The global risk score profile was also constructed and it was
discovered that the risk scores of most dysregulated LncMRTs were
concentrated in a small scale. The detail risk score profile of top
10 dysregulated LncMRTs were identified and it was found that some
risk scores did not change while others exhibited large changes
(Fig. 3B). For instance, the
difference in PCCs between IS patients and matched controls was
bigger than the P-values in the dysregulated LncMRT SP1/long
intergenic non-protein coding RNA (LINC)00475/interferon regulatory
factor 4. However, the change in those P-values was larger compared
with the PCC values in the dysregulated LncMRT SP1/UBAC2 anti-sense
RNA 1 (UBAC2-AS1)/synaptic functional regulator FMR1 (FMR1).
Therefore, it was inferred that there are several complex patterns
of these dysregulated LncMRTs.
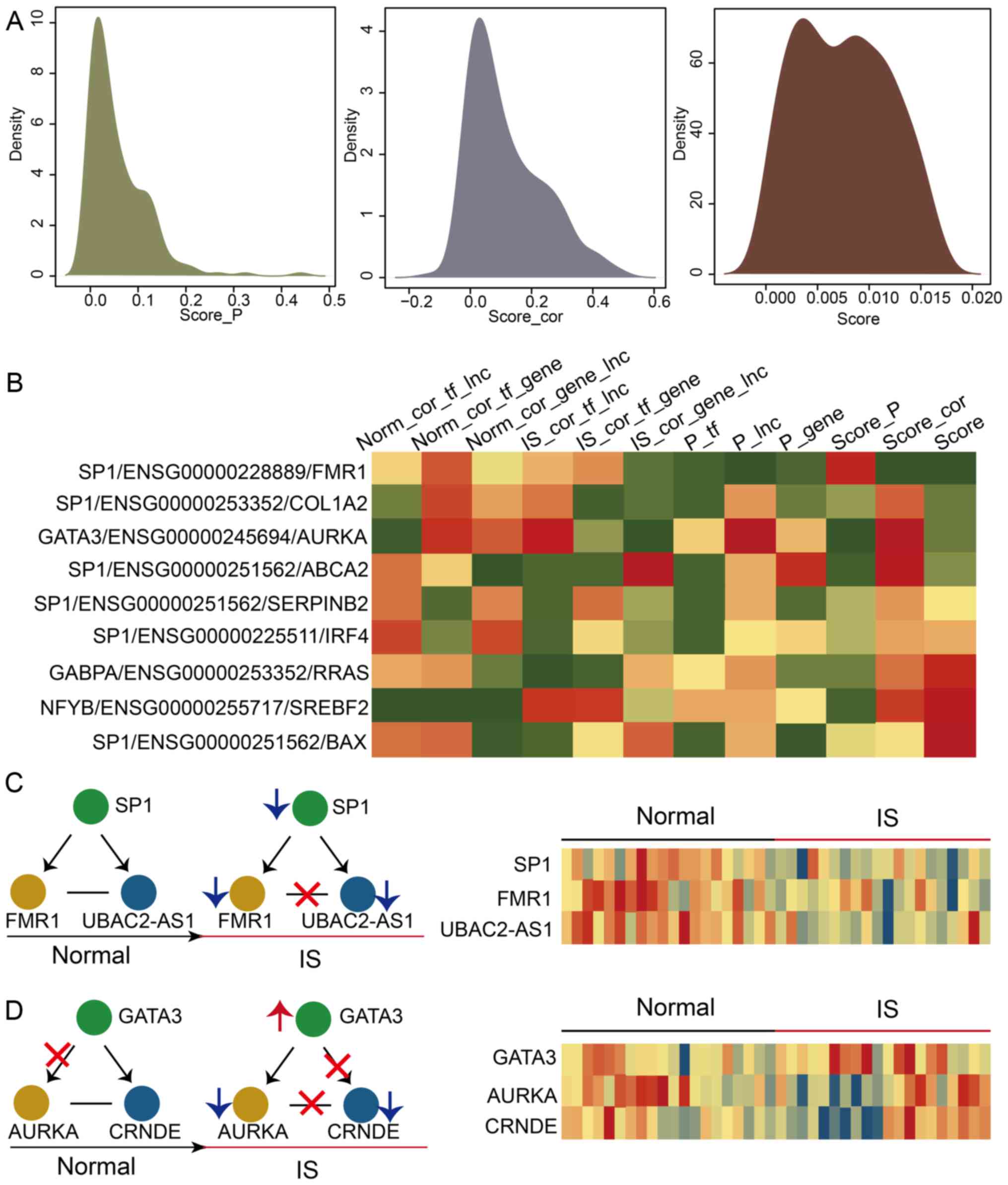 | Figure 3Construction of a functional score
profile for the dysregulated LncMRTs in IS. (A) The density
distribution curves of RSdif, RSPCC and integrated scores. (B) The
activity score profile for the top dysregulated LncMRTs in IS. (C)
The dysregulated patterns of LncMRTs (SP1/FMR1/UBAC2-AS1) in IS.
The heatmap presented the expression of molecules. The crosses
represent regulated interactions that were not established (no
co-expression was observed). Red and blue arrows represent up- and
downregulated transcription factors, lncRNAs and genes. (D) The
dysregulated patterns of LncMRTs (SP1/FMR1/UBAC2-AS1) in IS
(GATA3/AURKA/CRNDE). LncMRT, long non-coding RNA-mediated
regulatory triplet; IS, ischemic stroke; RSdif, risk scores of
differential expression P-values; RSPCC, risk scores of Pearson
Correlation Coefficients; SP1, transcription factor Sp1; FMR1,
synaptic functional regulator FMR1; GATA3, trans-acting
T-cell-specific transcription factor GATA-3; AURKA, aurora kinase
A; CRNDE, colorectal neoplasia differentially expressed;
UBAC2-AS1UBAC2, antisense RNA 1. |
In the dysregulated LncMRT SP1/UBAC2-AS1/FMR1, the
interaction between FMR1 and UBAC2-AS1 was absent in IS patients,
and all the molecules were downregulated (Fig. 3C). In the dysregulated LncMRT
trans-acting T-cell-specific transcription factor GATA-3
(GATA3)/colorectal neoplasia differentially expressed
(CRNDE)/aurora kinase A (AURKA), the interaction between GATA3 and
AURKA was present in IS patients (Fig. 3D). However, the other two
interactions were both absent in IS patients. The TF GATA3 was
upregulated and the gene AURKA and the lncRNA CRNDE were both
downregulated. Collectively, the results show that interactions
were absent or present in IS patients and demonstrate complex
features.
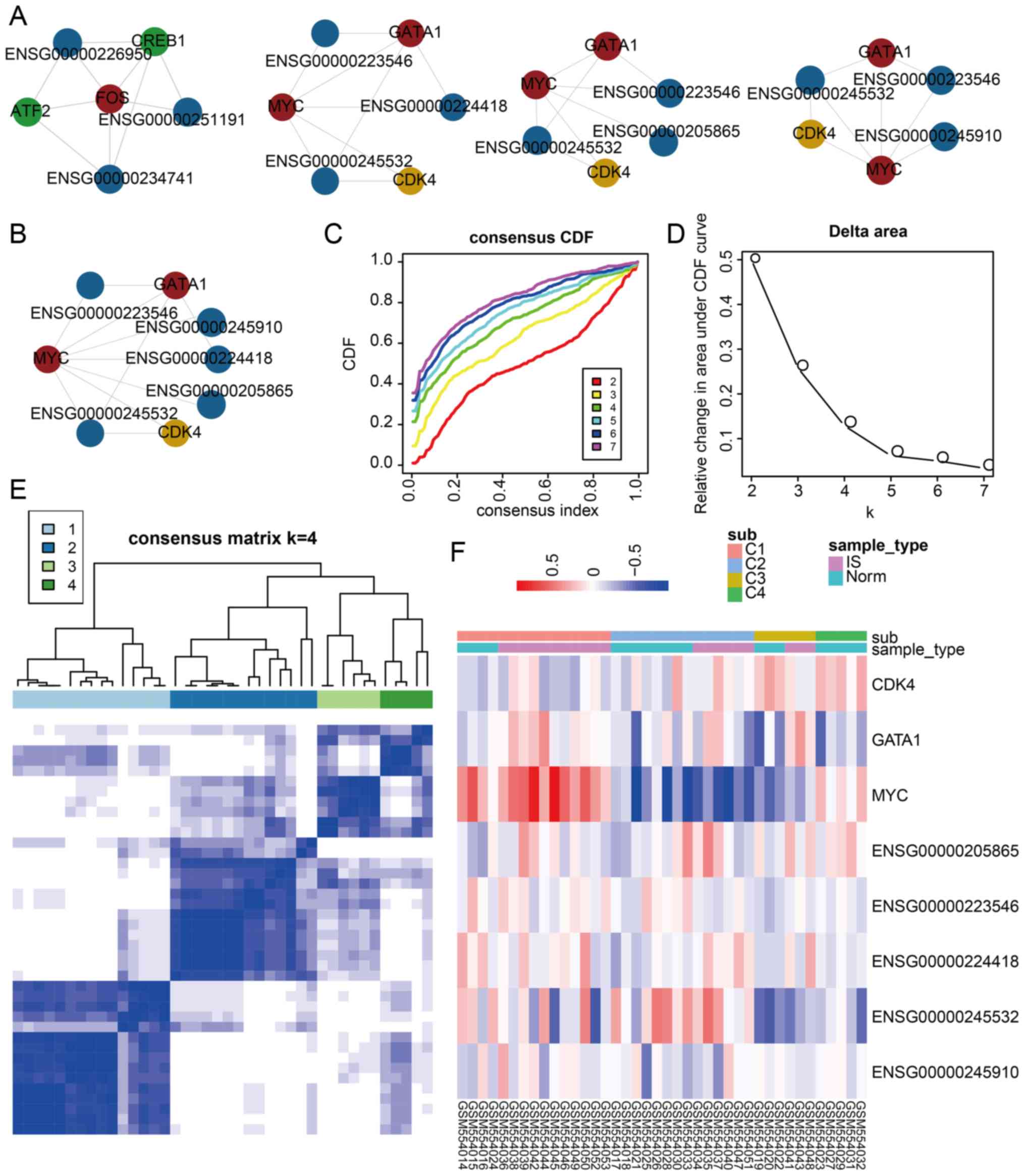
IS-related core clusters could be
regarded as specific biomarkers to distinguish IS patient and
matched control samples
A core cluster analysis was performed to further
reveal the communication between these three types of molecules in
the dysregulated LncMRT network for IS. A total of four core
clusters were identified and each core cluster contained a certain
number of TFs, genes and lncRNAs (Fig. 4A). The authors also discovered
that the last three core clusters shared most of their molecules
and may have similar functions. The last three core clusters were
combined to construct an integrated core cluster containing two
TFs, one gene and five lncRNAs (Fig.
4B). The expression profiles of the TFs, genes and lncRNAs in
each cluster were used to classify IS patient and matched control
samples following consensus clustering, which was used to determine
whether the LncMRTs could be regarded as special classifiers for
IS. The integrated core cluster could split all samples into
diverse groups. A total of four groups were identified, according
to the CDF and relative change in area under the CDF curve plot
(Fig. 4C and D). Each sample
group had a consensus expression pattern and could be distinguished
clearly (Fig. 4E). Most samples
could be classified accurately (Chi-square test, P=0.035); this was
especially true for the last group (C4), in which all matched
control samples were verified (Fig.
4F). The first core cluster could also distinguish IS patient
and matched control samples (Chi-square test, P=0.027).
Collectively, all the above results indicated that integrating the
expression profiles of dysregulated LncMRTs may identify specific
biomarkers that distinguish IS patient and matched control
samples.
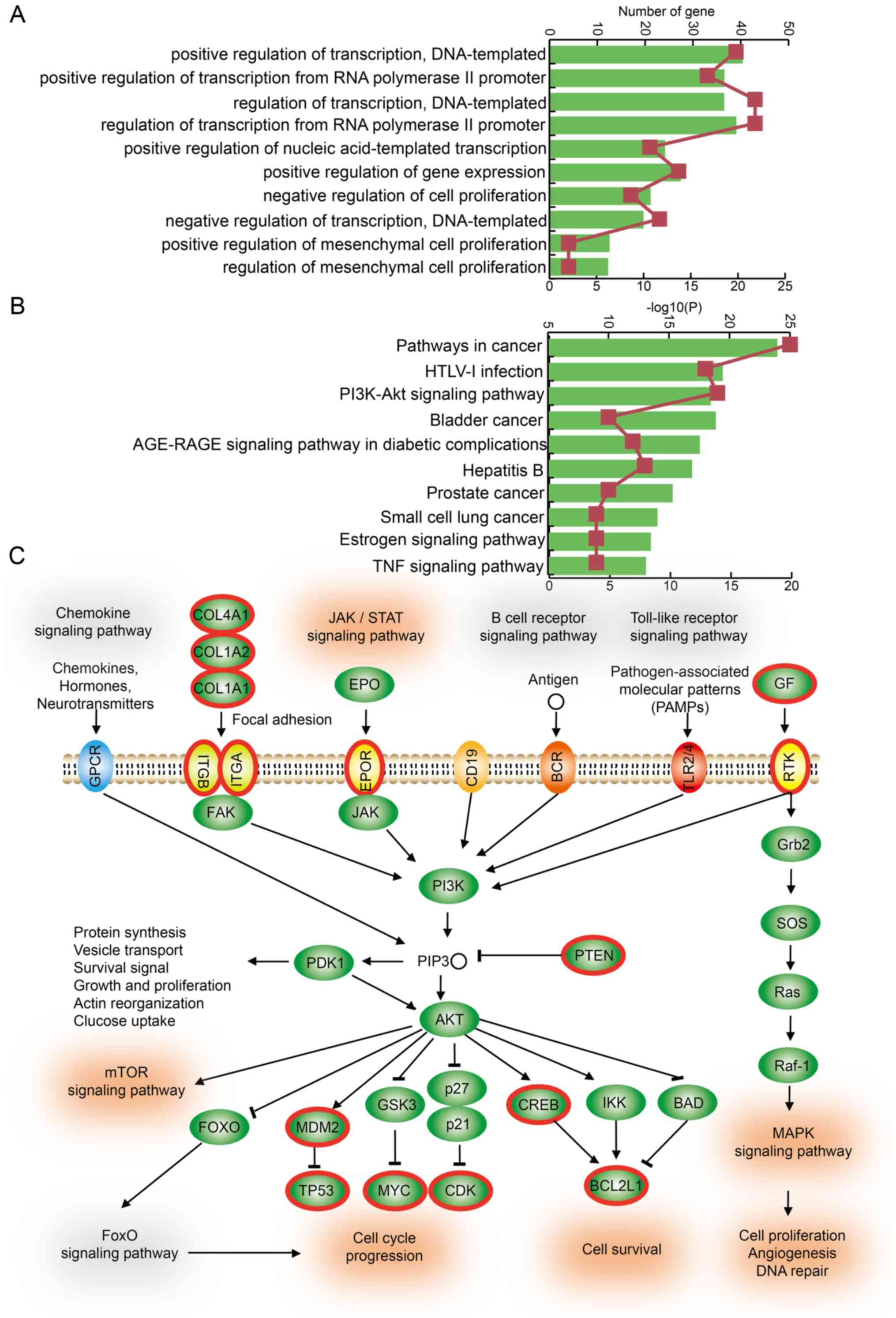
Dysregulated LncMRTs in IS patients are
associated with critical biological functions and the PI3K/Akt
signaling pathway
The GO enrichment analyses were performed based on
all TFs and genes in dysregulated LncMRTs in IS. These genes were
slightly enriched in some critical biological functions such as
positive regulation of transcription, creating DNA templates,
positive regulation of gene expression and positive or negative
regulation of mesenchymal cell proliferation (Fig. 5A). Mesenchymal cells were
associated with recovery from IS and provided neurological
protection (26). In addition,
KEGG pathway enrichment analyses were also performed and several
key pathways were enriched, such as the PI3K/Akt signaling pathway,
the advanced glycation endproducts (AGE)-receptor for AGE signaling
pathway, the estrogen signaling pathway and the tumor necrosis
factor signaling pathway (Fig.
5B). The PI3K/Akt signaling pathway is an intracellular
signaling pathway important in regulating the cell cycle. Quercetin
can decrease cell apoptosis in IS rat brains and the mechanism may
be related to the activation of the PI3K/Akt signaling pathway
(27). The PI3K/Akt signaling
pathway contributes to neuronal survival after IS (28). In the current study, 19 TFs and
genes in the dysregulated LncMRTs were involved in this pathway
(Fig. 5C). For example, PTEN is a
key gene in the PI3K/Akt signaling pathway and was also found to be
a negative regulator of neuronal cell survival (29). PTEN formed a dysregulated LncMRT
with HOX Transcript Antisense RNA (HOTAIR) and transcription factor
AP-2-α (TFAP2A). In summary, these results suggest that LncMRTs
associated with IS could play their roles by participating in the
regulation of the PI3K/Akt signaling pathway.
Discussion
Detecting transcriptome changes is a major challenge
of studying the mechanism of and treatment for IS. The expression
levels of coding and non-coding transcripts were frequently
regulated by each other. This cross-talk regulation among TFs,
genes and lncRNAs could form regulated triplets, and may be
functional motifs for IS. In the current study, an integrated and
computational approach was developed using experimentally verified
interactions and expression profiles of TFs, genes and lncRNAs to
explore the LncMRTs in IS.
In the current study, multiple dysregulated patterns
emerged from the analysis of TF, gene and lncRNA expression, and
variations in interactions in these LncMRTs. In some of the
dysregulated LncMRTs, almost all molecules and interactions changed
in IS patients compared with matched controls. In other
dysregulated LncMRTs, only an individual molecule and interaction
exhibited a clear change in IS patients. There were two major types
of interaction change: i) The regulatory interactions were present
in matched controls, but absent in IS patients; and ii) the
regulatory interactions were absent in matched controls, but
present in IS patients. Similar patterns and phenomena also
appeared in other types of regulatory motifs, such as competing
endogenous (ce)RNAs in cancer (30). Collectively, the results show that
the regulatory interactions were absent or present in LncRMTs in IS
patients compared with matched controls. The disturbance of these
LncRMTs may contribute to the development of IS.
A previous study suggested that some other types of
integrated and regulated motifs could be regarded as specific
disease- and survival-associated biomarkers. For example,
lncRNA-mediated ceRNAs could be viewed as prognostic biomarkers for
breast cancer (31). In the
current study, an integrated core cluster, including TFs GATA1 and
MYC, the gene cyclin-dependent kinase 4, and lncRNAs family with
sequence similarity 99 member B, LINC00630, STK24-AS1, NEAT1 and
small nucleolar RNA host gene 6, was constructed. The integrated
core cluster could split all the samples into four groups and may
be a specific biomarker profile for IS. In addition, this study was
undertaken to explore biomarkers in peripheral blood samples of IS
patients and matched controls. The finding would be more meaningful
if it could be tested in other types of samples, such as spinal
cord fluid.
A key pathway named the PI3K/Akt signaling pathway
was found by functional analyses of TFs and genes in the
dysregulated LncMRTs of IS. The activation of PI3K correlates with
increased cell survival and this effect is largely mediated through
the activation of the serine/threonine kinase Akt (32). PTEN plays a key regulatory role in
the cross-talk between the cell survival PI3K/Akt signaling pathway
and the pro-death JNK signaling pathway, which raises a new
possibility that agents targeting the phosphatase PTEN may expand
the therapeutic options of agents used to protect neurons from IS
(33). PTEN, TFAP2A and HOTAIR
formed a dysregulated LncMRT in IS patients, which suggests that
PTEN could play its role in the PI3K/Akt signaling pathway by
forming LncMRT in IS patients.
In conclusion, a dysregulated LncMRT network for IS
was constructed and analyzed. The risk scores were used to evaluate
the activity of each dysregulated LncMRT in IS. Multiple
dysregulated patterns of LncMRTs in IS were also found. In
addition, the core LncMRT cluster could distinguish IS patient and
matched control samples, and could be viewed as a specific
biomarker profile for IS. The functional analysis showed the
association between dysregulated LncMRTs and the PI3K/Akt signaling
pathway in IS patients. Collectively, the results of the present
study provide novel insights into the mechanisms underlying the
function of lncRNAs in IS.
Supplementary Data
Acknowledgments
Not applicable.
Funding
The current study was supported by the National
Natural Science Foundation of China (grant nos. 81820108014,
81771361, 81571166, 81801190, 81701190 and 81701155), the applied
technique research and development project of Harbin (grant no.
2016RAXYJ067) and the Postdoctoral project of Heilongjiang Province
(grant no. LBH-Z17138).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CYZ, ZHX, WLH and CLY conceived and designed the
present study. CYZ, WJJ, LXY and KXT performed the experiments and
analyzed the data. BCR, LS, LJ, SXS, WN and TK validated and
improved the computational approach in the present study. CYZ and
BCR wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Deb P, Sharma S and Hassan KM:
Pathophysiologic mechanisms of acute ischemic stroke: An overview
with emphasis on therapeutic significance beyond thrombolysis.
Pathophysiology. 17:197–218. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Feigin VL, Lawes CM, Bennett DA,
Barker-Collo SL and Parag V: Worldwide stroke incidence and early
case fatality reported in 56 population-based studies: A systematic
review. Lancet Neurol. 8:355–369. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bejot Y, Caillier M, Ben Salem D, Couvreur
G, Rouaud O, Osseby GV, Durier J, Marie C, Moreau T and Giroud M:
Ischaemic stroke subtypes and associated risk factors: A French
population based study. J Neurol Neurosurg Psychiatry.
79:1344–1348. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Quarta G, Stanzione R, Evangelista A,
Zanda B, Sciarretta S, Di Angelantonio E, Marchitti S, Di Murro D,
Volpe M and Rubattu S: A protective role of a cholesteryl ester
transfer protein gene variant towards ischaemic stroke in
Sardinians. J Intern Med. 262:555–561. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
O'Connell GC, Petrone AB, Treadway MB,
Tennant CS, Lucke-Wold N, Chantler PD and Barr TL: Machine-learning
approach identifies a pattern of gene expression in peripheral
blood that can accurately detect ischaemic stroke. NPJ Genom Med.
1:160382016. View Article : Google Scholar
|
|
6
|
Vemuganti R: All's well that transcribes
well: Non-coding RNAs and post-stroke brain damage. Neurochem Int.
63:438–449. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
St Laurent G, Wahlestedt C and Kapranov P:
The landscape of long noncoding RNA classification. Trends Genet.
31:239–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He W, Wei D, Cai, Chen S, Li S and Chen W:
Altered long non-coding RNA transcriptomic profiles in ischemic
stroke. Hum Gene Ther. 29:719–732. 2018. View Article : Google Scholar
|
|
9
|
Zhu R, Liu X and He Z: Long non-coding RNA
H19 and MALAT1 gene variants in patients with ischemic stroke in a
northern Chinese Han population. Mol Brain. 11:582018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hou XX and Cheng H: Long non-coding RNA
RMST silencing protects against middle cerebral artery occlusion
(MCAO)-induced ischemic stroke. Biochem Biophys Res Commun.
495:2602–2608. 2018. View Article : Google Scholar
|
|
11
|
Latchman DS: Transcription factors: An
overview. Int J Biochem Cell Biol. 29:1305–1312. 1997. View Article : Google Scholar
|
|
12
|
Jiang H, Li T, Qu Y, Wang X, Li B, Song J,
Sun X, Tang Y, Wan J, Yu Y, et al: Long non-coding RNA SNHG15
interacts with and stabilizes transcription factor Slug and
promotes colon cancer progression. Cancer Lett. 425:78–87. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sawaya AP, Pastar I, Stojadinovic O,
Lazovic S, Davis SC, Gil J, Kirsner RS and Tomic-Canic M: Topical
mevastatin promotes wound healing by inhibiting the transcription
factor c-Myc via the glucocorticoid receptor and the long
non-coding RNA Gas5. J Biol Chem. 293:1439–1449. 2018. View Article : Google Scholar :
|
|
14
|
Yu Y, Zhang M, Wang N, Li Q, Yang J, Yan
S, He X, Ji G and Miao L: Epigenetic silencing of tumor suppressor
gene CDKN1A by oncogenic long non-coding RNA SNHG1 in
cholangiocarci-noma. Cell Death Dis. 9:7462018. View Article : Google Scholar
|
|
15
|
Sepe R, Pellecchia S, Serra P, D'Angelo D,
Federico A, Raia M, Cortez Cardoso Penha R, Decaussin-Petrucci M,
Del Vecchio L, Fusco A and Pallante P: The long non-coding RNA
RP5-1024C241 and its associated-gene MPPED2 are down-regulated in
human thyroid neoplasias and act as tumour suppressors. Cancers
(Basel). 10. pp. E1462018, View Article : Google Scholar
|
|
16
|
Zhou L, Xu DY, Sha WG, Shen L and Lu GY:
Long non-coding RNA MALAT1 interacts with transcription factor
Foxo1 to regulate SIRT1 transcription in high glucose-induced HK-2
cells injury. Biochem Biophys Res Commun. 503:849–855. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ning S, Zhao Z, Ye J, Wang P, Zhi H, Li R,
Wang T, Wang J, Wang L and Li X: SNP@lincTFBS: An integrated
database of polymorphisms in human LincRNA transcription factor
binding sites. PLoS One. 9:e1038512014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wingender E, Dietze P, Karas H and Knuppel
R: TRANSFAC: A database on transcription factors and their DNA
binding sites. Nucleic Acids Res. 24:238–241. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yi Y, Zhao Y, Li C, Zhang L, Huang H, Li
Y, Liu L, Hou P, Cui T, Tan P, et al: RAID v2.0: An updated
resource of RNA-associated interactions across organisms. Nucleic
Acids Res. 45:D115–D118. 2017. View Article : Google Scholar :
|
|
20
|
Krug T, Gabriel JP, Taipa R, Fonseca BV,
Domingues-Montanari S, Fernandez-Cadenas I, Manso H, Gouveia LO,
Sobral J, Albergaria I, et al: TTC7B emerges as a novel risk factor
for ischemic stroke through the convergence of several genome-wide
approaches. J Cereb Blood Flow Metab. 32:1061–1072. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wilkerson MD and Hayes DN:
ConsensusClusterPlus: A class discovery tool with confidence
assessments and item tracking. Bioinformatics. 26:1572–1573. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kuleshov MV, Jones MR, Rouillard AD,
Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM,
Lachmann A, et al: Enrichr: A comprehensive gene set enrichment
analysis web server 2016 update. Nucleic Acids Res. 44:W90–W97.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
DeGregori J and Johnson DG: Distinct and
overlapping roles for E2F family members in transcription,
proliferation and apoptosis. Curr Mol Med. 6:739–748.
2006.PubMed/NCBI
|
|
24
|
Chen S, Wang M, Yang H, Mao L, He Q, Jin
H, Ye ZM, Luo XY, Xia YP and Hu B: LncRNA TUG1 sponges microRNA-9
to promote neurons apoptosis by up-regulated Bcl2l11 under
ischemia. Biochem Biophys Res Commun. 485:167–173. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Tang X, Liu K, Hamblin MH and Yin
KJ: Long noncoding RNA Malat1 regulates cerebrovascular pathologies
in ischemic stroke. J Neurosci. 37:1797–1806. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gutiérrez-Fernández M, Rodríguez-Frutos B,
Alvarez-Grech J, Vallejo-Cremades MT, Expósito-Alcaide M, Merino J,
Roda JM and Díez-Tejedor E: Functional recovery after hematic
administration of allogenic mesenchymal stem cells in acute
ischemic stroke in rats. Neuroscience. 175:394–405. 2011.
View Article : Google Scholar
|
|
27
|
Yao RQ, Qi DS, Yu HL, Liu J, Yang LH and
Wu XX: Quercetin attenuates cell apoptosis in focal cerebral
ischemia rat brain via activation of BDNF-TrkB-PI3K/Akt signaling
pathway. Neurochem Res. 37:2777–2786. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao H, Sapolsky RM and Steinberg GK:
Phosphoinositide-3-kinase/akt survival signal pathways are
implicated in neuronal survival after stroke. Mol Neurobiol.
34:249–270. 2006. View Article : Google Scholar
|
|
29
|
Shi GD, OuYang YP, Shi JG, Liu Y, Yuan W
and Jia LS: PTEN deletion prevents ischemic brain injury by
activating the mTOR signaling pathway. Biochem Biophys Res Commun.
404:941–945. 2011. View Article : Google Scholar
|
|
30
|
Wang P, Ning S, Zhang Y, Li R, Ye J, Zhao
Z, Zhi H, Wang T, Guo Z and Li X: Identification of
lncRNA-associated competing triplets reveals global patterns and
prognostic markers for cancer. Nucleic Acids Res. 43:3478–3489.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fan CN, Ma L and Liu N: Systematic
analysis of lncRNA-miRNA-mRNA competing endogenous RNA network
identifies four-lncRNA signature as a prognostic biomarker for
breast cancer. J Transl Med. 16:2642018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kohn AD, Takeuchi F and Roth RA: Akt, a
pleckstrin homology domain containing kinase, is activated
primarily by phosphory-lation. J Biol Chem. 271:21920–21926. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang QG, Wu DN, Han D and Zhang GY:
Critical role of PTEN in the coupling between PI3K/Akt and JNK1/2
signaling in ischemic brain injury. FEBS Lett. 581:495–505. 2007.
View Article : Google Scholar : PubMed/NCBI
|