Introduction
Asthma is a severe chronic inflammatory disease with
an increasing prevalence worldwide (1,2).
The pathogenesis of asthma involves complicated factors, including
immunity, infection, environmental factors and genetic inheritance
(3). Airway remodeling, a typical
characteristic of asthma, is manifested by airway wall thickening,
subepithelial fibrosis, increased smooth muscle mass, angiogenesis
and increased mucous glands (4,5).
MicroRNAs (miRNAs/miRs) are non-coding RNAs of 20-25
nucleotides that cannot be further translated into proteins, but
can suppress gene expression by binding to the 3′-untranslated
region (3′-UTR) of target mRNAs (6,7).
Accumulating research has shown that miRNAs participate in numerous
biological processes, such as cell proliferation (8), differentiation (9), apoptosis (10) and epithelial-mesenchymal
transition (EMT) (11).
Additionally, a previous study has revealed the important role of
miRNAs in mouse models of asthma (12). miR-106b-5p has been associated
with glioma tumorigenesis (13),
lung cancer (14), chronic
myeloid leukemia (15), breast
cancer (16,17) and hepatocellular carcinoma
(18). However, the molecular
mechanisms of miR-106b-5p in asthma remain unclear.
The purpose of the present study was to investigate
the function of miR-106b-5p in TGF-β1-induced pulmonary fibrosis
and EMT, the therapeutic effect of miR-106b-5p in asthma and the
underlying mechanisms.
Materials and methods
Cell culture and chemicals
Human bronchial epithelial cells (BEAS-2B) were
purchased from the American Type Culture Collection and were
cultivated in DMEM supplemented with 10% FBS (both Gibco; Thermo
Fisher Scientific, Inc.), penicillin (100 U/ml) and streptomycin
(100 mg/ml) in a humidified incubator containing 5% CO2
at 37°C. BEAS-2B cells were treated with TGF-β1 (Abcam; 10 ng/ml)
at 37°C for 24 h.
Ovalbumin (OVA)-induced murine asthma
model
A total of 10 BALB/c male mice (6 weeks old; 18-20
g), obtained from Beijing Vital River Laboratory Animal Technology
Co. Ltd. Mice were housed in plastic boxes with a 12-h light/dark
cycle and constant temperature (19-23°C) and humidity (55±10%).
Food and water were supplied ad libitum. Mice were
randomized into two groups: The control group and the OVA group,
each group containing 5 mice. On day 0 and day 14, the mice of the
OVA group were sensitized with 20 µg OVA (Sigma-Aldrich;
Merck KGaA) with 1 mg aluminum hydroxide (Thermo Fisher Scientific,
Inc.) adsorbed in 200 µl PBS by intraperitoneal injection.
On days 21-23, the mice of the OVA group were challenged through
the airway with 1% OVA (dissolved in PBS) for 30 min using an
ultrasonic nebulizer (INQUA NEB Plus; PARI GmbH). The mice of the
control group were treated with PBS. All the mice were euthanized
by cervical dislocation on day 24. The present study was approved
by the Nanjing Medical University Animal Experimental Ethics
Committee (approval no. 2005020).
Plasmids, small interfering RNAs (siRNAs)
and miRNA mimic or antimiR
The transcriptional start site of human sine oculis
homeobox homolog 1 (SIX1) promoter was set as +1. The promoter of
SIX1 DNA fragment −351 to +100 was inserted into the pGL3-Basic
vector (Promega Corporation) and named pGL3-451. The
transcriptional binding sites of SIX1 were predicted using the
JASPAR database (version 5.0; jaspar.genereg.net) (19). A series of plasmids with mutations
of E2F transcription factor 1 (E2F1)-binding sites were synthesized
from TsingKe Biological Technology. According to the binding sites,
a series of pGL3-mut plasmids were generated, named mut-E2F1-A,
mut-E2F1-B and mut-E2F1-A+B. The mutated sequence of the E2F1-A
binding site (−64 ATA GGC GCC GCC-53) was 5′-ATA TTA TAA GCC-3′,
and the mutated sequence of the E2F1-B binding site (+41 CGG GCG
GGA GG +51) was 5′-CGG ATA GGT GG-3′. The overexpression plasmid
pENTER-E2F1 (pE2F1) and the corresponding control plasmid pENTER
(TsingKe Biological Technology) were supplied by our lab (20). The double-stranded siRNAs
(Table I) were synthesized and
purified by high-performance chromatography by Shanghai GenePharma
Co., Ltd. The sequences of the miR mimics and antimiR (both
Guangzhou RiboBio Co., Ltd.) are listed in Table I. The type of negative controls
used was non-targeting.
 | Table ISequences used for siRNAs, miR mimic
and antimiR. |
Table I
Sequences used for siRNAs, miR mimic
and antimiR.
| Name | Sequence
(5′-3′) |
|---|
| siE2F1 | Sense:
CACUGAAUCUGACCACCAATT |
| Antisense:
UUGGUGGUCAGAUUCAGUGTT |
| siSIX1 | Sense:
GCAUCAGCUCCAAGACUCUTT |
| Antisense:
AGAGUCUUGGAGCUGAUGCTT |
| siNC | Sense:
UUCUCCGAACGUGUCACGUTT |
| Antisense:
ACGUGACACGUUCGGAGAATT |
| miR-106b-5p
mimic | Sense:
UAAAGUGCUGACAGUGCAGAU |
| Antisense:
AUCUGCACUGUCAGCACUUUA |
| mimic NC | Sense:
UUUGUACUACACAAAAGUACUG |
| Antisense:
CAGUACUUUUGUGUAGUACAAA |
| antimiR-106b-5p
antimiR-NC |
AUCUGCACUGUCAGCACUUUA |
|
CAGUACUUUUGUGUAGUACAAA |
Transient transfections and luciferase
assays
Transfections were conducted in BEAS-2B cells using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.). A total of 100 nM antimiR-106b-5p and 50 nM
miR-106b-5p mimics was used for transfection at 37°C for 48 h,
after which cells were harvested for subsequent experimentation.
Cells were seeded into 96-well plates (1×104
cells/well). A total of 100 ng of the promoter reporter plasmids
(pGL3-451 and pGL3-mut) together with the pRL-TK plasmid (used as
an internal control reporter vector; 4 ng; Promega Corporation)
were co-transfected into cells as an internal control using
Lipofectamine 3000 at 37°C for 48 h according to the manufacturer's
protocol. For siRNA and overexpression assays, pGL3-451 or pGL3-mut
plasmids were co-transfected with siE2F1 (50 nM) or pENTER-E2F1
(100 ng) into cells. After 24 h from transfection, promoter
activity was assessed using a Dual-Luciferase Reporter Assay System
(Promega Corporation) and normalized to the activity of pRL-TK
(Renilla luciferase activity). The E2F1 wild-type (WT)
3′-UTR was cloned into the pmiR-RB-reporter (TsingKe Biological
Technology) and the mutated putative miR-106b-5p binding site in
the E2F1 3′-UTR was cloned into the pmiR-RB-reporter and named E2F1
mutant (MUT) 3′-UTR. The SIX1 WT 3′-UTR was cloned into the
pmiR-RB-reporter and named pmiR-RB-SIX1 plasmid (TsingKe Biological
Technology) and cells were co-transfected using three different
concentrations (50, 100 and 150 nM) of miR-106b-5p mimic. The
results were representative of at least three independent
experiments conducted in triplicate.
Reverse transcription-quantitative
(RT-qPCR)
Total RNA was extracted from mouse tissues and cell
lines using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), and then reverse-transcribed into first strand
cDNA using the PrimeScript RT Master Mix Perfect Real Time kit
(Takara Biotechnology Co., Ltd.) according to the manufacturer's
protocol. qPCR was conducted in a LightCycler480II (Roche
Diagnostics) with TB Green technology (Takara Biotechnology Co.,
Ltd.). The thermocycling conditions were as follows: Initial
denaturation for 30 sec at 95°C, followed by 40 cycles of
denaturation for 5 sec at 95°C, annealing for 30 sec at 55°C and
extension for 30 sec at 72°C. The total mRNA levels were analyzed
in triplicate with β-actin as a normalized standard, while miR
expression was normalized to U6. The relative expression levels
were evaluated using the 2−ΔΔCq method (21). The specific primers (Guangzhou
RiboBio Co., Ltd.) are listed in Table II.
| Table IIPrimers used for reverse
transcription-quantitative PCR. |
Table II
Primers used for reverse
transcription-quantitative PCR.
| Gene | Forward primer
(5′-3′) | Reverse-primer
(5′-3′) |
|---|
| E2F1 |
AGCGGCGCATCTATGACATC |
GTCAACCCCTCAAGCCGTC |
| SIX1 |
AAGGAGAAGTCGAGGGGTGT |
TGCTTGTTGGAGGAGGAGTT |
| β-actin |
AAAGACCTGTACGCCAACAC |
GTCATACTCCTGCTTGCTGAT |
| E-cadherin |
CGAGAGCTACACGTTCACGG |
GGGTGTCGAGGGAAAAATAGG |
| N-cadherin |
TCAGGCGTCTGTAGAGGCTT |
ATGCACATCCTTCGATAAGACTG |
| Vimentin |
GACGCCATCAACACCGAGTT |
CTTTGTCGTTGGTTAGCTGGT |
| Fibronectin |
CGGTGGCTGTCAGTCAAAG |
AAACCTCGGCTTCCTCCATAA |
| Collagen IV |
GGACTACCTGGAACAAAAGGG |
GCCAAGTATCTCACCTGGATCA |
| α-SMA |
AAGAGGAAGACAGCACAGCTC |
GATGGATGGGAAAACAGCC |
| miR-106b-5p |
CTGGAGTAAAGTGCTGACAGTG |
GTGCAGGGTCCGAGGT |
| U6 |
GCTTCGGCAGCACATATACTAAAAT |
CGCTTCACGAATTTGCGTGTCAT |
Hematoxylin and eosin (H& E) staining
and immunohistochemistry (IHC)
Tissues for H&E and IHC were collected from
asthmatic mice model and control mice. The lungs of mice were
harvested and fixed in 4% paraformaldehyde for 24 h at room
temperature and then embedded in paraffin using a standard protocol
after dehydration. Subsequently, 4-µm-thick sections of
embedded lung tissue were mounted onto slides and stained with
H&E for 5 min at room temperature to identify tissue
inflammation. Tissue sections were viewed with a light microscope
(magnification, ×100). The expression levels of E2F1 and SIX1 were
evaluated by semi-quantitative IHC according to a previous study
(22). After paraffin removal,
sections were incubated in 0.01 mol/l citric acid buffer (pH 6.0)
in a microwave for 15 min at 95°C for antigen recovery.
Subsequently, sections were washed with xylene and rehydrated in a
descending alcohol series. The sections were then cooled and
incubated in 3 g/l H2O2 for 30 min at room
temperature to inactivate endogenous peroxidase, then blocked with
1:10 normal goat serum (Gibco; Thermo Fisher Scientific, Inc.) at
room temperature for 30 min. Subsequently, the supernatant was
discarded and primary anti-mouse E2F1 (1:400; cat. no. ab179445;
Abcam) and SIX1 (1:500; cat. no. 10709-1-AP; ProteinTech Group,
Inc.) antibodies were added overnight at 4°C, followed by
biotinylated goat anti-rabbit secondary antibody (1:500; cat. no.
SA00001-2; ProteinTech Group, Inc.) for 30 min at room temperature
and streptavidin-horseradish peroxidase (1:500; cat. no. A0303;
Beyotime Institute of Biotechnology) for 30 min at room
temperature. The stained cells were then immobilized and observed
under a light microscope (magnification, ×200).
Western blotting
Cell lysis buffer (Beyotime Institute of
Biotechnology) containing 0.1 mM phenylmethylsulfonyl fluoride (a
protease inhibitor) and phosphatase inhibitors (Nanjing KeyGen
Biotech Co., Ltd.), was used to lyse the cells and tissues. Protein
concentrations were measured using a bicinchoninic acid assay.
Protein samples (30-50 µg/lane) were separated via 8 and 12%
SDS-PAGE and transferred to PVDF membranes, which were subsequently
incubated with 5% dry milk in TBS-T saline [0.25 M Tris-HCl (pH
7.6), 0.19 M NaCl and 0.1% Tween 20] for 2 h at room temperature to
block non-specific binding. The protein blots were incubated
over-night at 4°C with primary antibodies against GAPDH (1:4,000;
ProteinTech Group, Inc.; cat. no. 60004-1-Ig), E2F1 (1:1,000;
Abcam; cat. no. ab179445), SIX1 (1:1,000; ProteinTech Group, Inc.;
cat. no. 10709-1-AP), collagen IV (1:2,000; Abcam; cat. no.
ab182744), fibronectin (1:1,000; ProteinTech Group, Inc.; cat. no.
15613-1-AP), α-smooth muscle actin (SMA; 1:2,000; Abcam; cat. no.
ab7817), E-cadherin (1:4,000; Abcam; cat. no. ab40772), N-cadherin
(1:4,000; Abcam; cat. no. ab76011) and vimentin (1:1,000; Abcam;
cat. no. ab92547) diluted with primary antibody dilution buffer
(Beyotime Institute of Biotechnology). Subsequently, the membranes
were washed thrice with TBS-T and treated with HRP-conjugated goat
anti-rabbit IgG (1:3,000; ProteinTech Group, Inc.; cat. no.
SA00001-2) or anti-mouse IgG (1:3,000; ProteinTech Group, Inc.;
cat. no. SA00001-1) diluted with secondary antibody dilution buffer
(Beyotime Institute of Biotechnology) at room temperature for 2 h.
The blots were then developed by incubation in a chemiluminescence
substrate (SuperSignal West Femto Maximum Sensitivity Substrate;
Thermo Fisher Scientific, Inc.) and exposed to X-ray films.
Chromatin immunoprecipitation (ChIP)
assay
The ChIP assay was performed using the EZ-Magna Chip
A kit (EMD Millipore; cat. no. 17-10086) following the
manufacturer's instructions. A total of 1×107 BEAS-2B
cells were fixed with 1% formaldehyde for 10 min at room
temperature, and stopped by addition of glycine to a final
concentration of 0.125 M. Fixed cells were harvested in 2 ml PBS
buffer with protease inhibitors. Cells were pelleted using
centrifugation (800 × g at 4°C for 5 min) and suspended in 0.5 ml
nuclear lysis buffer. Cells were sonicated with a 0.25-inch
diameter probe for 7W, 15 sec twice at 4°C and span at 10,000 × g
at 4°C for 10 min to remove insoluble material. For each
immunoprecipitation, 450 µl lysates was used. Samples were
spun, and the supernatants were incubated at 4°C for 3 h with
either no antibody, anti-IgG control antibody (1.0 µg; EMD
Millipore) or anti-E2F1 antibody (10.0 µg; Abcam; cat. no.
ab179445) to be tested. Immune complexes were recovered by adding
20 µl blocked protein A/G beads and incubated at 4°C
overnight. Pellet protein A/G magnetic beads were isolated using a
magnetic separator. DNA fragment extraction from beads was
performed using the ChIP assay kit. For ChIP-qPCR analysis, the
purified DNA from input or immunoprecipitated samples were assayed
by qPCR with SYBR Green I Master Mix (Takara Bio, Inc.) after
reverse cross-linking and DNA purification. The primers used were
as follows: Forward, 5′-CTT CCT TCT CCT CCC ACC ACT CC-3′ and
reverse, 5′-CCG GCG CAC TCA GTA GCC TTT-3′.
Statistical analysis
Statistical analyses were conducted using SPSS 22.0
(IBM Corp.). All data were presented as the mean ± SD from three
independent experiments. The differences among multiple groups were
analyzed using one-way ANOVA with repeated measures followed by
Tukey's post-hoc test, while differences between two groups by
Student's unpaired t-test. The correlation between various factors
was analyzed using Pearson's correlation analysis. TargetScanHuman
v7.2 (http://www.targetscan.org/vert_72/) and StarBase v2.0
(http://starbase.sysu.edu.cn/starbase2/) were used to
predict miRNA-targeting genes. P<0.05 was considered to indicate
a statistically significant difference.
Results
miR-106b-5p expression is downregulated
and SIX1 expression is upregulated in mice with asthma and
TGF-β1-induced BEAS-2B cells
To investigate whether miR-106b-5p expression was
different in vivo, a mouse model of asthma was established
in the present study. Fig. 1A
shows the treatment schedule of asthmatic mice model and the
control group. The difference was further verified by hematoxylin
and eosin staining of lung sections (Fig. 1B). The control group exhibited a
very small amount of inflammatory cell infiltration, without
thickening of bronchial wall (Fig.
1Ba). A moderate number of neutrophils and a small amount of
eosinophil infiltration was observed at the bronchial wall and
surrounding tissue of the OVA group, with slightly thickened wall
(Fig. 1Bb). RT-qPCR was performed
to estimate miR-106b-5p expression in the mouse model with airway
remodeling induced by repetitive OVA challenge and in control mice.
miR-106b-5p expression was significantly decreased in asthmatic
mice compared with in control mice (Fig. 1C). Yang et al (23) demonstrated that SIX1
downregulation effectively suppressed airway inflammation and
reversed airway remodeling in mice with asthma. Hence, the present
study investigated whether there was an association between
miR-106b-5p and SIX1. Therefore, mRNA and protein expression levels
of SIX1 were also analyzed (Fig. 1D
and F), and the correlation between miR-106b-5p and SIX1
expression was determined (Fig.
1E). SIX1 expression was significantly increased in the asthma
group compared with in the control group and was negatively
correlated with miR-106-5p expression. Furthermore, SIX1 expression
in the mouse lung tissues was evaluated using IHC, revealing that
SIX1 staining displayed higher intensity in lung tissues from the
asthmatic mice group compared with those from the control mice
(Fig. 1G). Additionally, SIX1
expression in the IHC analysis was quantified, revealing that SIX1
expression was significantly increased in the asthma group compared
with in the control group (Fig.
1K). In vitro, miR-106b-5p expression was analyzed in
BEAS-2B cells after treatment with TGF-β1 (10 ng/ml) for 6, 12, 24
and 48 h, revealing that miR-106b-5p expression was significantly
decreased in TGF-β1-induced BEAS-2B cells compared with the control
group at 24 and 48 h (Fig. 1H).
SIX1 expression was significantly increased in BEAS-2B cells
treated with TGF-β1 (10 ng/ml) for 24 h (Fig. 1I and J).
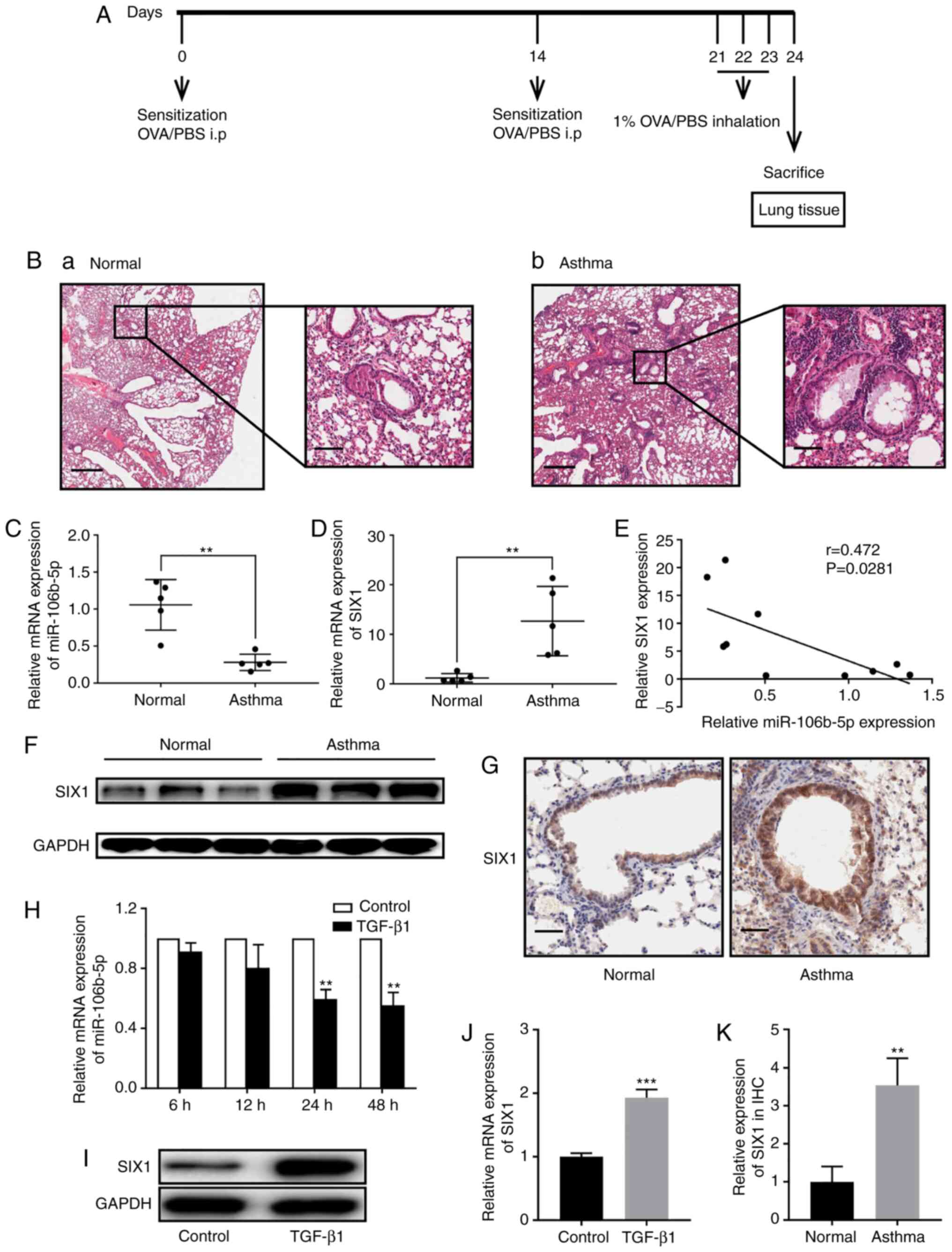 | Figure 1miR-106b-5p and SIX1 expression in
asthmatic mice and TGF-β1-induced BEAS-2B cells. (A) Treatment
schedule of asthmatic mice model and control group. (B) Lung
tissues in (a) normal and (b) asthmatic mice model (scale bar, 500
and 100 µm). Relative expression levels of (C) miR-106b-5p
and (D) SIX1 in asthmatic (n=5) and normal mice (n=5) were measured
by RT-qPCR. (E) Pearson's correlation analysis between miR-106b-5p
and SIX1 expression. (F) SIX1 protein expression in asthmatic and
normal mice. (G) SIX1 expression in the lung tissues of asthmatic
and normal mice was evaluated via IHC (scale bar, 50 µm).
(H) After TGF-β1 (10 ng/ml) treatment for 6, 12, 24 and 48 h,
miR-106b-5p expression in BEAS-2B cells was analyzed by RT-qPCR.
(I) Western blotting and (J) RT-qPCR were performed to detect the
protein and mRNA expression levels of SIX1, respectively, in
BEAS-2B cells after treatment with or without TGF-β1 (10 ng/ml) for
24 h. (K) Quantification of SIX1 expression according to IHC. Data
are presented as the mean ± SD of three independent experiments.
**P<0.01; ***P<0.001. miR, microRNA;
SIX1, SIX1, sine oculis homeobox homolog 1; RT-qPCR, reverse
transcription-quantitative PCR; IHC, immunohistochemistry. |
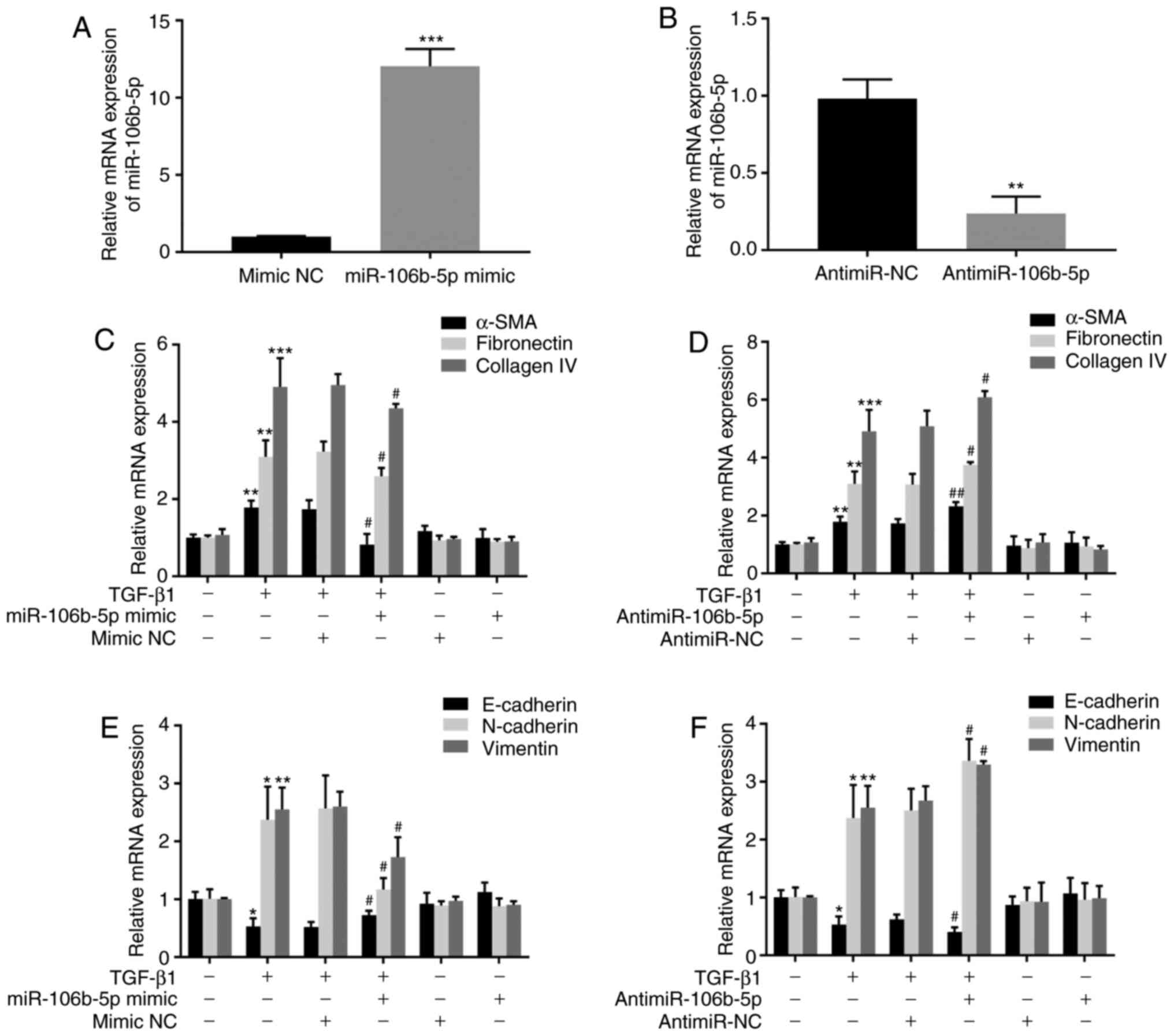
miR-106b-5p inhibits TGF-β1-induced
fibrosis and EMT in BEAS-2B cells
To further explore the function of miR-106b-5p in
TGF-β1-stimulated fibrosis and EMT in BEAS-2B cells, miR-106b-5p
mimics or antimiR-106b-5p were transfected into TGF-β1-stimulated
BEAS-2B cells. Firstly, RT-qPCR was employed to detect miR-106b-5p
expression after transfection. In BEAS-2B cells, a 12.04-fold
increase or a 4.13-fold decrease was detected in miR-106b-5p mRNA
expression after transfection with the miR-106b-5p mimic or
antimiR-106b-5p, respectively (Fig.
2A and B), indicating a high efficiency of transfection for
subsequent experiments. Secondly, the mRNA expression levels of
α-SMA, fibronectin and collagen IV were assessed to define the
function of miR-106b-5p in TGF-β1-stimulated fibrosis in BEAS-2B
cells. TGF-β1 treatment increased the expression levels of α-SMA,
fibronectin and collagen IV in BEAS-2B cells. The data suggested
that compared with TGF-β1 treatment alone, the overexpression of
miR-106b-5p suppressed α-SMA, fibronectin and collagen IV in
TGF-β1-induced BEAS-2B cells, while miR-106b-5p-knockdown
aggravated TGF-β1-stimulated fibrosis (Fig. 2C and D). In addition, the results
indicated that the overexpression or knockdown of miR-106b-5p did
not exert effects on fibrosis without TGF-β1 treatment (Fig. 2C and D). Thirdly, the mRNA
expression levels of EMT-associated markers, including E-cadherin,
N-cadherin and vimentin, were detected by RT-qPCR to identify
whether miR-106b-5p affected EMT in TGF-β1-stimulated BEAS-2B
cells. TGF-β1 treatment increased the expression levels of the
mesenchymal markers (N-cadherin and vimentin) and decreased the
expression levels of the epithelial marker E-cadherin in BEAS-2B
cells (Fig. 2E and F). The data
suggested that compared with TGF-β1 treatment alone, miR-106b-5p
overexpression in TGF-β1-induced cells negatively regulated the
expression levels of the mesenchymal markers (N-cadherin and
vimentin) and positively regulated the expression levels of the
epithelial marker E-cadherin, while miR-106b-5p-knockdown
aggravated TGF-β1-stimulated EMT (Fig. 2E and F). Additionally, the
overexpression or inhibition of miR-106b-5p alone did not affect
the basal gene expression levels in BEAS-2B cells (Fig. 2E and F).
miR-106b-5p does not bind to the
consensus sequences in the 3′-UTR of SIX1 mRNA
To explore the association between miR-106b-5p and
SIX1, western blotting and RT-qPCR were employed to measure SIX1
expression in miR-106b-5p-mimic-transfected or
antimiR-106b-5p-transfected BEAS-2B cells. SIX1 expression was
significantly decreased after miR-106b-5p mimic transfection and
significantly increased after antimiR-106b-5p transfection,
compared with their respective negative controls (Fig. 3A and B). miRNAs function by
downregulating downstream genes by guiding the cytoplasmic
RNA-induced silencing complexes targeting the 3′-UTR of mRNAs to
suppress their translation or induce their degradation (6). Thus, the present study explored the
potential effects of miR-106b-5p on the inhibition of mRNA function
by binding to consensus sequences in the 3′-UTR of SIX1. However,
the effects of miR-106b-5p on SIX1 mRNA targets could not be
determined using TargetScanHuman v7.2. To further investigate the
influence of miR-106b-5p on SIX1, the pmiR-RB-SIX1 reporter plasmid
and three different concentrations of miR-106b-5p mimic were
transfected into BEAS-2B cells. Subsequently, the dual-luciferase
assay detection kit revealed that miR-106b-5p did not have a
regulatory effect on the SIX1 3′-UTR (Fig. 3C and D).
 | Figure 3Association between miR-106b-5p and
SIX1. (A) miR-106b-5p and SIX1 mRNA expression in
miR-106b-5p-mimic- (50 nM) or antimiR-106b-5p-transfected (100 nM)
BEAS-2B cells. (B) Western blotting was performed to detect the
protein expression levels of SIX1 after transfecting BEAS-2B cells
with miR-106b-5p-mimic or antimiR-106b-5p. (C) Structure of
SIX1-3′-UTR plasmid, pmiR-RB-reporter construct in TM-SIX1 mRNA
3′-UTR of a reporter plasmid containing the SV40 promoter-driven
fluorescence reporter, downstream of the SIX1 mRNA 3′-UTR
construct. (D) Luciferase reporter plasmids containing the
wild-type SIX1-3′UTR were co-transfected into BEAS-2B cells with
miR-106b-5p (50, 100 or 150 nM) to detect the luciferase activity.
Data are presented as the mean ± SD of three independent
experiments. *P<0.05, **P<0.01 and
***P<0.001 vs. respective NCs. NS, not significant;
miR, microRNA; SIX1, SIX1, sine oculis homeobox homolog 1; NC,
negative control; UTR, untranslated region. |
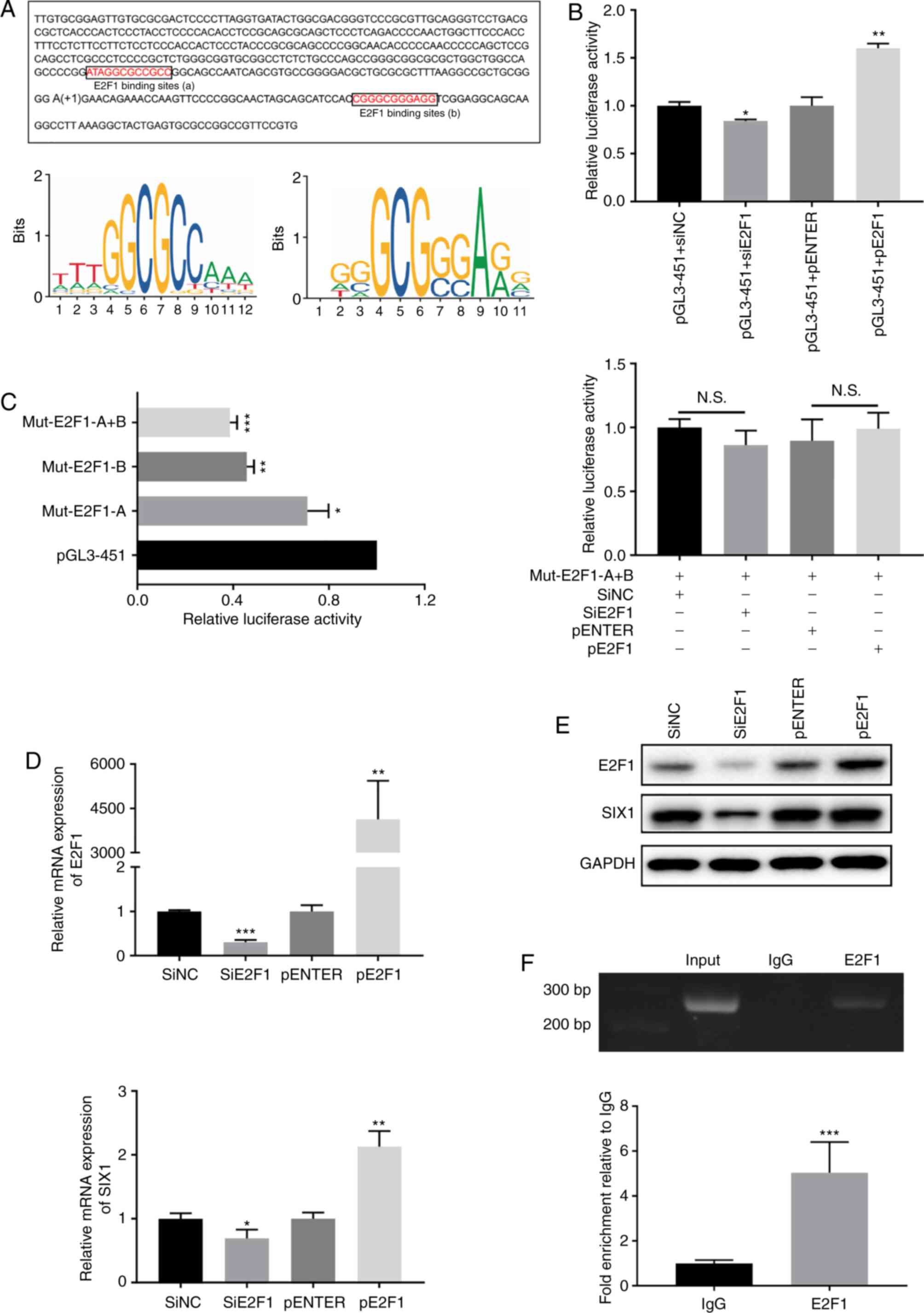
miR-106b-5p directly targets E2F1
E2F1 was identified as a target gene of miR-106b-5p
using TargetScan and StarBase v2.0. Hence, the mRNA and protein
expression levels of E2F1 were analyzed in asthmatic and control
mice, revealing that E2F1 expression was significantly increased in
the asthma group compared with in the control group (Fig. 4A and B). Additionally, E2F1
expression in the mouse lung tissues was evaluated using IHC,
revealing that E2F1 staining displayed a higher intensity in lung
tissues from the asthmatic mice group compared with those from the
control group (Fig. 4C). E2F1
expression in the IHC analysis was also quantified, revealing that
E2F1 expression was significantly increased in the asthma group
compared with in the control group (Fig. 4D). Furthermore, E2F1 expression
was significantly increased in BEAS-2B cells treated with 10 ng/ml
for 24 h (Fig. 4F). To
investigate the association between miR-106b-5p and E2F1, the E2F1
WT and MUT 3′-UTRs were cloned into the pmiR-RB-reporter (Fig. 4E). The results demonstrated that
the miR-106b-5p mimic significantly suppressed luciferase activity
in BEAS-2B cells treated with the E2F1 WT 3′-UTR, but did not have
influence on BEAS-2B cells treated with the E2F1 MUT 3′-UTR
(Fig. 4E). Subsequently, western
blotting and RT-qPCR were employed to measure E2F1 expression in
miR-106b-5p-mimic- or antimiR-106b-5p-transfected BEAS-2B cells,
revealing that E2F1 expression was significantly decreased after
miR-106b-5p mimic transfection and significantly increased after
antimiR-106b-5p transfection (Fig. 4G
and H), compared with their respective negative controls.
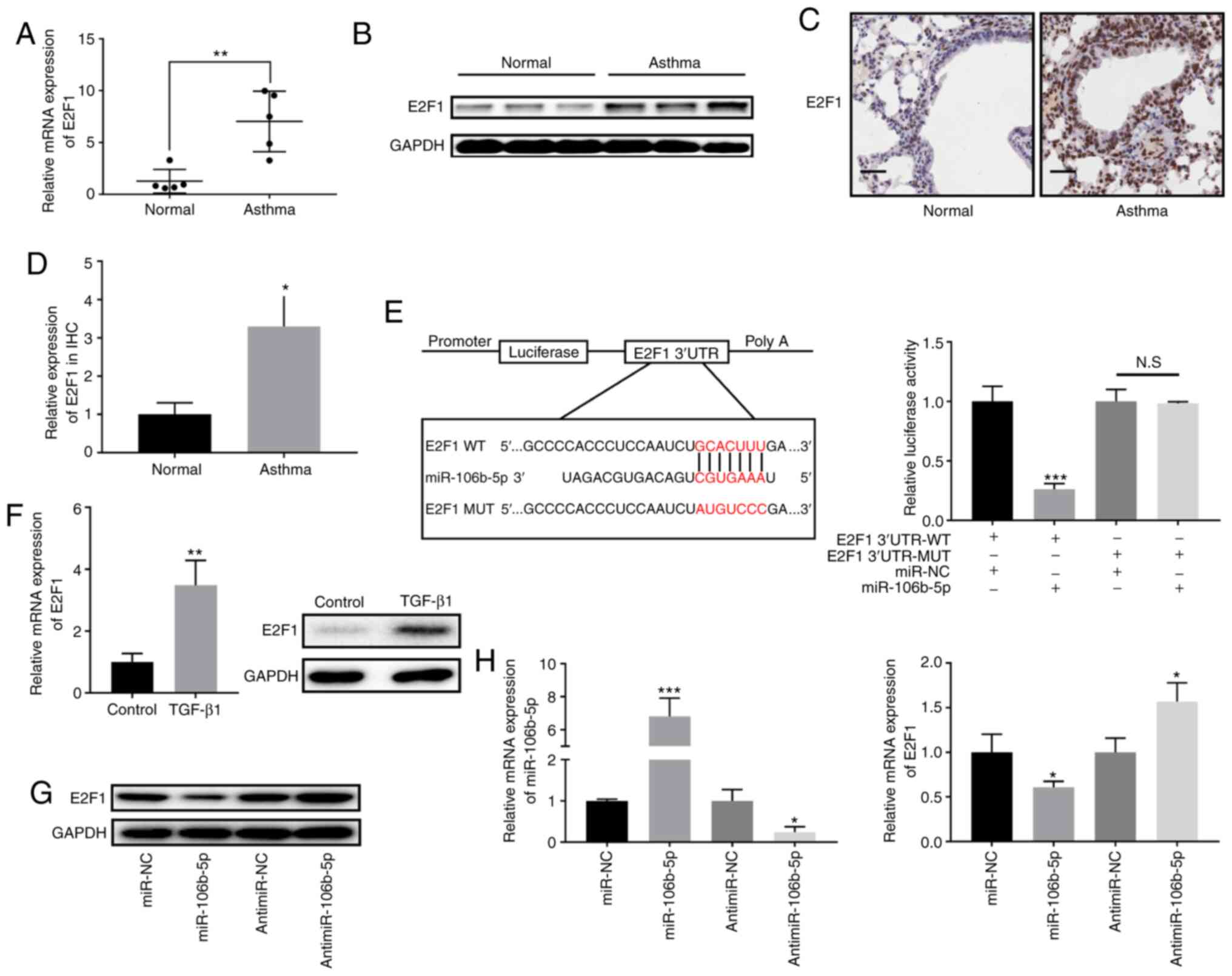 | Figure 4Association between miR-106b-5p and
E2F1. (A) Relative E2F1 expression in asthmatic (n=5) and normal
mice (n=5) was measured by RT-qPCR. (B) E2F1 protein expression in
asthmatic and normal mice. (C) E2F1 expression in the lung tissues
of asthmatic and normal mice was evaluated via IHC (scale bar, 50
µm). (D) Quantification of E2F1 expression according to IHC.
(E) Schematic illustration of the miR-106b-5p binding site in E2F1
and the site mutagenesis (left panel). Luciferase reporter plasmids
containing WT or MUT E2F1-3′-UTR were co-transfected into BEAS-2B
cells with miR-106b-5p or miR-NC (right panel). (F) RT-qPCR and
western blotting was performed to detect the mRNA and protein
expression levels, respectively, of E2F1 in BEAS-2B cells after
treatment with or without TGF-β1 (10 ng/ml) for 24 h. (G) E2F1
protein expression and (H) miR-106b-5p and E2F1 mRNA expression in
miR-106b-5p-mimic- or antimiR-106b-5p-transfected BEAS-2B cells.
Data are presented as the mean ± SD of three independent
experiments. *P<0.05, **P<0.01 and
***P<0.001 vs. respective NCs. RT-qPCR, reverse
transcription-quantitative PCR; NS, not significant; miR, microRNA;
E2F1, E2F transcription factor 1; NC, negative control; UTR,
untranslated region; WT, wild-type; MUT, mutant; IHC,
immunohistochemistry. |
E2F1 regulates SIX1 at the
transcriptional level
To explore whether E2F1 could regulate SIX1
transcription directly, a sequence from -351 to +100 upstream of
the human SIX1 promoter was cloned into pGL3-451. By scanning this
promoter region using the JASPAR database, two potential binding
sites and the key nucleotides of E2F1 were found (Fig. 5A). After co-transfecting pGL3-451
with plasmids containing siE2F1 or pE2F1, the luciferase activity
of SIX1 was significantly increased by E2F1 overexpression and
significantly decreased by siE2F1 (Fig. 5B). Subsequently, a series of
plasmids with point mutations of E2F1 binding sites were cloned and
were then transfected into BEAS-2B cells. As demonstrated by
Fig. 5C, E2F1-A and E2F1-B
mutations in BEAS-2B cells significantly inhibited the promoter
activity, as well as E2F1-A+B mutations. Additionally, when siE2F1
or pE2F1 were co-transfected with the mutations of E2F1-A+B into
BEAS-2B cells, there were no significant changes in luciferase
activity. To further explore the association between E2F1 and SIX1,
E2F1 was overexpressed or inhibited in BEAS-2B cells by siE2F1 or
pE2F1 transfection (Fig. 5D and
E). The results indicated that the mRNA and protein expression
levels of SIX1 were decreased with siE2F1 and increased with pE2F1,
compared with their respective negative controls (Fig. 5D and E). Additionally, the CHIP
assay performed in BEAS-2B cells further suggested that E2F1 could
bind to the promoter region of SIX1 in vitro (Fig. 5F).
E2F1- and SIX1-knockdown inhibits
TGF-β1-induced fibrosis and EMT in BEAS-2B cells
To further clarify the role of E2F1, E2F1 expression
was inhibited in BEAS-2B cells by siE2F1 transfection (Fig. 6A). siE2F1 decreased the
TGF-β1-stimulated expression levels of fibronectin, α-SMA and
collagen IV in BEAS-2B cells, indicating that E2F1-knockdown
inhibited TGF-β1-stimulated fibrosis (Fig. 6B and D). Furthermore, siE2F1
ameliorated TGF-β1-stimulated increases in the expression levels of
vimentin and N-cadherin, and TGF-β1-stimulated decreases in
E-cadherin expression (Fig. 6C and
E). The present results indicated that E2F1 silencing exhibited
a similar phenotype to overexpressed miR-106b-5p in BEAS-2B cells.
Additionally, siSIX1 was transfected in BEAS-2B cells to explore
the function of SIX1 on fibrosis and EMT in asthma. RT-qPCR and
western blotting were employed to measure SIX1 expression in
siSIX1-transfected cells compared with in siNC-transfected cells
(Fig. 6F). Western blot analysis
revealed that inhibition of SIX1 reversed TGF-β1-induced
upregulation of fibronectin, α-SMA, collagen IV, N-cadherin and
vimentin proteins, and TGF-β1-induced downregulation of E-cadherin
protein in BEAS-2B cells (Fig. 6G and
H). The current results demonstrated that SIX1-knockdown
impeded fibrosis and EMT in TGF-β1-induced BEAS-2B cells.
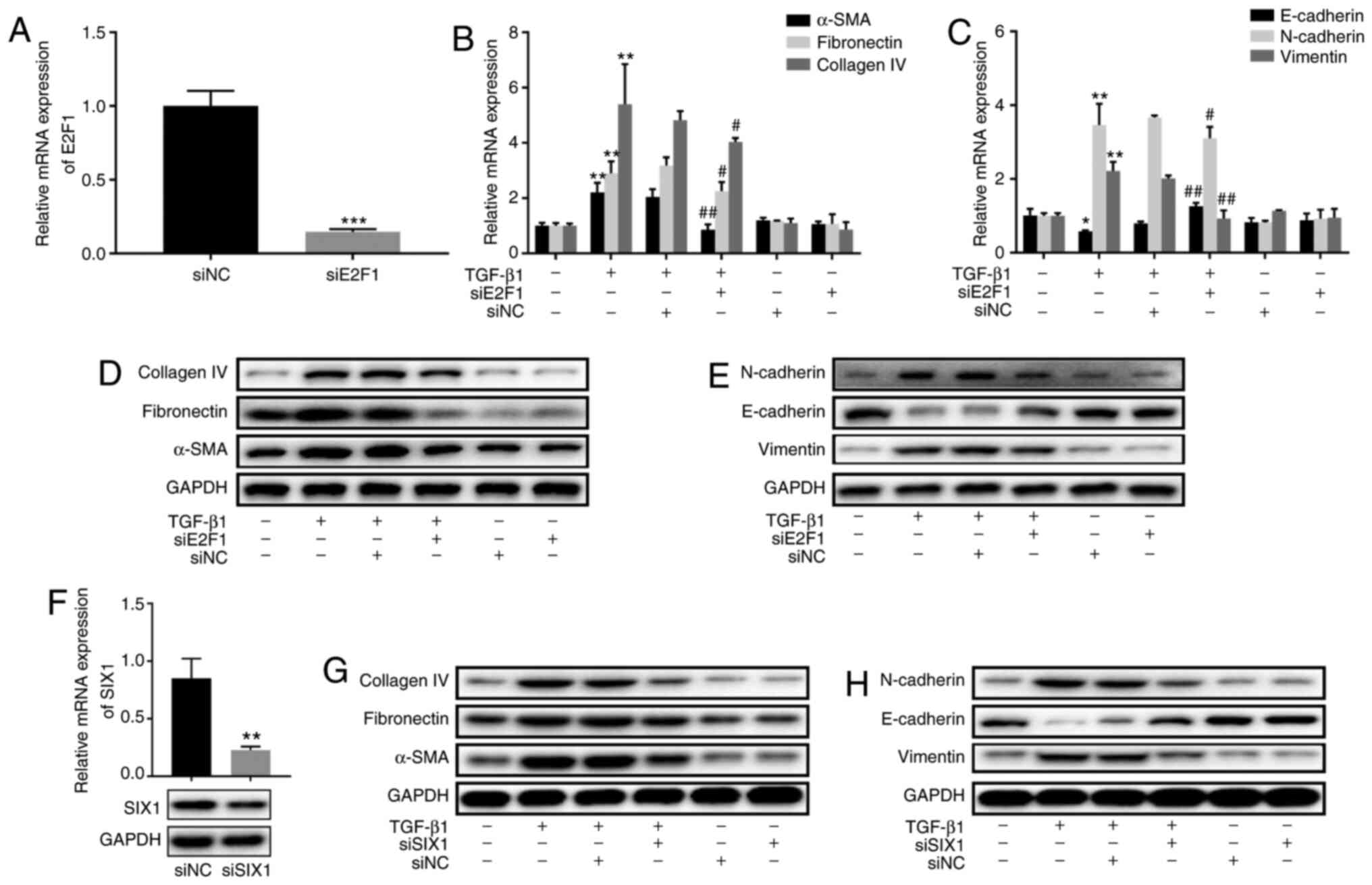 | Figure 6Effect of E2F1 and SIX1 on
TGF-β1-induced fibrosis and EMT in BEAS-2B cells. (A) E2F1
expression following siNC or siE2F1 transfection in BEAS-2B cells
was determined by RT-qPCR. (B) RT-qPCR was performed to assess the
expression levels of fibrosis-associated proteins α-SMA,
fibronectin and collagen IV in siE2F1-transfected BEAS-2B cells
after treatment with or without TGF-β1 for 24 h. (C) RT-qPCR was
employed to detect the mRNA expression levels of EMT-associated
proteins E-cadherin, N-cadherin and vimentin in siE2F1-transfected
BEAS-2B cells after treatment with or without TGF-β1 for 24 h. (D)
Protein expression levels of fibrosis-associated proteins α-SMA,
fibronectin and collagen IV in siE2F1-transfected BEAS-2B cells
after treatment with or without TGF-β1 for 24 h. (E) Protein
expression levels of EMT-associated proteins E-cadherin, N-cadherin
and vimentin in siE2F1-transfected BEAS-2B cells after treatment
with or without TGF-β1 for 24 h. (F) SIX1 expression in BEAS-2B
cells was determined by RT-qPCR and western blotting. (G) Protein
expression levels of fibrosis-associated proteins α-SMA,
fibronectin and collagen IV in siSIX1-transfected BEAS-2B cells
after treatment with or without TGF-β1 for 24 h. (H) Protein
expression levels of EMT-associated proteins E-cadherin, N-cadherin
and vimentin in siSIX1-transfected BEAS-2B cells after treatment
with or without TGF-β1 for 24 h. Data are presented as the mean ±
SD of three independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. control.
#P<0.05 and ##P<0.01 vs. siNC+TGF-β1.
miR, microRNA; E2F1, E2F transcription factor 1; SIX1, sine oculis
homeobox homolog 1; NC, negative control; si, small interfering;
RT-qPCR, reverse transcription-quantitative PCR; EMT,
epithelial-mesenchymal transition; SMA, smooth muscle actin. |
miR-106b-5p negatively regulates SIX1 via
E2F1
To detect whether miR-106b-5p could negatively
regulate SIX1 via E2F1, miR-106b-5p mimic and antimiR-106b-5p were
co-transfected with pGL3-451 or pGL3-451 with mutation of E2F1
binding sites. The luciferase assay revealed that miR-106b-5p
overexpression significantly inhibited luciferase activity in
pGL3-451, but not in pGL3-451 with mutation of E2F1 binding sites
(Fig. 7A). Meanwhile,
miR-106b-5p-knockdown significantly promoted luciferase activity in
pGL3-451, but not in pGL3-451 with mutation of E2F1 binding sites
(Fig. 7A). Additionally, western
blotting revealed that the miR-106b-5p-mediated decrease in SIX1
expression was partially rescued by co-transfection with pE2F1
(Fig. 7B). Subsequently, E2F1
binding to SIX1 promoter was examined in miR-106b-5p-mimic- and
antimiR-106b-5p-transfected BEAS-2B cells. The level of E2F1
binding to SIX1 promoter was decreased in
miR-106b-5p-mimic-transfected BEAS-2B cells, but increased after
antimiR-106b-5p transfection (Fig.
7C). Overall, the present results suggested that miR-106b-5p
could negatively regulate SIX1 expression in BEAS-2B cells via
E2F1.
Discussion
Airway remodeling, an inevitable outcome of severe
bronchial asthma (24), remains
irreversible in severe asthma, despite advances in asthma treatment
(25). Airway remodeling is
caused by persistent damage to the airway epithelium and is
manifested by subepithelial fibrosis, smooth muscle cell
proliferation, mucus cell metaplasia and excessive deposition of
extracellular matrix (ECM) (26).
It has been previously demonstrated that airway remodeling can
impair lung function of patients with asthma (27).
EMT has been demonstrated to contribute to airway
remodeling, causing chronic inflammatory airway diseases such as
asthma and chronic obstructive pulmonary disorder (28-30). During EMT, epithelial cells
gradually lose their epithelial features and acquire mesenchymal
characteristics, leading to airway remodeling and inflammation in
asthma (31). Additionally, in
patients with severe asthma, EMT decreases the sensitivity of
airway epithelial cells to drug treatment and glucocorticoid
therapy (32). In EMT, epithelial
cell-cell adhesions are disrupted and mesenchymal
membrane-associated proteins, such as N-cadherin and α-SMA, are
upregulated, whereas epithelial adhesion molecules such as
E-cadherin exhibit the opposite changes (11). Cell migration, alteration of ECM
deposition and differentiation are other important molecular
mechanisms involved in EMT (33).
EMT may be caused by signaling molecules such as TGF-β1, bone
morphogenetic proteins, epidermal growth factor, hepatocyte growth
factor and fibroblast growth factor (33). TGF-β1 is a growth factor secreted
by airway epithelial cells and infiltrating immune cells (34). Studies have revealed that patients
with asthma have higher levels of TGF-β1 in bronchoalveolar lavage
fluid and bronchial biopsy compared with the normal control group
(35,36). A high level of TGF-β1 helps
epithelial cells to transform fibroblasts into myofibroblasts,
which contributes to the development of EMT (28,37,38).
miR-106b-5p serves a vital role in cancer
progression, such as in glioma tumorigenesis (13), lung cancer (14), chronic myeloid leukemia (15), breast cancer (16,17) and hepatocellular carcinoma
(18). However, to the best of
our knowledge, there is no evidence on the function of miR-106b-5p
in fibrosis and EMT in asthma. In the present study, miR-106b-5p
expression was downregulated in asthmatic mice and TGF-β1-induced
BEAS-2B cells, which highlighted that miR-106b-5p may inhibit
TGF-β1-stimulated fibrosis in BEAS-2B cells. Subsequently,
miR-106b-5p was found to inhibit the mesenchymal markers vimentin
and N-cadherin, and promote the epithelial marker E-cadherin in
TGF-β1-induced BEAS-2B cells. The opposite results were found after
inhibiting miR-106b-5p. The present results suggested that E2F1, as
a direct target for miR-106b-5p, may increase SIX1 expression by
binding to its promoter region. Additionally, SIX1 was demonstrated
to be a TGF-β1-inducible gene. To the best of our knowledge, the
present study was the first to identify the involvement of the
miR-106b-5p/E2F1/SIX1 signaling pathway in asthma. Therefore,
miR-106b-5p may be a potential therapeutic target for asthma.
Furthermore, the current data suggested that
miR-106b-5p directly targeted the 3′-UTR of the E2F1 mRNA. Although
E2F1 has been demonstrated to promote pathological liver fibrosis
(39), its role in asthma has not
been evaluated. Hence, the exact function of E2F1 in asthma was
explored in the present study. It was revealed that the expression
levels of the EMT-associated markers vimentin and N-cadherin were
downregulated, while E-cadherin expression was upregulated in
TGF-β1-induced BEAS-2B cells transfected with siE2F1. Additionally,
the current data revealed that E2F1-knockdown effectively prevented
TGF-β1-induced fibrosis in vitro, which was then reversed by
co-transfection with the antimiR-106b-5p, indicating the critical
role of E2F1 in preventing airway remodeling in asthma.
The present study further revealed that E2F1, a
classical transcription factor, positively regulated SIX1 at the
transcriptional level. Western blotting and RT-qPCR results
demonstrated that E2F1 acted as a transcriptional inducer to
enhance SIX1 expression. CHIP and luciferase assays further
confirmed this hypothesis.
In conclusion, the present study identified that
miR-106b-5p inhibited fibrosis and EMT via the
miR-106b-5p/E2F1/SIX1 signaling pathway in TGF-β1-induced BEAS-2B
cells. Therefore, miR-106b-5p may be a potential therapeutic target
for asthma.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81970579 and
81972954).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SL mainly designed the experiments and wrote the
manuscript. XC and XW contributed to performing the cell
experiments and reviewing the manuscript. SZ, XD and JC designed
and conducted the animal experiments. SL, XC, XW, SZ, XD and JC
contributed to data analysis. GZ mainly constructed the idea of
this article and provided administrative, technical and material
support. SL, XC and GZ were responsible for confirming the
authenticity of the data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was approved by the Nanjing Medical
University Animal Experimental Ethics Committee (approval no.
2005020).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Ellwood P, Asher MI, Billo NE, Bissell K,
Chiang CY, Ellwood EM, El-Sony A, García-Marcos L, Mallol J, Marks
GB, et al: The Global Asthma Network rationale and methods for
Phase I global surveillance: Prevalence, severity, management and
risk factors. Eur Respir J. 49:16016052017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prasad B, Nyenhuis SM, Imayama I, Siddiqi
A and Teodorescu M: Asthma and obstructive sleep apnea overlap:
What has the evidence taught Us? Am J Respir Crit Care Med.
201:1345–1357. 2020. View Article : Google Scholar
|
|
3
|
Woodcock A, Vestbo J, Bakerly ND, New J,
Gibson JM, McCorkindale S, Jones R, Collier S, Lay-Flurrie J, Frith
L, et al: Effectiveness of fluticasone furoate plus vilanterol on
asthma control in clinical practice: An open-label, parallel group,
randomised controlled trial. Lancet. 390:2247–2255. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sharma P, Yi R, Nayak AP, Wang N, Tang F,
Knight MJ, Pan S, Oliver B and Deshpande DA: Bitter taste receptor
agonists mitigate features of allergic asthma in mice. Sci Rep.
7:461662017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Breton JD, Heydet D, Starrs LM, Veldre T
and Ghildyal R: Molecular changes during TGFβ-mediated lung
fibroblast-myofibroblast differentiation: Implication for
glucocorticoid resistance. Physiol Rep. 6:e136692018. View Article : Google Scholar
|
|
6
|
Aquino-Jarquin G: Emerging role of
CRISPR/Cas9 technology for MicroRNAs editing in cancer research.
Cancer Res. 77:6812–6817. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Giroux P, Bhajun R, Segard S, Picquenot C,
Charavay C, Desquilles L, Pinna G, Ginestier C, Denis J, Cherradi N
and Guyon L: miRViz: A novel webserver application to visualize and
interpret microRNA datasets. Nucleic Acids Res. 48:W252–W261. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu L, Xu J, Liu J, Zhang H, Sun C, Wang Q,
Shi C, Zhou X, Hua D, Luo W, et al: The novel chromatin
architectural regulator SND1 promotes glioma proliferation and
invasion and predicts the prognosis of patients. Neuro Oncol.
21:742–754. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu G, Li Y, Ma Y, Lu J, Chen Y, Jiang Q,
Qin Q, Zhao L, Huang Q, Luo Z, et al: Long noncoding RNA LINC00511
contributes to breast cancer tumourigenesis and stemness by
inducing the miR-185-3p/E2F1/Nanog axis. J Exp Clin Cancer Res.
37:2892018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang CJ, Zhu CC, Xu J, Wang M, Zhao WY,
Liu Q, Zhao G and Zhang ZZ: Correction to: The lncRNA UCA1 promotes
proliferation, migration, immune escape and inhibits apoptosis in
gastric cancer by sponging anti-tumor miRNAs. Mol Cancer.
18:1292019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Du X, Tu Y, Liu S, Zhao P, Bao Z, Li C, Li
J, Pan M and Ji J: LINC00511 contributes to glioblastoma
tumorigenesis and epithelial-mesenchymal transition via
LINC00511/miR-524-5p/YB1/ZEB1 positive feedback loop. J Cell Mol
Med. 24:1474–1487. 2020. View Article : Google Scholar
|
|
12
|
Chiba Y and Misawa M: MicroRNAs and their
therapeutic potential for human diseases: MiR-133a and bronchial
smooth muscle hyperresponsiveness in asthma. J Pharmacol Sci.
114:264–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu F, Gong J, Huang W, Wang Z, Wang M,
Yang J, Wu C, Wu Z and Han B: MicroRNA-106b-5p boosts glioma
tumorigensis by targeting multiple tumor suppressor genes.
Oncogene. 33:4813–4822. 2014. View Article : Google Scholar
|
|
14
|
Chen Z, Chen X, Lei T, Gu Y, Gu J, Huang
J, Lu B, Yuan L, Sun M and Wang Z: Integrative analysis of NSCLC
identifies LINC01234 as an oncogenic lncRNA that interacts with
HNRNPA2B1 and regulates miR-106b biogenesis. Mol Ther.
28:1479–1493. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang J, Yin Z, Li Y, Liu Y, Huang G, Gu C
and Fei J: The identification of long non-coding RNA H19 target and
its function in chronic myeloid leukemia. Mol Ther Nucleic Acids.
19:1368–1378. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Z, Li TE, Chen M, Pan JJ and Shen KW:
miR-106b-5p contributes to the lung metastasis of breast cancer via
targeting CNN1 and regulating Rho/ROCK1 pathway. Aging (Albany NY).
12:1867–1887. 2020. View Article : Google Scholar
|
|
17
|
Lee J, Kim HE, Song YS, Cho EY and Lee A:
miR-106b-5p and miR-17-5p could predict recurrence and progression
in breast ductal carcinoma in situ based on the transforming growth
factor-beta pathway. Breast Cancer Res Treat. 176:119–130. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gu H, Gu S, Zhang X, Zhang S, Zhang D, Lin
J, Hasengbayi S and Han W: miR-106b-5p promotes aggressive
progression of hepatocellular carcinoma via targeting RUNX3. Cancer
Med. 8:6756–6767. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fornes O, Castro-Mondragon JA, Khan A, van
der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M,
Baranašić D, et al: JASPAR 2020: Update of the open-access database
of transcription factor binding profiles. Nucleic Acids Res.
48:D87–D92. 2020.
|
|
20
|
Feng DD, Cao Q, Zhang DQ, Wu XL, Yang CX,
Chen YF, Yu T, Qi HX and Zhou GP: Transcription factor E2F1
positively regulates interferon regulatory factor 5 expression in
non-small cell lung cancer. OncoTargets Ther. 12:6907–6915. 2019.
View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Young K and Morrison H: Quantifying
microglia morphology from photomicrographs of immunohistochemistry
prepared tissue using imagej. J Vis Exp. 57648:2018.
|
|
23
|
Yang ZC, Yi MJ, Shan YC, Wang C, Ran N,
Jin LY, Fu P, Feng XY, Xu L and Qu ZH: Targeted inhibition of Six1
attenuates allergic airway inflammation and remodeling in asthmatic
mice. Biomed Pharmacother. 84:1820–1825. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Olafsdottir TA, Theodors F, Bjarnadottir
K, Bjornsdottir US, Agustsdottir AB, Stefansson OA, Ivarsdottir EV,
Sigurdsson JK, Benonisdottir S, Eyjolfsson GI, et al: Eighty-eight
variants highlight the role of T cell regulation and airway
remodeling in asthma pathogenesis. Nat Commun. 11:3932020.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bertolini F, Carriero V, Bullone M, Sprio
AE, Defilippi I, Sorbello V, Gani F, Di Stefano A, Massimo Luigi
and Ricciardolo F: Correlation of matrix-related airway remodeling
and bradykinin B1 receptor expression with fixed airflow
obstruction in severe asthma. Allergy. Dec 7–2020.Epub ahead of
print. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vignola AM, Kips J and Bousquet J: Tissue
remodeling as a feature of persistent asthma. J Allergy Clin
Immunol. 105:1041–1053. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ward C, Pais M, Bish R, Reid D, Feltis B,
Johns D and Walters EH: Airway inflammation, basement membrane
thickening and bronchial hyperresponsiveness in asthma. Thorax.
57:309–316. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hackett TL, Warner SM, Stefanowicz D,
Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick
SM, Bai TR and Knight DA: Induction of epithelial-mesenchymal
transition in primary airway epithelial cells from patients with
asthma by transforming growth factor-beta1. Am J Respir Crit Care
Med. 180:122–133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y, Jia M, Yan X, Cao L, Barnes PJ,
Adcock IM, Huang M and Yao X: Increased neutrophil
gelatinase-associated lipocalin (NGAL) promotes airway remodelling
in chronic obstructive pulmonary disease. Clin Sci (Lond).
131:1147–1159. 2017. View Article : Google Scholar
|
|
30
|
Gorowiec MR, Borthwick LA, Parker SM,
Kirby JA, Saretzki GC and Fisher AJ: Free radical generation
induces epithelial-to-mesenchymal transition in lung epithelium via
a TGF-β1-dependent mechanism. Free Radic Biol Med. 52:1024–1032.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hackett TL: Epithelial-mesenchymal
transition in the pathophysiology of airway remodelling in asthma.
Curr Opin Allergy Clin Immunol. 12:53–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ijaz T, Pazdrak K, Kalita M, Konig R,
Choudhary S, Tian B, Boldogh I and Brasier AR: Systems biology
approaches to under-standing epithelial mesenchymal transition
(EMT) in mucosal remodeling and signaling in asthma. World Allergy
Organ J. 7:132014. View Article : Google Scholar
|
|
33
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ito J, Harada N, Nagashima O, Makino F,
Usui Y, Yagita H, Okumura K, Dorscheid DR, Atsuta R, Akiba H and
Takahashi K: Wound-induced TGF-β1 and TGF-β2 enhance airway
epithelial repair via HB-EGF and TGF-α. Biochem Biophys Res Commun.
412:109–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chakir J, Shannon J, Molet S, Fukakusa M,
Elias J, Laviolette M, Boulet LP and Hamid Q: Airway
remodeling-associated mediators in moderate to severe asthma:
Effect of steroids on TGF-beta, IL-11, IL-17, and type I and type
III collagen expression. J Allergy Clin Immunol. 111:1293–1298.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamaguchi M, Niimi A, Matsumoto H, Ueda T,
Takemura M, Matsuoka H, Jinnai M, Otsuka K, Oguma T, Takeda T, et
al: Sputum levels of transforming growth factor-beta1 in asthma:
Relation to clinical and computed tomography findings. J Investig
Allergol Clin Immunol. 18:202–206. 2008.PubMed/NCBI
|
|
37
|
Warburton D, Schwarz M, Tefft D,
Flores-Delgado G, Anderson KD and Cardoso WV: The molecular basis
of lung morphogenesis. Mech Dev. 92:55–81. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Holgate ST, Davies DE, Puddicombe S,
Richter A, Lackie P, Lordan J and Howarth P: Mechanisms of airway
epithelial damage: Epithelial-mesenchymal interactions in the
pathogenesis of asthma. Eur Respir J Suppl. 44:24s–29s. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Y, Xu N, Xu J, Kong B, Copple B, Guo
GL and Wang L: E2F1 is a novel fibrogenic gene that regulates
cholestatic liver fibrosis through the Egr-1/SHP/EID1 network.
Hepatology. 60:919–930. 2014. View Article : Google Scholar : PubMed/NCBI
|