1. Introduction
Ciliopathies comprise a heterogeneous group of
genetic disorders caused by structural or functional disruption of
cilia, or by abnormal cilia biogenesis (1,2).
The two main subcategories, namely motile and immotile/primary
ciliopathies, both involve disruption of the cilium, and also share
several causal genes (3-5). However, clinically, they are quite
different; while motile ciliopathies (Kartagener syndrome and
primary ciliary dyskinesia) are characterized by pulmonary disease,
infertility, situs inversus or reversal of organ laterality
(6), primary ciliopathies
include a wide class of diseases that range from organ-specific
disorders to pleiotropic syndromes with multiorgan involvement.
These distinct phenotypes may be explained through the structural
differences between primary and motile cilia, as well as their
distinct functions (7).
The aim of the present review was to comprehensively
describe the primary ciliopathies, focusing on genetic
heterogeneity, diagnosis and clinical aspects, with a brief
overview of their biological basis.
2. Cilium
Structure
Motile cilia have been observed in protozoa since
the early microscopy era (8).
Unlike motile cilia, which are concentrated in clusters and line
the respiratory tract, fallopian tubes, the efferent ductules of
the testis and brain ventricles (9), the primary cilium is a single
hair-like organelle, with variable length (1-9 μm) (10), projecting from the apical surface
of almost all types of cells, with certain exceptions (lymphocytes,
granulocytes, hepatocytes and acinar cells) (11). Primary cilia are dynamic
organelles that are assembled in the G0/G1
cell cycle stage and become disassembled with the onset of cell
division (12).
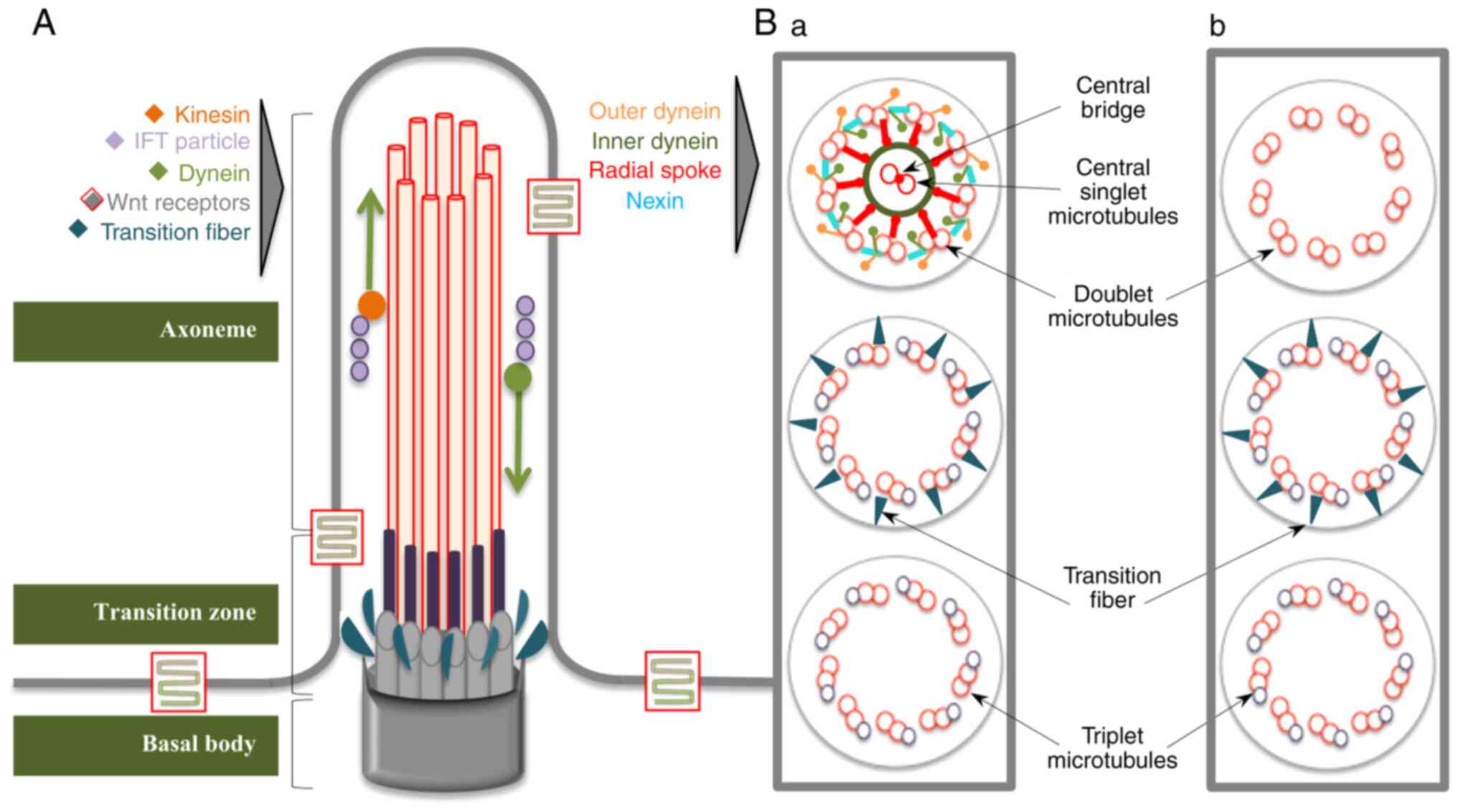
Both types of cilia are structurally composed of a
microtubule backbone, termed the axoneme, surrounded by matrix and
covered by the ciliary membrane, which is continuous with the
plasma membrane (Fig. 1). At the
base of this ensemble, a specialized centriole, referred to as the
basal body (BB), docks the cilium to the cell (13). The axoneme of the primary cilium
consists of 9 outer doublet microtubules, (9 + 0 type), while
motile cilia possess an extra inner pair of microtubules,
reinforced by nexin bridges (9 + 2 type) and an accessory structure
involved in motility, formed of dynein arms and radial spokes
(14). Each doublet contains a
complete microtubule (A tubule) and one incomplete microtubule (B
tubule), which are composed of tubulin protofilaments and are
attached to each other through tektins and Ca-binding ribbon
proteins (15). The BB structure
contains 9 radially arranged microtubule triplets (A, B and C) and
no central pair. The A and B tubules expand into the proximal
segment of the cilium and are connected to the ciliary membrane by
Y fibers, constituting a distinctive subcompartment known as the
transition zone. Proximal to the Y fibers are the transitional
fibers, which help to anchor the BB to the plasma membrane
(16). The BB is responsible for
the configuration of the microtubule scaffold and coordinates the
ciliary trafficking pathway; thus, it is involved, together with
transitional fibers, in ciliogenesis (17). Surrounding the transitional
fibers are numerous strings of particles, acting as a selective
filter for intraflagellar transport (IFT) molecules, known as the
ciliary necklace (18,19). In the distal region of the
cilium, the backbone contains a single microtubule fiber (A
tubule), which delimits the ciliary tip, a proteic zone with cell
type-specific structure and function (20,21). In addition to this classical
structure of the cilia, there is evidence showing the existence of
motile 9 + 0 cilia, covering the node, which are responsible for
left-right asymmetry, or sensory 9 + 2 cilia, which are observed in
the inner ear cells (22-26).
Function of the primary cilium
Since their discovery in the kidneys and the thyroid
gland by Zimmermann in 1898, primary cilia have been considered as
vestigial organelles, without a specific function, due to their
lack of motility and their absence in several cells during mitosis
(27). In 1975, Webber and Lee
(28) raised the hypothesis of a
possible sensory role of mammalian nephron cilia, by comparing them
to those in sensory tissues. This hypothesis was confirmed in 2000
in a study by Pazour et al (29), which presented experimental
evidence showing the physiological function of the primary cilium.
Once the implication of primary cilia in human diseases was
demonstrated (30), the
awareness of the significance of this organelle increased.
Subsequently, a number of studies demonstrated the complex roles of
primary cilia as mechanoreceptors, chemoreceptors and osmosensors
(31-34).
A highly specialized process occurring in the
ciliary compartment is the IFT: A bidirectional movement during
which a protein complex (IFT particle) is shuttled along the
microtubule backbone from the BB to the tip of the cilium (through
kinesin-anterograde transport) and back (facilitated by
dynein-retrograde transport) (35). As the synthesis of proteins
essential for the development of cilia is not possible inside the
ciliary compartment and proteins are carried through IFT, the
importance of IFT in ciliogenesis must be emphasized, as well as
its involvement in the delivery of signals from the cilium to the
cell, higlighting its significant role in cilia-mediated signaling
pathways (36).
Furthermore, >25 receptors and ion channels have
been localized to the ciliary membrane, where a growing number of
extracellular signals are received and transduced by the ciliary
ensemble, facilitating certain signaling pathways that control the
development of organs, as well as behavioral processes.
Particularly important primary cilia-related signaling pathways
include the following: Wingless (Wnt), Hedgehog (Hh), receptor
tyrosine kinase (RTK), G-protein coupled receptors (GPCRs), Notch,
transforming growth factor-β (TGF-β), mechanistic target of
rapamycin (mTOR) and Salvador-Warts-Hippo (SWH) signaling. In
addition, other signaling pathways that have been linked to primary
cilia include extracellular matrix protein-mediated signaling,
transient receptor potential channel-mediated signaling,
vasopressin signaling in renal epithelial cells, somatostatin,
serotonin and melanin-concentrating hormone signaling (37,38).
The Wnt signaling pathway comprises a large family
of secreted, cysteine-rich proteins, acting as a network of signal
transduction pathways that are responsible for embryonic
development, as well as tissue homeostasis and regeneration in
adults (39). At least three
signaling pathways have been described: The canonical Wnt pathway
(or Wnt/β-catenin pathway), and the non-canonical planar cell
polarity (PCP) and Wnt/Ca2+ pathways. The primary cilium
and BB were found to be required for the regulation of both
canonical and non-canonical Wnt signaling pathways. Canonical Wnt
signaling acts through its end effectors as co-transcriptional
factors, together with the T-cell factor/lymphoid enhancer factor 1
family of proteins, and co-activates the expression of Wnt target
genes to modulate the cell cycle, leading to cell differentiation,
proliferation, adhesion and migration, and tissue development
(40). Canonical Wnt signaling
appears to be directly or indirectly implicated in the formation of
almost all organ systems during embryogenesis; it has been shown to
be involved in anterior head fold formation and neuroectodermal
patterning, in controlling further posterior patterning, as well as
in the genesis and development of the heart, lungs, kidney, eyes,
skin, blood cells and bone (41,42). In addition, the essential role of
the Wnt pathway in stem cell renewal has been highlighted (42,43). The non-canonical PCP pathway
appears to act independently on transcription and plays a key role
in the modification and rearrangement of the actin cytoskeleton.
Moreover, the molecular constituents of this pathway were shown to
randomize the orientation of polarized epithelial cells and to
coordinate the morphology and convergent extension of dorsal
mesodermal and ectodermal cells during gastrulation and neural tube
closure (44). Yet, the role of
cilia in canonical Wnt signal transduction is controversial,
although several studies have suggested the importance of primary
cilia in the decrease of canonical Wnt signaling (45-47). By contrast, the contribution of
the integrity of primary cilia to the non-canonical PCP Wnt pathway
is well established. Movement of the BB to the apical cell surface
and centriolar position are essential for the establishment of cell
polarity; thus, defects in ciliary proteins implicated in
ciliogenesis and BB migration lead to various PCP errors (48). The Wnt/Ca2+ pathway
shares a number of components with the PCP, but has been described
as a separate pathway, which stimulates intracellular
Ca2+ release from the endoplasmic reticulum.
Ca2+ waves are hypothesized to serve as a key modulator
in early pattern formation during embryo gastrulation. The
Wnt/Ca2+ pathway regulates embryogenesis in a complex
manner, including promoting ventral cell fate, negative regulation
of dorsal axis development, regulation of tissue separation and
convergent extension movements during gastrulation, as well as
heart formation. The Wnt/Ca2+ pathway also functions as
a critical modulator of both the canonical and PCP pathways
(44). Wnt signaling also
regulates a number of other signaling pathways that have not been
yet completely elucidated, but appear to be linked with myogenesis,
axonal guidance, neuronal migration and synaptogenesis (49,50).
Another key signaling pathway that was demonstrated
to be essential for a variety of developmental processes is the Hh
signaling pathway, described through its three Hh homologues:
Desert Hh, Indian Hh (Ihh) and Sonic Hh (Shh). The Shh pathway, the
most extensively investigated signaling pathway, functions due to
the synergy of several molecule/proteins acting as transmembrane
receptors, namely Patched homolog 1, Smoothened (SMO) and GLI
transcription factors, leading to the transcription of Hh target
genes (51). The role of Shh
proteins emerges during embryonic development and morphogenesis,
controlling left-right asymmetry, dorso-ventral axes and distal
limb patterning. Moreover, proliferation of hematopoietic, retinal
and neural stem cells, as well as development of epithelial tissues
during organogenesis, appear to be modulated by Shh (52). Primary cilia are essential for
the transduction of Hh signaling, playing a dual role through
positive and negative regulation. It has been shown that abnormal
cilia may lead to either loss-of-function Hh phenotypes in the
neural tube, or gain-of-function Hh phenotypes in the limbs,
indicating that the Hh pathway may play an important role in
primary cilia biogenesis (38).
The migration, proliferation, differentiation and
apoptosis of cells are also controlled by another cilia-related
pathway, the platelet-derived growth factor receptor-α (PDGFRα)
pathway (53,54). PDGFRα belongs to the large family
of RTK transmembrane receptors and is required for activation of
the Ras-Mek1/2-Erk1/2 pathway, thus causing axonemal
reestablishment, cell cycle progression and chemotaxis (55). The development of numerous cells
and tissues, including neurons, oligodendrocytes, astrocytes,
alveolar smooth muscle cells, cardiac fibroblasts and bone cells,
relies on PDGFα signaling (56).
PDGFRα, which is bound to the membrane of the primary cilium,
regulates cytoskeletal reorganization to drive directional
migration of fibroblasts in wound healing. Defects in primary cilia
lead to abnormal wound healing. Moreover, disassembly of cilia,
which allows the centriole to participate in mitosis during cell
cycle progression, is modulated by PGFRα signaling (57). Along with this signaling, other
RTK signaling pathways have recently been described, including
EGFR, which plays an important role in mechanosensation and the
migration of kidney epithelial cells or airway smooth muscle cells,
and insulin-like growth factor receptor, which is involved in
preadipocyte differentiation (37).
GPCRs comprise a large family of transmembrane
receptors divided into six classes (A-F), for which >30
receptors belonging to the A (rhodopsin-like receptors), B
(secretin receptor family) and F (frizzled/SMO-component of Shh
signaling) classes are found on the ciliary membrane. Among these
receptors, opsin, olfactory, serotonin (HTR6), somatostatin
(SSTR3), vasopressin (V2R), dopamine (D1R, D2R and D5R) and
prostaglandin (EP4) receptors are involved in a wide spectrum of
cellular and physiological processes, including photoreception,
olfactory sensation, feeding behavior, pain sensation, osmotic
function in kidney cells, physiological function in cardiac
myocytes, neuronal processes and energy homeostasis (58,59). GPCRs are involved in neuronal or
retinal cilia function and control the length of primary cilia or
ciliogenesis. Conversely, absence or shortening of primary cilia
may interfere with normal brain development, interneuron
connectivity, gonadotropin hormone release at the nerve terminals
or the sensory potential of cells (48).
In addition to these main signaling cascades, an
increasing number of pathways have been associated with primary
cilia. It has been concluded that the Notch signaling is involved
in the physiology of primary cilia. Notch3 receptor, which is
localized in the ciliary membrane, is activated by presenilin 2, a
ciliary BB enzyme, thereby regulating epidermal cell proliferation
and differentiation (60). Loss
of primary cilia or knockdown of IFT molecules may result in
diminished Notch activation, leading to decreased cell
proliferation and differentiation defects. In the neuroepithelia of
the developing neural tube, activation of Notch signaling leads to
increased primary cilium length, as well as accumulation of Smo
molecules within the primary cilium. This interplay between the
Notch and Shh pathways in primary cilia may specify ventral cell
fate in the developing neural tube (38).
TGF-β signaling has been recently associated with
cilia, whereas TGF-β1 and TGF-β2 receptors are located on the
ciliary tip. Primary cilia use diverse methods to regulate TGF-β
pathways, through SMAD2/3 and ERK1/2 activation by TGF stimulation,
modulating various cellular processes, such as the differentiation
of cardiomyocytes, osteocytes and myofibroblasts. Moreover,
endothelial primary cilia, which act as flow sensors in the blood
vessels, inhibit the endothelial-to-mesenchymal transition, and
this process is related to attenuation of the TGF-β signaling.
There is also strong evidence regarding the impairment of
mechanosensation and maturation in human osteoblasts due to
shortening of primary ciliary length through TGF-β signaling
(61,62).
The SWH pathway controls organ size and cell
proliferation through a core of serine/threonine-kinases that
interact with nephrocystin 4 or Crumbs 3 receptors located in the
cilium. One of the major components of SWH signaling, MST1/2, which
is localized to the BB, has been found to be crucial for primary
cilia biogenesis, with loss of MST1/2 leading to defects in
ciliogenesis (63).
It has also been demonstrated that the primary
cilium regulates mTOR signaling, which plays a pivotal role in
metabolism and cell proliferation, thereby determining cell size,
through the Lkb1 tumor suppressor, AMP-activated protein kinase and
folliculin. In epithelial primary cilia, the mTOR pathway is
upregulated by polycystin-1 through the tuberin protein, thus being
involved in cyst formation (64,65).
Brain-derived neurotrophic factor signaling, which
is involved in neuronal development, synaptic plasticity, satiety
and weight control, has recently been proposed to be linked with
the BBS4 protein and primary cilia (66).
3. Ciliopathies
Given the notable complexity of interconnected
signaling pathways in cilia, the role of the primary cilium as a
cellular hub is becoming increasingly obvious; its clinical
importance emerges from the consequences of its structural or
functional defects, which lead to a broad category of disorders,
collectively termed as ciliopathies.
The term 'ciliopathy' is most likely attributed to
immotile or primary cilia-related disorders, and it has been
recently allocated to certain conditions that have long been known
as separate clinical entities (67). The first ciliophathy ever defined
was Bardet-Biedl syndrome (BBS) in 2003 (68), although this disease had been
known since 1866, when Laurence and Moon (69) first described the phenotype,
including retinitis pigmentosa, mental retardation, hypogonadism
and spastic paraplegia, in four cases. Decades later, a similar
phenotype, consisting of obesity, retinal dystrophy, polydactyly
and cognitive problems with learning difficulties, was reported in
1920 by Bardet and in 1922 by Biedl (70).
Clinical overlaps in primary
ciliopathies
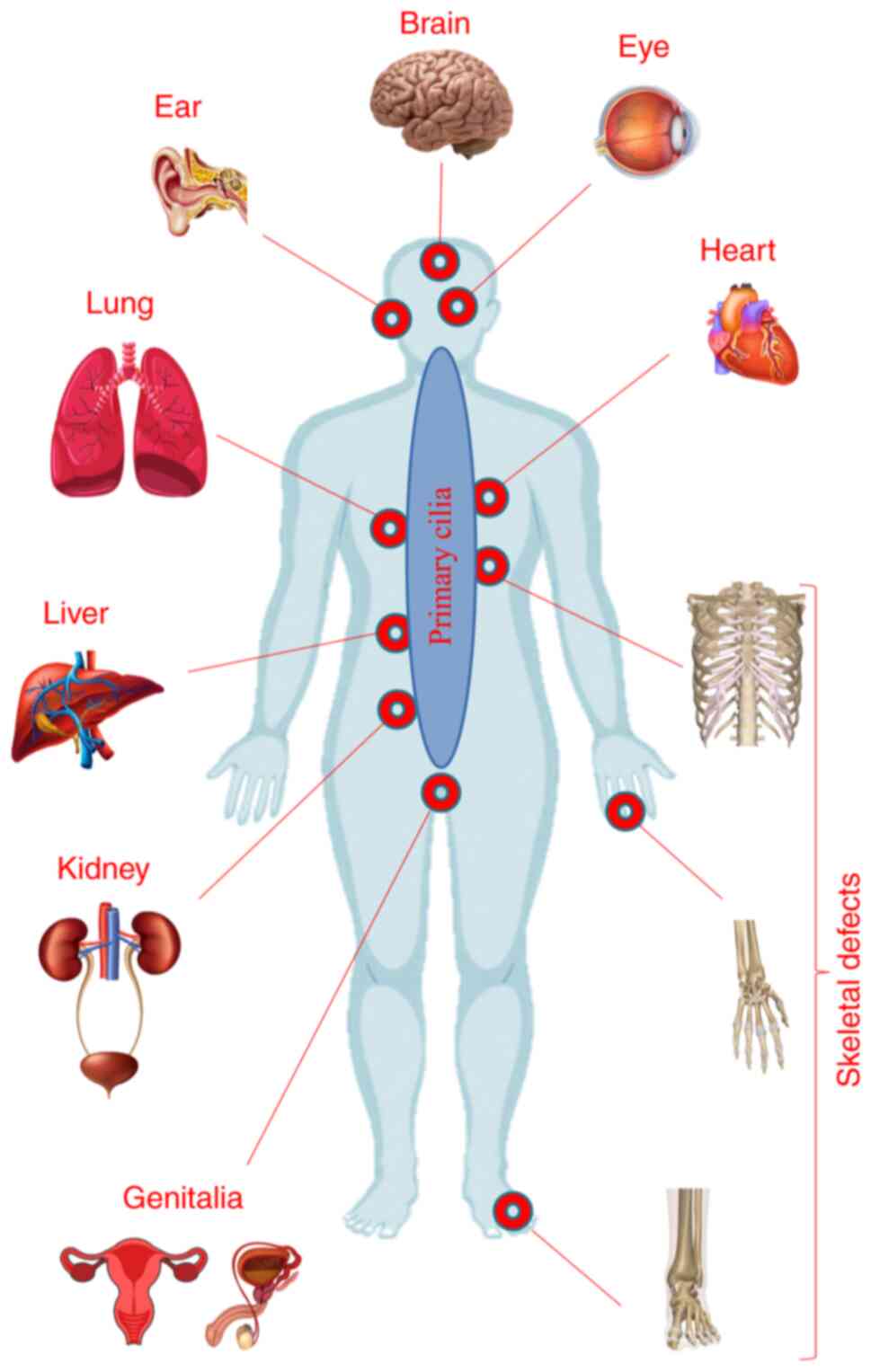
As a result of the presence of primary cilia in
nearly all tissues and organs, impairment of their structure or
function may result in a vast group of phenotypes, ranging from
single organ impairment to complex systemic disorders (Fig. 2). In addition to their isolated
involvement, the kidney and the eyes (retina) are also implicated
in defining the heterogeneous pattern of primary ciliopathies, with
the participation of other organs, including the brain, skeletal
system and liver (71).
Additional system contributions in defining the ciliopathic
clinical picture are summarized in Table I.
 | Table IAdditional clinical features of
ciliopathiesa. |
Table I
Additional clinical features of
ciliopathiesa.
| Type of system | Clinical
feature |
|---|
| Cardiovascular | Atrial or/and
ventricular septal defects, dilated cardiomyopathy, hypertrophic
cardiomyopathy and valvular defects |
| Respiratory | Breathing
abnormalities, respiratory insufficiency, pulmonary hypoplasia,
atelectatic lungs and interstitial fibrosis |
| Endocrine | Panhypopituitarism,
growth hormone deficiency, hypothyroidism, diabetes mellitus and
hypogonadism |
| Genital | Genital hypoplasia,
micropenis and ambiguous genitalia |
| Pancreatic | Pancreatic
dysgenesis, pancreatic fibrosis and cystic pancreas |
| Aural | Sensorial hearing
loss |
Renal manifestations
Renal impairment is the most common sign in primary
ciliopathies, histologically characterized by renal cysts, a
thickened and irregular tubular basement membrane, and interstitial
fibrosis. Clinically, two frequently observed categories have been
defined: Polycystic kidney disease (PKD) and nephronophthisis
(NPHP). Both entities are characterized by a progressive decline in
renal function, eventually leading to renal failure (72,73). The onset of the diseases varies.
Some signs could be detected prenatally due to the presence of
oligohydramnios and enlarged kidneys, or shortly after birth due to
the occurrence of severe hypertension or respiratory insufficiency
(74,75). During childhood, the symptoms of
renal disease are unspecific and may include polydipsia, polyuria,
secondary enuresis and urinary concentration defects. Poor growth
may occur due to chronic dehydration. As a result of renal
insufficiency and its progression to end-stage renal failure, new
complications may develop, including anemia, metabolic acidosis,
anorexia and/or hypertension (76). Renal ultrasound examination shows
large, normal-sized or small kidneys, with increased echogenicity,
loss of corticomedullary differentiation and the presence of renal
cysts (76,77). Dysplastic, lobulated or horseshoe
kidneys, kidney malrotation and renal agenesis are less frequently
encountered in ciliopathic disorders (78).
Liver manifestations
Liver cysts, liver fibrosis and ductal plate
malformation with abnormal bile ducts may be summarized as liver
fibrocystic diseases, and they are often found, in addition to PKD,
in primary ciliopathies (79).
The liver disease can remain asymptomatic, or it can lead to
complications, particularly portal hypertension and esophageal
varices, cholangitis or cholestasis (80). End-stage hepatic disease
requiring transplantation has also been reported in some patients
(81,82). The cardinal symptom is
hepatomegaly, which can be associated with elevated serum levels of
hepatic enzymes, or liver hyperechogenicity on abdominal ultrasound
(83).
Ocular manifestations
Retinal dystrophy (with both rod and cone
photoreceptor involvement) is commonly encountered in primary
ciliopathic disorders. The clinical manifestations of visual
impairment range from night blindness, color blindness and loss of
peripheral vision, to progressive visual loss and complete
blindness (84). Disruptions of
ocular motility, such as oculomotor apraxia and nystagmus, are also
frequently described (85,86). Additional ocular defects include
strabismus, amblyopia, astigmatism, congenital cataracts and
coloboma (78).
Central nervous system (CNS)
manifestations
The major neuroimaging finding, which characterizes
a distinct group of diseases referred to as Joubert syndrome (JS)
and related disorders, is the 'molar tooth sign' (MTS), comprising
cerebellar vermis hypoplasia or aplasia, with enlargement of the
fourth ventricle, thickened and horizontalized superior cerebellar
peduncles and a deepened interpeduncular fossa (87,88). Neurological abnormalities may
also include Dandy-Walker malformation (DWM), ventriculomegaly,
periventricular nodular heterotopia, hydrocephalus,
encephalocele/meningocele, polymicrogyria, absence of the pituitary
gland, corpus callosum defects and morphological brainstem
abnormalities (83,89-91). A wide range of clinical signs may
be observed, such as hypotonia, ataxia, developmental delay,
intellectual disability (ID), impaired or absent speech, behavioral
disturbances such as hyperactivity and aggressiveness, and
self-mutilation (92,93).
Skeletal manifestations
Clinical manifestations of the skeletal system may
vary from mild phenotypes, such as polydactyly, to severe
deformities, possibly leading to death. Polydactyly of the hands
and/or feet, which is usually post-axial, but may also be pre-axial
and, in some cases, central or mesoaxial, is present in most
individuals with cilia-related disorders (94-96). In addition to polydactyly, the
hands and feet may be affected to various degrees by oligodactyly,
syndactyly, camptodactyly, brachydactyly, carpal and tarsal
shortening, short long bones, rhizomelic micromelia, fibular
aplasia or limb agenesis (97,98). Truncal skeletal defects may
include a constrictive thoracic cage, with shortened and
horizontalized ribs, which may be life-threatening in some cases;
abnormal or absent clavicles, small scapulae and scoliosis may also
be observed (99).
Cranioskeletal characteristics include craniosynostosis, macro- or
microcephaly, head shape anomalies, frontal bossing, a prominent
forehead, bitemporal narrowing, cleft palate, zygomatic arch
hypoplasia, maxillary hypoplasia and micrognathia (100).
Diagnosis of primary ciliopathies
Clinical diagnosis
Given the numerous overlapping features and marked
genetic heterogeneity, considerable efforts have been made to
diagnose and classify ciliopathies, in order to optimize clinical
management of the patients and improve the accuracy of genetic
counseling.
For some of these diseases with severe phenotypes
leading to a high mortality rate in utero or during the
perinatal period, a prenatal diagnosis is possible in the presence
of pathognomonic ultrasonographic signs in conjunction with
α-fetoprotein testing of the amniotic fluid and DNA testing of the
fetus (101). Early in the
pregnancy (weeks 8-11), ultrasonographic screening can detect
certain fetal malformations, such as an enlarged cisterna magna or
encephalocele (102). Between
11 and 14 weeks of pregnancy, enlarged polycystic kidneys or
polydactyly may be detected (103), while later in the second
trimester, other brain anomalies (e.g., DWM and hydrocephalus)
(104) and severe skeletal
anomalies (e.g., rhizomelic shortening of the long bones and
hypoplastic thoracic cage) may be identified (105). Fetal MRI can detect MTS at 27
weeks of pregnancy (106).
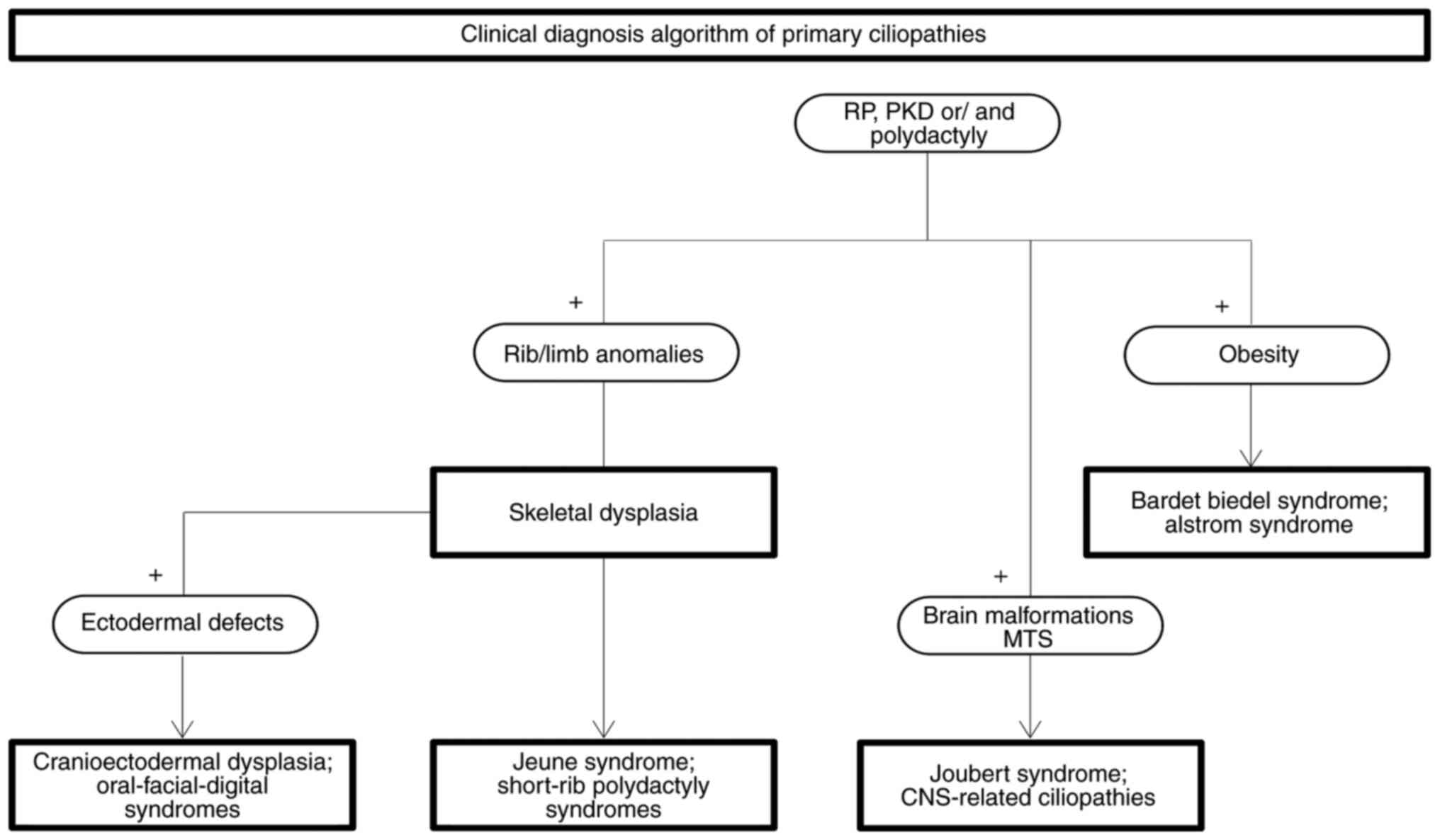
Postnatally, Beales and Kenny proposed a clinical
diagnosis algorithm starting with the presence of renal and retinal
involvement and/or polydactyly (Fig.
3). Adding limb or rib abnormalities to this core of clinical
manifestations may easily direct the diagnosis to ciliary skeletal
dysplasias. Furthermore, identification of ectodermal defects in
this group suggests the diagnosis of oral-facial-digital syndrome
(OFDS) or cranioectodermal dysplasia (CED), while their absence
indicates the diagnosis of short-rib polydactyly syndrome (SRPS).
The detection of MTS and other CNS abnormalities should raise the
suspicion of JS or JS-related disorders, whereas the presence of
obesity points towards the diagnosis of BBS or Alström syndrome
(ALMS) (107).
Genetics and molecular diagnosis
Since the description of the first ciliopathy gene,
BBS6, by two distinct research groups in 2000, due to the advances
in genomic sequencing technologies, a number of genes have been
associated with ciliary phenotypes (108,109). Only in the last 5 years,
through the intensive use of specific gene panels, whole exome and
whole genome sequencing >100 new ciliary genes have been
identified. At present, there are >190 known genes associated
with recognized ciliopathies, of which >140 genes (Table SI) are implicated in primary
ciliopathies. Other candidate genes (>240), of which the protein
products have been shown to be associated with cilia function or
structure, may be involved in either new or confirmed primary or
motile ciliopathies (110).
Ciliopathies are considered to be Mendelian disorders (111), although a plethora of evidence
has also indicated a non-Mendelian pattern of inheritance, or even
environmental contribution to defining the phenotype. Genetic locus
heterogeneity, copy number variants (112,113), oligogenicity (114,115), multiple allelism (116-119) and transposon-mediated
mutagenesis (120) have been
described, highlighting the marked complexity of the genetic
mechanisms responsible for ciliopathic phenotypes. Moreover, the
severity or variability of the phenotypes is suggested to be
modulated by the pattern of ciliary gene expression and its effect
on protein function (null, truncating or hypomorphic) (121), by epistatic interactions
(122,123) and by genetic modifiers or
stochastic effects (45,111,124-128).
Classification of primary
ciliopathies
Several types of ciliopathies have been recognized,
considering the level to which an organ is affected for defining
their phenotype.
Retinal ciliopathies
Retinal ciliopathies include clinical entities
manifesting as retinal degeneration, and they are caused by
defective morphogenesis or dysfunction of specialized sensory cilia
from the retina that form the outer segment of photoreceptors.
Proteins, such as rhodopsin or ambient lighting-dependent proteins,
are trafficked along these specialized primary cilia by means of
IFT particles. Impairment of IFT leads to the accumulation of
rhodopsin, defects in outer segment development and cell death,
which result in the phenotype of retinal degeneration (129,130). Among non-syndromic retinal
ciliopathies, the ocular phenotype ranges from the most common
retinitis pigmentosa [Mendelian Inheritance in Man (MIM), 26800]
(47), which initially manifests
as night blindness, followed by loss of peripheral vision, due to
the impairment of rod photoreceptor function, and can progress to
complete blindness (84,131) to the most severe congenital
retinal dystrophy, Leber congenital amaurosis (LCA; MIM, 204000),
which frequently results in blindness within the first year of
life. Visual loss is usually accompanied by sensory nystagmus,
amaurotic pupillary response and absent electroretinogram signs.
Photophobia, high refractive errors, keratoconus and enophthalmos
are often seen in LCA. Involvement of the retina may range from
normal, to retinal degeneration, retinal aplasia or biochemical
dysfunction (dysplasia) (132).
Overlapping with these two disorders, other ocular dystrophies have
also been described as retinal ciliopathies: Cone dystrophy (MIM,
304020), characterized by visual loss and color vision defects,
cone-rod dystrophy (MIM, 120970), characterized by photophobia,
abnormal color vision, night and peripheral vision loss, and
macular dystrophy (MIM, 300834), characterized by loss of color and
sharp vision (133).
Progressive retinal degeneration and sensorineural hearing loss are
the first symptoms found in ALMS (MIM, 203800); these are
accompanied later in childhood by obesity and diabetes mellitus.
Additional features, such as cardiomyopathy, epilepsy, respiratory
disturbance and renal or endocrine dysfunction, support the
classification of these disorders as syndromic retinal ciliopathies
(134). A rare combination of
retinal and renal ciliopathies characterizes Senior-Løken syndrome
(SLSN; MIM, 266900), with a specific clinical presentation
consisting of retinal dystrophy and NPHP. Consequently, SLSN is
considered by some studies as a syndromic retino-ciliopathy or, by
others, as a renal (NPHP-related) ciliopathy (135,136).
Renal ciliopathies
Renal ciliopathies encompass a group of disorders,
the hallmark of which is kidney disease, including autosomal
dominant polycystic kidney disease (ADPKD; MIM, 173900), autosomal
recessive polycystic kidney disease (ARPKD; MIM, 263200) and NPHP
(MIM, 256100). In the kidney, epithelial primary cilia lining the
nephron tubules and collecting ducts act as sensory antennae
sensitive to urine composition, osmolarity and flow. Defects in
several signaling pathways, such as G-protein signaling, mTOR or
Wnt, induced by decreased of flow-mediated intracellular calcium
concentration, may lead to cyst formation. Moreover, disruption of
the balance between canonical and non-canonical Wnt signaling may
affect the polarity of epithelial tubular cells, also resulting in
cyst formation (137).
ADPKD and ARPKD are different, not only due to the
inheritance pattern, but also based on the microscopic and
ultrasonographic appearance of the cysts, associated organ
anomalies, age at onset, severity and prognosis (138). While ADPKD is characterized by
large cysts originating from the distal nephrons and collecting
ducts, which grow in volume and number with age, and by the
presence of cysts in the liver, pancreas or other epithelial
organs, intracranial aneurysms and mitral valve prolapse (139), the cysts in ARPKD are small,
originate from the distal tubules and collecting ducts and display
a salt-and-pepper pattern, and the liver is always affected by
fibrosis (138). In contrast to
ADPKD, which starts in late adulthood and slowly progresses to
end-stage renal disease (ESRD), ARPKD is more severe, with
antenatal onset and diagnosis during late pregnancy or at birth,
leading to increased perinatal death rate (30-50%). Death occurs as
a consequence of respiratory insufficiency due to pulmonary
hypoplasia and thoracic compression by the extremely expanded
kidneys (75). NPHP, which is
characterized by corticomedullary cysts, atrophy and interstitial
fibrosis resulting in nephron disintegration, is the main cause of
ESRD in children (140). The
severity and, subsequently, the progression to ESRD, depend on the
clinical variant, namely the infantile, juvenile or adolescent
variant. The infantile variant is the most severe, with prenatal
manifestations consisting of oligohydramnios and bilateral enlarged
cystic kidneys. Thus, ESRD develops in the first year of life. The
first symptoms of the classical juvenile form, which is
characterized by renal interstitial fibrosis and inflammation, with
progression to tubular atrophy and small cyst formation, develop
during the first decade of life and ESRD occurs at the mean age of
13 years (74). NPHP may be
limited to the kidneys or may be part of other ciliopathic
conditions, such as Joubert/COACH syndrome, SLSN, BBS,
Meckel-Gruber syndrome (MKS) or skeletal disorders (141). BBS (MIM, 209900) is the most
extensively investigated ciliopathy, and it has provided valuable
data for the entire spectrum of human cilia-related disorders due
to its overlapping characteristics at the level of phenotype,
genotype, protein-protein interactions and participation in
signaling pathways (142). BBS
is a multisystem disorder, but renal impairment is its most
prominent cause of morbidity and mortality. The major clinical
characteristics, including retinal dystrophy, obesity, post-axial
polydactyly, renal anomalies, cognitive impairment and
hypogonadism, are suggestive of the diagnosis. The presence of four
of those characteristics, or association of three primary
characteristics with two secondary features is considered as
sufficient for clinical diagnosis (143). Secondary features include
speech delay, developmental delay, diabetes and congenital heart
disease. BBS is characterized by marked clinical variability, which
cannot be fully attributed to the 24 genes identified to date
(144). MKS (MIM, 249000),
which displays renal (cystic kidney dysplasia) as well as
neurological [occipital encephalocele (OE)] manifestations, may be
considered as either a renal or a CNS-related ciliopathy. Hepatic
fibrosis completes the specific clinical triad of this condition,
although polydactyly is often considered as the 4th pathognomonic
feature (145). MKS has a
heterogeneous, severe phenotype, which is not compatible with life,
with death occurring in utero or shortly after birth. Renal
dysfunction may often lead to oligohydramnios or anhydramnios.
Apart from OE, which is the most frequent finding, additional CNS
malformations found in MKS include olfactory bulb dysgenesis, optic
nerve hypoplasia, agenesis of the corpus callosum,
holoprosencephaly, cerebellar hypoplasia or total anencephaly.
Cleft lip and palate, shortening of the long bones, congenital
heart defects and pulmonary hypoplasia may further complicate the
clinical picture (45,101).
CNS-related ciliopathies
CNS-related ciliopathies comprise a group of
conditions, the hallmark of which is the MTS, which is required for
diagnosis. Impairment of the Wnt pathway, which is a major
signaling pathway involved in cerebellar development, may be
responsible for defective cerebellar vermis hypoplasia, one of the
components of MTS. In addition to this pathway, other neuronal
primary cilium-specific pathways are required for normal brain
development, regulating neuronal fate, proliferation, migration and
differentiation. Dysregulation of these pathways, including Shh,
PDGFRα and GRCR, may manifest with malformations during cortical
development or midline defects, which are often found in JS or
CNS-related ciliopathies (71).
Depending on the additional clinical characteristics, JS (MIM,
213300) has been classified into several groups as follows: i) Pure
or classic JS, characterized by hypotonia, developmental delay,
abnormal eye movements, breathing abnormalities, ataxia and ID; ii)
JS with ocular defects, including retinal dystrophy or LCA; iii) JS
with renal defects (NPHP); iv) JS with oculorenal defects, also
named cerebello-oculorenal syndrome, comprising SLSN (retinal
dystrophy, LCA and NPHP) associated with MTS, and Dekaban-Arima
syndrome (cerebrooculohepatorenal syndrome) characterized by
chorioretinal coloboma or retinal dystrophy, PKD, MTS and hepatic
fibrosis in some cases; v) JS with congenital hepatic fibrosis; vi)
JS with congenital hepatic fibrosis and associated chorioretinal
coloboma, also known as COACH syndrome; and vii) JS with
orofaciodigital defects, including a lobulated or bifid tongue,
hamartomas, cleft lip and/or palate and polydactyly, also known as
orofaciodigital syndrome type VI (83,87). To date, ~40 causative genes
covering >90% of clinical subjects have been identified
(146-152).
Ciliopathies with skeletal
involvement
This group of disorders is characterized by variable
severity, ranging from mild to severe or even lethal phenotypes.
Two subgroups have been distinguished: Those with major skeletal
system involvement, including craniofacial, thoracic cage and long
bone involvement, known as short-rib thoracic dysplasias (SRTDs),
with or without polydactyly or ciliary condrodysplasias, and OFDS,
with milder involvement of the skeletal system (153).
Development of the cartilage and bones is a complex
process that is modulated mainly by the IFT and Hh pathways.
Disruption of Ihh signaling in chondral primary cilia affects
chondrocyte maturation during the ossification process.
Consequently, various skeletal abnormalities, including
polydactyly, shortening of the ribs or long bones and craniofacial
abnormalities, may occur (154,155). Dysregulation of IFT, which is
involved in the trafficking of the transmembrane SMO receptor, a
signal transducer in Hh signaling, may lead to premature
differentiation and decreased proliferation of chondrocytes,
manifesting as specific SRTDs and defects of long bone growth
plates (156).
There are >19 types of SRTDs, classified based on
phenotype severity, radiological findings and confirmation of
genetic defects (100,157). Chondroectodermal dysplasia or
Ellis-van-Creveld syndrome (EVC; MIM, 2255000), Weyers acrodental
dysostosis (WAD; MIM, 193530) and Sensenbrenner syndrome or CED
(MIM, 218330) are the milder disorders in this group. EVC is
characterized by disproportionate short limb dwarfism, short ribs,
polydactyly, cardiac malformations and ectodermal defects affecting
the hair, teeth and nails (158-160). WAD is an allelic disorder to
EVC, but displays a milder phenotype, consisting of moderate short
stature, postaxial polydactyly, and nail and dental anomalies, and
is inherited in an autosomal dominant manner (161). CED is characterized by
craniofacial abnormalities, such as sagittal craniosynostosis,
leading to dolichocephaly, frontal bossing and dental defects, in
conjunction with skeletal abnormalities (short stature, rhizomelic
limbs, brachydactyly and narrow thorax) and ectodermal anomalies
(thin/sparse hair, hypoplastic nails and skin laxity) (162,163). Kidney involvement (NPHP
progressing to renal failure) and liver involvement (ranging from
asymptomatic hepatomegaly to acute cholangitis, liver cirrhosis and
severe cholestasis) are common findings in CED (164).
The second group with more severe phenotypes is
comprised of Jeune asphyxiating thoracic dystrophy (JATD; MIM,
208500) and conorenal syndrome or Mainzer-Saldino syndrome (MZSDS;
MIM, 266920). The specific presentation of JATD includes a
constrictive thoracic cage and secondary respiratory distress due
to restrictive pulmonary hypoplasia. Respiratory distress is the
main cause of mortality in ~60% of the patients (100). Additional skeletal findings may
include a short stature, short limbs with irregular metaphyses,
cone-shaped epiphyses in the hands, foot polydactyly, a shortened
ilium and a trident-shaped acetabulum. Retinal degeneration,
NPHP-like or cystic renal disease, pancreatic and liver involvement
or brain malformations are occasionally found in patients with JATD
(100,165). MZSDS is characterized by the
triad of retinal dystrophy, renal disease (typically NPHP) and
phalangeal cone-shaped epiphyses (166). The thorax is less narrow
compared with that in patients with JATD. Short stature, hepatic
fibrosis and cerebellar ataxia are variable traits that may be
observed in MZSDS (166,167).
The last subtype is the perinatally lethal SRPS, the
core features of which include a constrictive thoracic cage,
significantly shortened long bones, polydactyly, brahydactyly and
pelvic abnormalities (100).
Different types have been characterized, based mainly on
radiological findings: SRPS types I (Saldino-Noonan syndrome) and
III (Verma-Naumoff syndrome) (MIM, 613091); SRP type II or Majewski
syndrome (MIM, 263520); SRPS type IV or Beemer-Langer syndrome
(MIM, 269860); and SRPS type V (MIM, 614091) (98). In addition to skeletal
abnormalities, involvement of the brain, heart, kidneys, liver,
pancreas and genitalia have often been recorded in SRPS. In some
cases, facial dysmorphism may also be observed (100,165).
Apart from the typical manifestations, some
'unusual' features may be observed in each group, further expanding
and complicating the phenotype; these include atlantoaxial
instability and spinal cord compression (168,169), short irregularly bent ribs,
hypoplastic and bent mesomelic bones, short campomelic long bones,
undermineralized bones (170,171), OE or MTS (172).
OFDS (MIM, 311200) describes a heterogeneous group
of diseases caused by defects in ~18 genes (173-175). Clinical manifestations include
anomalies of the face (micrognathia, hypertelorism, telecanthus,
cleft lips and low-set ears), the oral cavity (gingival frenulae,
lingual hamartomas, cleft/lobulated tongue and cleft palate) and
the digits (polydactyly, brachydactyly, oligodactyly and bifid
digits), associated with an extensive spectrum of additional
features affecting the CNS, the kidneys, the heart or the eyes,
outlining the 13 forms described to date (173,176). In addition to renal
involvement, which is commonly found in OFDS, a series of features
overlapping with other ciliopathies (JS, SRPS and EVC) have been
reported, such as MTS identified in OFD types 4, 6 and 14, and
tibial abnormalities observed in OFD types 4, 8 and 12 (177,178).
Other unclassified subtypes have also been
described, which are characterized, in addition to the typical
features, by fused kidneys (179), tetralogy of Fallot (179,180), coarctation of the aorta
(181), corpus callosum
agenesis (179), cerebellar
vermis hypoplasia, DW malformation, ID, 12th rib hypoplasia
(174) and short mesoaxial
phalanges (182).
4. Conclusions
Increasing use of whole exome sequencing has enabled
the discovery of new causal genes in ciliopathies. Combined efforts
have been made in the fields of proteomics, cell biology and model
organisms to link the genes with their phenotypic effect. Taken
together, all these studies have improved our knowledge on
recognized ciliopathies, confirmed the proposed cilia-related
disorders or identified new ciliary diseases.
Since Baker and Beales (183) proposed 72 conditions as
candidates for ciliopathic disorders in 2009, several have been
included in the group of known ciliopathies (Table II), increasing their number to
35 (184-202).
| Table IINewly defined ciliopathies. |
Table II
Newly defined ciliopathies.
| MIM ID | Disease name | Gene name | Protein
localization | (Refs.) |
|---|
| 616287 | Lethal congenital
contracturesyndrome; hypomyelination neuropathy-arthrogryposis
syndrome | ADCY6 | Axoneme | (184) |
| 243605 | Stromme syndrome;
lethal fetal brain malformation-duodenal atresia-bilateral renal
hypoplasia syndrome; microcephaly | CENPF | Basal body | (185,186) |
| 135150 | Birt-Hogg-Dubé
syndrome | FLCN | Basal body;
axoneme | (187) |
| 201000 | Carpenter
syndrome | RAB23 | Axoneme | (200,201) |
| 616897 | Complex lethal
osteochondrodysplasia | TAPT1 | Basal body | (189) |
| NO MIM ID | A novel syndrome
with multiple congenital malformations and developmental delay | USP9X | Axoneme | (190) |
| 601707 | Curry-Jones
syndrome | SMO | Axoneme | (191) |
| 607131 |
Al-Gazali-Bakalinova syndrome | KIF7 | Axoneme | (192) |
| 236680 614120 | Hydrolethalus | HYLS1; KIF7; | Basal body;
axoneme; basal body | (193-195) |
| 175700 | Greig
cephalopolysyndactyly syndrome | GLI3 | Axoneme (tip) | (196) |
| 612651 | Lethal
endocrine-cerebro-osteodysplasia syndrome | ICK | IFT | (197) |
| NO MIM ID | Pituitary stalk
interruption syndrome | GPR161 | Axoneme | (198) |
| 300707 |
Syndactyly-telecanthus-anogenital and
renal malformations syndrome | FAM58A | Probably
cytosolic | (202) |
By contrast, other conditions were excluded from the
list of possible or likely ciliopathies following the determination
of their genetic background, including Kabuki syndrome (type 1 MIM,
147920; type 2 MIM, 300867) following identification of its causal
genes, MLL2 (MIM, 602113) and KDM6A (MIM, 300128) (203,204), or Neu-Laxova syndrome (type 1:
MIM, 256520; type 2: MIM, 616038) due to the discovery of its
causal genes, PHGDG (MIM, 606879) and PSAT1 (MIM, 610936) (205,206).
The delineation of the ciliary proteome and its
interaction with extraciliary molecules opens new perspectives in
reclassifying cilia-related disorders. Thus, the disorders
characterized by ciliopathy-overlapping phenotypes, the causal
genes of which are not expressed in the ciliary assembly, but
interfere with ciliogenesis or the cilia signaling network, have
been termed ciliopathy-like disorders. A representative example is
Cohen syndrome (MIM, 216550), which is defined by obesity,
developmental delay, retinal degeneration and intermittent
neutropenia (207), and is
caused by mutations in VPS13B (MIM, 607817) (208). The expression product, which is
localized to the Golgi apparatus, may impair processing of ciliary
components (207).
Townes-Brocks syndrome (MIM, 107480), which is characterized by
hearing impairment, PKD, ESRD, imperforate anus and digit
malformations (209), is caused
by mutations in the SALL1 (MIM, 602218) gene, which encodes a
zinc-finger transcription factor; its interaction with two
ciliogenesis suppressors, CEP97 (MIM, 615864) and CCP110 (MIM,
609544), leads to cilia formation and function impairment (210). For these ciliopathy-like
conditions, Reiter and Leroux (110) proposed the term second-order
ciliopathies, whereas first-order ciliopathies are defined as
disorders in which disease-associated proteins are expressed in the
primary ciliary compartment. Although applying this classification
is seemingly straightforward, unexpected evidence has uncovered the
possibility of a condition being either first- or second-order,
thus complicating the picture. One such example is MKS, a
well-known ciliopathy caused by mutations in genes encoding
proteins that are localized in the transition zone (101). A recent study identified a new
causal gene for MKS, TXNDC15 (MIM, 617778), a non-ciliary gene, the
bi-allelic mutations of which lead to abnormal cilia biogenesis
(211).
Numerous conditions remain to be elucidated, either
due to the fact that the genetic cause has not been uncovered or
since the pathophysiological mechanism underlying the phenotype
remains elusive. Predicting organ involvement and, consequently,
phenotype severity based on genetic defects also represents a major
challenge.
Future research will hopefully provide new insights
that may help reorganize and further elucidate the striking field
of ciliopathies.
Supplementary Data
Availability of data and materials
Not applicable.
Authors' contributions
IOF collect the data, wrote the manuscript,
prepared the figures and the tables. MBu wrote the manuscript. MBa
revised and approved the manuscript. All authors read and approved
the final manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
The authors are grateful to Dr Costel Darie,
Associate Professor of Chemistry and Biomolecular Science, Clarkson
University (Potsdam, NY, USA) for reviewing this work.
References
|
1
|
Badano JL, Mitsuma N, Beales PL and
Katsanis N: The ciliopathies: An emerging class of human genetic
disorders. Annu Rev Genomics Hum Genet. 7:125–148. 2006. View Article : Google Scholar
|
|
2
|
Waters AM and Beales PL: Ciliopathies: An
expanding disease spectrum. Pediatr Nephrol. 26:1039–1056. 2011.
View Article : Google Scholar
|
|
3
|
Moore A, Escudier E, Roger G, Tamalet A,
Pelosse B, Marlin S, Clément A, Geremek M, Delaisi B, Bridoux AM,
et al: RPGR is mutated in patients with a complex X linked
phenotype combining primary ciliary dyskinesia and retinitis
pigmentosa. J Med Genet. 43:326–333. 2006. View Article : Google Scholar
|
|
4
|
Budny B, Chen W, Omran H, Fliegauf M,
Tzschach A, Wisniewska M, Jensen LR, Raynaud M, Shoichet SA, Badura
M, et al: A novel X-linked recessive mental retardation syndrome
comprising macrocephaly and ciliary dysfunction is allelic to
oral-facial-digital type I syndrome. Hum Genet. 120:171–178. 2006.
View Article : Google Scholar
|
|
5
|
Moalem S, Keating S, Shannon P, Thompson
M, Millar K, Nykamp K, Forster A, Noor A and Chitayat D: Broadening
the ciliopathy spectrum: Motile cilia dyskinesia, and
nephronophthisis associated with a previously unreported homozygous
mutation in the INVS/NPHP2 gene. Am J Med Genet A. 161A:1792–1796.
2013. View Article : Google Scholar
|
|
6
|
Shapiro AJ, Zariwala MA, Ferkol T, Davis
SD, Sagel SD, Dell SD, Rosenfeld M, Olivier KN, Milla C, Daniel SJ,
et al: Diagnosis, monitoring, and treatment of primary ciliary
dyskinesia: PCD foundation consensus recommendations based on state
of the art review. Pediatr Pulmonol. 51:115–132. 2016. View Article : Google Scholar
|
|
7
|
Tobin JL and Beales PL: The nonmotile
ciliopathies. Genet Med. 11:386–402. 2009. View Article : Google Scholar
|
|
8
|
Leeuwenhoek AV: Observations, communicated
to the publisher by Mr. Antony van Leewenhoeck, in a dutch letter
of the 9th Octob. 1676. Here English'd: Concerning little animals
by him observed in rain-well-seaand snow water; as also in water
wherein pepper had lain infused. Philosophical Transactions.
12:821–831. 1677. View Article : Google Scholar
|
|
9
|
Lee L: Mechanisms of mammalian ciliary
motility: Insights from primary ciliary dyskinesia genetics. Gene.
473:57–66. 2011. View Article : Google Scholar
|
|
10
|
Guo J, Higginbotham H, Li J, Nichols J,
Hirt J, Ghukasyan V and Anton ES: Developmental disruptions
underlying brain abnormalities in ciliopathies. Nat Commun.
6:78572015. View Article : Google Scholar
|
|
11
|
Chang CF, Schock EN, Attia AC, Stottmann
RW and Brugmann SA: The ciliary baton: Orchestrating neural crest
cell development. Curr Top Dev Biol. 111:97–134. 2015. View Article : Google Scholar
|
|
12
|
Mirvis M, Stearns T and James Nelson W:
Cilium structure, assembly, and disassembly regulated by the
cytoskeleton. Biochem J. 475. pp. 2329–2353. 2018, View Article : Google Scholar
|
|
13
|
Marshall WF and Nonaka S: Cilia: Tuning in
to the cell's antenna. Curr Biol. 16:R604–R614. 2006. View Article : Google Scholar
|
|
14
|
Roberts AJ, Kon T, Knight PJ, Sutoh K and
Burgess SA: Functions and mechanics of dynein motor proteins. Nat
Rev Mol Cell Biol. 14:713–726. 2013. View Article : Google Scholar
|
|
15
|
Linck R, Fu X, Lin J, Ouch C, Schefter A,
Steffen W, Warren P and Nicastro D: Insights into the structure and
function of ciliary and flagellar doublet microtubules: Tektins,
Ca2+-binding proteins, and stable protofilaments. J Biol
Chem. 289:17427–17444. 2014. View Article : Google Scholar
|
|
16
|
Reiter JF, Blacque OE and Leroux MR: The
base of the cilium: Roles for transition fibres and the transition
zone in ciliary formation, maintenance and compartmentalization.
EMBO Rep. 13:608–618. 2012. View Article : Google Scholar
|
|
17
|
Vertii A, Hung HF, Hehnly H and Doxsey S:
Human basal body basics. Cilia. 5:132016. View Article : Google Scholar
|
|
18
|
Satir P and Christensen ST: Overview of
structure and function of mammalian cilia. Annu Rev Physiol.
69:377–400. 2007. View Article : Google Scholar
|
|
19
|
Hu Q, Milenkovic L, Jin H, Scott MP,
Nachury MV, Spiliotis ET and Nelson WJ: A septin diffusion barrier
at the base of the primary cilium maintains ciliary membrane
protein distribution. Science. 329. pp. 436–439. 2010, View Article : Google Scholar
|
|
20
|
Fisch C and Dupuis-Williams P:
Ultrastructure of cilia and flagella-back to the future! Biol Cell.
103:249–270. 2011. View Article : Google Scholar
|
|
21
|
Czarnecki PG and Shah JV: The ciliary
transition zone: From morphology and molecules to medicine. Trends
Cell Biol. 22:201–210. 2012. View Article : Google Scholar
|
|
22
|
Nonaka S, Tanaka Y, Okada Y, Takeda S,
Harada A, Kanai Y, Kido M and Hirokawa N: Randomization of
left-right asymmetry due to loss of nodal cilia generating leftward
flow of extraembryonic fluid in mice lacking KIF3B motor protein.
Cell. 95:829–837. 1998. View Article : Google Scholar
|
|
23
|
Afzelius BA: Cilia-related diseases. J
Pathol. 204:470–477. 2004. View Article : Google Scholar
|
|
24
|
Dabdoub A and Kelley MW: Planar cell
polarity and a potential role for a Wnt morphogen gradient in
stereociliary bundle orientation in the mammalian inner ear. J
Neurobiol. 64:446–457. 2005. View Article : Google Scholar
|
|
25
|
Hirokawa N, Tanaka Y, Okada Y and Takeda
S: Nodal flow and the generation of left-right asymmetry. Cell.
125:33–45. 2006. View Article : Google Scholar
|
|
26
|
Hamada H: Roles of motile and immotile
cilia in left-right symmetry breaking. Etiology and Morphogenesis
of Congenital Heart Disease: From Gene Function and Cellular
Interaction to Morphology. Nakanishi T, Markwald RR, Baldwin HS,
Keller BB, Srivastava D and Yamagishi H: Springer; Tokyo: pp.
57–65. 2016, View Article : Google Scholar
|
|
27
|
Bloodgood RA: From central to rudimentary
to primary: The history of an underappreciated organelle whose time
has come. The primary cilium. Methods Cell Biol. 94:3–52. 2009.
|
|
28
|
Webber WA and Lee J: Fine structure of
mammalian renal cilia. Anat Rec. 182:339–343. 1975. View Article : Google Scholar
|
|
29
|
Pazour GJ, Dickert BL, Vucica Y, Seeley
ES, Rosenbaum JL, Witman GB and Cole DG: Chlamydomonas IFT88 and
its mouse homologue, polycystic kidney disease gene tg737, are
required for assembly of cilia and flagella. J Cell Biol.
151:709–718. 2000. View Article : Google Scholar
|
|
30
|
Pazour GJ, San Agustin JT, Follit JA,
Rosenbaum JL and Witman GB: Polycystin-2 localizes to kidney cilia
and the ciliary level is elevated in orpk mice with polycystic
kidney disease. Curr Biol. 12:R378–R380. 2002. View Article : Google Scholar
|
|
31
|
Gradilone SA, Masyuk AI, Splinter PL,
Banales JM, Huang BQ, Tietz PS, Masyuk TV and Larusso NF:
Cholangiocyte cilia express TRPV4 and detect changes in luminal
tonicity inducing bicarbonate secretion. Proc Natl Acad Sci USA.
104:19138–19143. 2007. View Article : Google Scholar
|
|
32
|
Mansini AP, Peixoto E, Jin S, Richard S
and Gradilone SA: The chemosensory function of primary cilia
regulates cholangiocyte migration, invasion, and tumor growth.
Hepatology. 69:1582–1598. 2019. View Article : Google Scholar
|
|
33
|
Masyuk AI, Gradilone SA, Banales JM, Huang
BQ, Masyuk TV, Lee SO, Splinter PL, Stroope AJ and Larusso NF:
Cholangiocyte primary cilia are chemosensory organelles that detect
biliary nucleotides via P2Y12 purinergic receptors. Am J Physiol
Gastrointest Liver Physiol. 295:G725–G734. 2008. View Article : Google Scholar
|
|
34
|
Praetorius HA and Spring KR: Bending the
MDCK cell primary cilium increases intracellular calcium. J Membr
Biol. 184:71–79. 2001. View Article : Google Scholar
|
|
35
|
Hou Y and Witman GB: Dynein and
intraflagellar transport. Exp Cell Res. 334:26–34. 2015. View Article : Google Scholar
|
|
36
|
Pedersen LB and Rosenbaum JL:
Intraflagellar transport (IFT) role in ciliary assembly, resorption
and signalling. Curr Top Dev Biol. 85:23–61. 2008. View Article : Google Scholar
|
|
37
|
Christensen ST, Clement CA, Satir P and
Pedersen LB: Primary cilia and coordination of receptor tyrosine
kinase (RTK) signalling. J Pathol. 226:172–184. 2012. View Article : Google Scholar
|
|
38
|
Wheway G, Nazlamova L and Hancock JT:
Signaling through the primary cilium. Front Cell Dev Biol. 6:82018.
View Article : Google Scholar
|
|
39
|
Veland IR, Awan A, Pedersen LB, Yoder BK
and Christensen ST: Primary cilia and signaling pathways in
mammalian development, health and disease. Nephron Physiol.
111:39–53. 2009. View Article : Google Scholar
|
|
40
|
Cardenas-Rodriguez M and Badano JL:
Ciliary biology: Understanding the cellular and genetic basis of
human ciliopathies. Am J Med Genet C Semin Med Genet. 151C:263–280.
2009. View Article : Google Scholar
|
|
41
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004. View Article : Google Scholar
|
|
42
|
Grigoryan T, Wend P, Klaus A and
Birchmeier W: Deciphering the function of canonical Wnt signals in
development and disease: Conditional loss- and gain-of-function
mutations of beta-catenin in mice. Genes Dev. 22:2308–2341. 2008.
View Article : Google Scholar
|
|
43
|
Reya T, Duncan AW, Ailles L, Domen J,
Scherer DC, Willert K, Hintz L, Nusse R and Weissman IL: A role for
Wnt signalling in self-renewal of haematopoietic stem cells.
Nature. 423:409–414. 2003. View Article : Google Scholar
|
|
44
|
Komiya Y and Habas R: Wnt signal
transduction pathways. Organogenesis. 4:68–75. 2008. View Article : Google Scholar
|
|
45
|
Abdelhamed ZA, Wheway G, Szymanska K,
Natarajan S, Toomes C, Inglehearn C and Johnson CA: Variable
expressivity of ciliopathy neurological phenotypes that encompass
Meckel-Gruber syndrome and Joubert syndrome is caused by complex
de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum Mol
Genet. 22:1358–1372. 2013. View Article : Google Scholar
|
|
46
|
Wheway G, Abdelhamed Z, Natarajan S,
Toomes C, Inglehearn C and Johnson CA: Aberrant Wnt signalling and
cellular over-proliferation in a novel mouse model of Meckel-Gruber
syndrome. Dev Biol. 377:55–66. 2013. View Article : Google Scholar
|
|
47
|
Lin F, Hiesberger T, Cordes K, Sinclair
AM, Goldstein LS, Somlo S and Igarashi P: Kidney-specific
inactivation of the KIF3A subunit of kinesin-II inhibits renal
ciliogenesis and produces polycystic kidney disease. Proc Natl Acad
Sci USA. 100:5286–5291. 2003. View Article : Google Scholar
|
|
48
|
Anvarian Z, Mykytyn K, Mukhopadhyay S,
Pedersen LB and Christensen ST: Cellular signalling by primary
cilia in development, organ function and disease. Nat Rev Nephrol.
15:199–219. 2019. View Article : Google Scholar
|
|
49
|
von Maltzahn J, Chang NC, Bentzinger CF
and Rudnicki MA: Wnt signaling in myogenesis. Trends Cell Biol.
22:602–609. 2012. View Article : Google Scholar
|
|
50
|
Salinas PC: Wnt signaling in the
vertebrate central nervous system: From axon guidance to synaptic
function. Cold Spring Harb Perspect Biol. 4:a0080032012. View Article : Google Scholar
|
|
51
|
Eggenschwiler JT and Anderson KV: Cilia
and developmental signaling. Annu Rev Cell Dev Biol. 23:345–373.
2007. View Article : Google Scholar
|
|
52
|
Varjosalo M and Taipale J: Hedgehog:
Functions and mechanisms. Genes Dev. 22:2454–2472. 2008. View Article : Google Scholar
|
|
53
|
Schneider L, Clement CA, Teilmann SC,
Pazour GJ, Hoffmann EK, Satir P and Christensen ST: PDGFRalphaalpha
signaling is regulated through the primary cilium in fibroblasts.
Curr Biol. 15:1861–1866. 2005. View Article : Google Scholar
|
|
54
|
Clement DL, Mally S, Stock C, Lethan M,
Satir P, Schwab A, Pedersen SF and Christensen ST: PDGFRα signaling
in the primary cilium regulates NHE1-dependent fibroblast migration
via coordinated differential activity of MEK1/2-ERK1/2-p90RSK and
AKT signaling pathways. J Cell Sci. 126:953–965. 2013.
|
|
55
|
Pala R, Alomari N and Nauli SM: Primary
cilium-dependent signaling mechanisms. Int J Mol Sci. 18:22722017.
View Article : Google Scholar
|
|
56
|
Heldin CH: Targeting the PDGF signaling
pathway in the treatment of non-malignant diseases. J Neuroimmune
Pharmacol. 9:69–79. 2014. View Article : Google Scholar
|
|
57
|
Nishimura Y, Kasahara K, Shiromizu T,
Watanabe M and Inagaki M: Primary cilia as signaling hubs in health
and disease. Adv Sci (Weinh). 6:18011382018. View Article : Google Scholar
|
|
58
|
Schou KB, Pedersen LB and Christensen ST:
Ins and outs of GPCR signaling in primary cilia. EMBO Rep.
16:1099–1113. 2015. View Article : Google Scholar
|
|
59
|
Hilgendorf KI, Johnson CT and Jackson PK:
The primary cilium as a cellular receiver: Organizing ciliary GPCR
signaling. Curr Opin Cell Biol. 39:84–92. 2016. View Article : Google Scholar
|
|
60
|
Ezratty EJ, Stokes N, Chai S, Shah AS,
Williams SE and Fuchs E: A role for the primary cilium in Notch
signaling and epidermal differentiation during skin development.
Cell. 145:1129–1141. 2011. View Article : Google Scholar
|
|
61
|
Clement CA, Ajbro KD, Koefoed K,
Vestergaard ML, Veland IR, Henriques de Jesus MP, Pedersen LB,
Benmerah A, Andersen CY, Larsen LA and Christensen ST: TGF-β
signaling is associated with endocytosis at the pocket region of
the primary cilium. Cell Rep. 3:1806–1814. 2013. View Article : Google Scholar
|
|
62
|
Vestergaard ML, Awan A, Warzecha CB,
Christensen ST and Andersen CY: Immunofluorescence microscopy and
mRNA analysis of human embryonic stem cells (hESCs) including
primary cilia associated signaling pathways. Methods Mol Biol.
1307:123–140. 2016. View Article : Google Scholar
|
|
63
|
Basten SG and Giles RH: Functional aspects
of primary cilia in signaling, cell cycle and tumorigenesis. Cilia.
2:62013. View Article : Google Scholar
|
|
64
|
Zhong M, Zhao X, Li J, Yuan W, Yan G, Tong
M, Guo S, Zhu Y and Jiang Y, Liu Y and Jiang Y: Tumor suppressor
folliculin regulates mTORC1 through primary Cilia. J Biol Chem.
291:11689–11697. 2016. View Article : Google Scholar
|
|
65
|
Boehlke C, Kotsis F, Patel V, Braeg S,
Voelker H, Bredt S, Beyer T, Janusch H, Hamann C, Gödel M, et al:
Primary cilia regulate mTORC1 activity and cell size through Lkb1.
Nat Cell Biol. 12:1115–1122. 2010. View Article : Google Scholar
|
|
66
|
Leitch CC and Zaghloul NA: BBS4 is
necessary for ciliary localization of TrkB receptor and activation
by BDNF. PLoS One. 9:e986872014. View Article : Google Scholar
|
|
67
|
Lee JE and Gleeson JG: A systems-biology
approach to understanding the ciliopathy disorders. Genome Med.
3:592011. View
Article : Google Scholar
|
|
68
|
Ansley SJ, Badano JL, Blacque OE, Hill J,
Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM,
et al: Basal body dysfunction is a likely cause of pleiotropic
Bardet-Biedl syndrome. Nature. 425:628–633. 2003. View Article : Google Scholar
|
|
69
|
Laurence JZ and Moon RC: Four cases of
'retinitis pigmentosa' occurring in the same family, and
accompanied by general imperfections of development. 1866. Obes
Res. 3:400–403. 1995. View Article : Google Scholar
|
|
70
|
Moore SJ, Green JS, Fan Y, Bhogal AK,
Dicks E, Fernandez BA, Stefanelli M, Murphy C, Cramer BC, Dean JC,
et al: Clinical and genetic epidemiology of Bardet-Biedl syndrome
in Newfoundland: a 22-year prospective, population-based, cohort
study. Am J Med Genet A. 132A:352–360. 2005. View Article : Google Scholar
|
|
71
|
Mitchison HM and Valente EM: Motile and
non-motile cilia in human pathology: From function to phenotypes. J
Pathol. 241:294–309. 2017. View Article : Google Scholar
|
|
72
|
Hildebrandt F and Zhou W:
Nephronophthisis-associated ciliopathies. J Am Soc Nephrol.
18:1855–1871. 2007. View Article : Google Scholar
|
|
73
|
Bergmann C: Early and severe polycystic
kidney disease and related ciliopathies: An emerging field of
interest. Nephron. 141:50–60. 2019. View Article : Google Scholar
|
|
74
|
Srivastava S, Molinari E, Raman S and
Sayer JA: Many Genes-One disease? Genetics of nephronophthisis
(NPHP) and NPHP-Associated disorders. Front Pediatr. 5:2872018.
View Article : Google Scholar
|
|
75
|
Bergmann C: Genetics of autosomal
recessive polycystic kidney disease and its differential diagnoses.
Front Pediatr. 5:2212018. View Article : Google Scholar
|
|
76
|
Salomon R, Saunier S and Niaudet P:
Nephronophthisis. Pediatr Nephrol. 24:2333–2344. 2009. View Article : Google Scholar
|
|
77
|
Srivastava S and Sayer JA:
Nephronophthisis. J Pediatr Genet. 3:103–114. 2014. View Article : Google Scholar
|
|
78
|
Jenkins D and Beales PL: Genes and
mechanisms in human ciliopathies. Emery and Rimoin's Principles and
Practice of Medical Genetics. Rimoin D, Pyeritz R and Korf B:
Academic Press; Oxford; pp. 1–36. 2013
|
|
79
|
Gunay-Aygun M: Liver and kidney disease in
ciliopathies. Am J Med Genet C Semin Med Genet. 151C:296–306. 2009.
View Article : Google Scholar
|
|
80
|
Brancati F, Iannicelli M, Travaglini L,
Mazzotta A, Bertini E, Boltshauser E, D'Arrigo S, Emma F, Fazzi E,
Gallizzi R, et al: MKS3/TMEM67 mutations are a major cause of COACH
Syndrome, a Joubert Syndrome related disorder with liver
involvement. Hum Mutat. 30:E432–E442. 2009. View Article : Google Scholar
|
|
81
|
Doherty D, Parisi MA, Finn LS, Gunay-Aygun
M, Al-Mateen M, Bates D, Clericuzio C, Demir H, Dorschner M, van
Essen AJ, et al: Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L)
cause COACH syndrome (Joubert syndrome with congenital hepatic
fibrosis). J Med Genet. 47:8–21. 2010. View Article : Google Scholar
|
|
82
|
Parisi MA: Clinical and molecular features
of Joubert syndrome and related disorders. Am J Med Genet C Semin
Med Genet. 151C:326–340. 2009. View Article : Google Scholar
|
|
83
|
Brancati F, Dallapiccola B and Valente EM:
Joubert Syndrome and related disorders. Orphanet J Rare Dis.
5:202010. View Article : Google Scholar
|
|
84
|
Berger W, Kloeckener-Gruissem B and
Neidhardt J: The molecular basis of human retinal and vitreoretinal
diseases. Prog Retin Eye Res. 29:335–375. 2010. View Article : Google Scholar
|
|
85
|
Chung DC and Traboulsi EI: Leber
congenital amaurosis: Clinical correlations with genotypes, gene
therapy trials update, and future directions. J AAPOS. 13:587–592.
2009. View Article : Google Scholar
|
|
86
|
Wente S, Schroder S, Buckard J, Büttel HM,
von Deimling F, Diener W, Häussler M, Hübschle S, Kinder S,
Kurlemann G, et al: Nosological delineation of congenital ocular
motor apraxia type Cogan: An observational study. Orphanet J Rare
Dis. 11:1042016. View Article : Google Scholar
|
|
87
|
Valente EM, Dallapiccola B and Bertini E:
Joubert syndrome and related disorders. Handb Clin Neurol.
113:1879–1888. 2013. View Article : Google Scholar
|
|
88
|
Poretti A, Snow J, Summers AC, Tekes A,
Huisman TAGM, Aygun N, Carson KA, Doherty D, Parisi MA, Toro C, et
al: Joubert syndrome: Neuroimaging findings in 110 patients in
correlation with cognitive function and genetic cause. J Med Genet.
54:521–529. 2017. View Article : Google Scholar
|
|
89
|
Romani M, Micalizzi A and Valente EM:
Joubert syndrome: Congenital cerebellar ataxia with the molar
tooth. Lancet Neurol. 12:894–905. 2013. View Article : Google Scholar
|
|
90
|
Poretti A, Boltshauser E, Loenneker T,
Valente EM, Brancati F, Il'yasov K and Huisman TA: Diffusion tensor
imaging in Joubert syndrome. AJNR Am J Neuroradiol. 28:1929–1933.
2007. View Article : Google Scholar
|
|
91
|
Valente EM, Brancati F and Dallapiccola B:
Genotypes and phenotypes of Joubert syndrome and related disorders.
Eur J Med Genet. 51:1–23. 2008. View Article : Google Scholar
|
|
92
|
Akizu N, Silhavy JL, Rosti RO, Scott E,
Fenstermaker AG, Schroth J, Zaki MS, Sanchez H, Gupta N, Kabra M,
et al: Mutations in CSPP1 lead to classical Joubert syndrome. Am J
Hum Genet. 94:80–86. 2014. View Article : Google Scholar
|
|
93
|
Khan S, Lin S, Harlalka GV, Ullah A, Shah
K, Khalid S, Mehmood S, Hassan MJ, Ahmad W, Self JE, et al: BBS5
and INPP5E mutations associated with ciliopathy disorders in
families from Pakistan. Ann Hum Genet. 83:477–482. 2019. View Article : Google Scholar
|
|
94
|
Srour M, Schwartzentruber J, Hamdan FF,
Ospina LH, Patry L, Labuda D, Massicotte C, Dobrzeniecka S,
Capo-Chichi JM, Papillon-Cavanagh S, et al: Mutations in C5ORF42
cause Joubert syndrome in the French Canadian population. Am J Hum
Genet. 90:693–700. 2012. View Article : Google Scholar
|
|
95
|
Verma PK and El-Harouni AA: Review of
literature: Genes related to postaxial polydactyly. Front Pediatr.
3:82015. View Article : Google Scholar
|
|
96
|
Marion V, Stutzmann F, Gerard M, De Melo
C, Schaefer E, Claussmann A, Hellé S, Delague V, Souied E, Barrey
C, et al: Exome sequencing identifies mutations in LZTFL1, a BBSome
and smoothened trafficking regulator, in a family with
Bardet--Biedl syndrome with situs inversus and insertional
polydactyly. J Med Genet. 49:317–321. 2012. View Article : Google Scholar
|
|
97
|
Figuera LE, Rivas F and Cantu JM:
Oral-facial-digital syndrome with fibular aplasia: A new variant.
Clin Genet. 44:190–192. 1993. View Article : Google Scholar
|
|
98
|
Kannu P, McFarlane JH, Savarirayan R and
Aftimos S: An unclassifiable short rib-polydactyly syndrome with
acromesomelic hypomineralization and campomelia in siblings. Am J
Med Genet A. 143A:2607–2611. 2007. View Article : Google Scholar
|
|
99
|
Schmidts M and Mitchison HM: Severe
skeletal abnormalities caused by defects in retrograde
intraflagellar transport dyneins. King SM: Academic Press; pp.
356–401. 2018
|
|
100
|
Schmidts M: Clinical genetics and
pathobiology of ciliary chondrodysplasias. J Pediatr Genet.
3:46–94. 2014.
|
|
101
|
Hartill V, Szymanska K, Sharif SM, Wheway
G and Johnson CA: Meckel-Gruber syndrome: An update on diagnosis,
clinical management, and research advances. Front Pediatr.
5:2442017. View Article : Google Scholar
|
|
102
|
Doherty D, Glass IA, Siebert JR, Strouse
PJ, Parisi MA, Shaw DW, Chance PF, Barr M Jr and Nyberg D: Prenatal
diagnosis in pregnancies at risk for Joubert syndrome by ultrasound
and MRI. Prenat Diagn. 25:442–447. 2005. View Article : Google Scholar
|
|
103
|
Sepulveda W, Sebire NJ, Souka A, Snijders
RJ and Nicolaides KH: Diagnosis of the Meckel-Gruber syndrome at
eleven to fourteen weeks' gestation. Am J Obstet Gynecol.
176:316–319. 1997. View Article : Google Scholar
|
|
104
|
Reece EA and Goldstein I: Early prenatal
diagnosis of hydrocephalus. Am J Perinatol. 14:69–73. 1997.
View Article : Google Scholar
|
|
105
|
den Hollander NS, Robben SG, Hoogeboom AJ,
Niermeijer MF and Wladimiroff JW: Early prenatal sonographic
diagnosis and follow-up of Jeune syndrome. Ultrasound Obstet
Gynecol. 18:378–383. 2001. View Article : Google Scholar
|
|
106
|
Poretti A, Brehmer U, Scheer I, Bernet V
and Boltshauser E: Prenatal and neonatal MR imaging findings in
oral-facial-digital syndrome type VI. AJNR Am J Neuroradiol.
29:1090–1091. 2008. View Article : Google Scholar
|
|
107
|
Beales PL and Kenny TD: Towards the
diagnosis of a ciliopathy. Ciliopathies: A Reference for
Clinicians. Oxford University Press; Oxford; pp. 1–7. 2013
|
|
108
|
Katsanis N, Beales PL, Woods MO, Lewis RA,
Green JS, Parfrey PS, Ansley SJ, Davidson WS and Lupski JR:
Mutations in MKKS cause obesity, retinal dystrophy and renal
malformations associated with Bardet-Biedl syndrome. Nat Genet. 26.
pp. 67–70. 2000, View
Article : Google Scholar
|
|
109
|
Slavotinek AM, Stone EM, Mykytyn K,
Heckenlively JR, Green JS, Heon E, Musarella MA, Parfrey PS,
Sheffield VC and Biesecker LG: Mutations in MKKS cause Bardet-Biedl
syndrome. Nat Genet. 26. pp. 15–16. 2000, View Article : Google Scholar
|
|
110
|
Reiter JF and Leroux MR: Genes and
molecular pathways underpinning ciliopathies. Nat Rev Mol Cell
Biol. 18:533–547. 2017. View Article : Google Scholar
|
|
111
|
Shaheen R, Szymanska K, Basu B, Patel N,
Ewida N, Faqeih E, Al Hashem A, Derar N, Alsharif H, Aldahmesh MA,
et al: Characterizing the morbid genome of ciliopathies. Genome
Biol. 17:2422016. View Article : Google Scholar
|
|
112
|
Lindstrand A, Davis EE, Carvalho CM,
Pehlivan D, Willer JR, Tsai IC, Ramanathan S, Zuppan C, Sabo A,
Muzny D, et al: Recurrent CNVs and SNVs at the NPHP1 locus
contribute pathogenic alleles to Bardet-Biedl syndrome. Am J Hum
Genet. 94:745–754. 2014. View Article : Google Scholar
|
|
113
|
Lindstrand A, Frangakis S, Carvalho CM,
Richardson EB, McFadden KA, Willer JR, Pehlivan D, Liu P,
Pediaditakis IL, Sabo A, et al: Copy-Number variation contributes
to the mutational load of Bardet-Biedl syndrome. Am J Hum Genet.
99:318–336. 2016. View Article : Google Scholar
|
|
114
|
Fauser S, Munz M and Besch D: Further
support for digenic inheritance in Bardet-Biedl syndrome. J Med
Genet. 40:e1042003. View Article : Google Scholar
|
|
115
|
Katsanis N: The oligogenic properties of
Bardet-Biedl syndrome. Hum Mol Genet. 13(Spec No 1): R65–R71. 2004.
View Article : Google Scholar
|
|
116
|
Katsanis N, Ansley SJ, Badano JL, Eichers
ER, Lewis RA, Hoskins BE, Scambler PJ, Davidson WS, Beales PL and
Lupski JR: Triallelic inheritance in Bardet-Biedl syndrome, a
Mendelian recessive disorder. Science. 293. pp. 2256–2259. 2001,
View Article : Google Scholar
|
|
117
|
Katsanis N, Eichers ER, Ansley SJ, Lewis
RA, Kayserili H, Hoskins BE, Scambler PJ, Beales PL and Lupski JR:
BBS4 is a minor contributor to Bardet-Biedl syndrome and may also
participate in triallelic inheritance. Am J Hum Genet. 71. pp.
22–29. 2002, View
Article : Google Scholar
|
|
118
|
Beales PL, Badano JL, Ross AJ, Ansley SJ,
Hoskins BE, Kirsten B, Mein CA, Froguel P, Scambler PJ, Lewis RA,
et al: Genetic interaction of BBS1 mutations with alleles at other
BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am J
Hum Genet. 72:1187–1199. 2003. View
Article : Google Scholar
|
|
119
|
Abu-Safieh L, Al-Anazi S, Al-Abdi L,
Hashem M, Alkuraya H, Alamr M, Sirelkhatim MO, Al-Hassnan Z,
Alkuraya B, Mohamed JY, et al: In search of triallelism in
Bardet-Biedl syndrome. Eur J Hum Genet. 20:420–427. 2012.
View Article : Google Scholar
|
|
120
|
Delvallée C, Nicaise S, Antin M, Leuvrey
AS, Nourisson E, Leitch CC, Kellaris G, Stoetzel C, Geoffroy V,
Scheidecker S, et al: A BBS1 SVA F retrotransposon insertion is a
frequent cause of Bardet-Biedl syndrome. Clin Genet. 99:318–324.
2021. View Article : Google Scholar
|
|
121
|
Davis EE and Katsanis N: The ciliopathies:
A transitional model into systems biology of human genetic disease.
Curr Opin Genet Dev. 22:290–303. 2012. View Article : Google Scholar
|
|
122
|
Badano JL, Kim JC, Hoskins BE, Lewis RA,
Ansley SJ, Cutler DJ, Castellan C, Beales PL, Leroux MR and
Katsanis N: Heterozygous mutations in BBS1, BBS2 and BBS6 have a
potential epistatic effect on Bardet-Biedl patients with two
mutations at a second BBS locus. Hum Mol Genet. 12. pp. 1651–1659.
2003, View Article : Google Scholar
|
|
123
|
Badano JL, Leitch CC, Ansley SJ,
May-Simera H, Lawson S, Lewis RA, Beales PL, Dietz HC, Fisher S and
Katsanis N: Dissection of epistasis in oligogenic Bardet-Biedl
syndrome. Nature. 439. pp. 326–330. 2006, View Article : Google Scholar
|
|
124
|
Slavotinek A and Biesecker LG: Genetic
modifiers in human development and malformation syndromes,
including chaperone proteins. Hum Mol Genet. 12(Spec No 1):
R45–R50. 2003. View Article : Google Scholar
|
|
125
|
Hildebrandt F, Benzing T and Katsanis N:
Ciliopathies. N Engl J Med. 364:1533–1543. 2011. View Article : Google Scholar
|
|
126
|
Novarino G, Akizu N and Gleeson JG:
Modeling human disease in humans: The ciliopathies. Cell.
147:70–79. 2011. View Article : Google Scholar
|
|
127
|
Zaki MS, Sattar S, Massoudi RA and Gleeson
JG: Co-occurrence of distinct ciliopathy diseases in single
families suggests genetic modifiers. Am J Med Genet A.
155A:3042–3049. 2011. View Article : Google Scholar
|
|
128
|
Khanna H, Davis EE, Murga-Zamalloa CA,
Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman
MI, Waseem N, Chakarova CF, et al: A common allele in RPGRIP1L is a
modifier of retinal degeneration in ciliopathies. Nat Genet.
41:739–745. 2009. View
Article : Google Scholar
|
|
129
|
Bujakowska KM, Liu Q and Pierce EA:
Photoreceptor cilia and retinal ciliopathies. Cold Spring Harb
Perspect Biol. 9:a0282742017. View Article : Google Scholar
|
|
130
|
Goldberg AF, Moritz OL and Williams DS:
Molecular basis for photoreceptor outer segment architecture. Prog
Retin Eye Res. 55:52–81. 2016. View Article : Google Scholar
|
|
131
|
Estrada-Cuzcano A, Roepman R, Cremers FP,
den Hollander AI and Mans DA: Non-syndromic retinal ciliopathies:
Translating gene discovery into therapy. Hum Mol Genet.
21:R111–R124. 2012. View Article : Google Scholar
|
|
132
|
den Hollander AI, Roepman R, Koenekoop RK
and Cremers FP: Leber congenital amaurosis: Genes, proteins and
disease mechanisms. Prog Retin Eye Res. 27:391–419. 2008.
View Article : Google Scholar
|
|
133
|
Adams NA, Awadein A and Toma HS: The
retinal ciliopathies. Ophthalmic Genet. 28:113–125. 2007.
View Article : Google Scholar
|
|
134
|
Marshall JD, Muller J, Collin GB, Milan G,
Kingsmore SF, Dinwiddie D, Farrow EG, Miller NA, Favaretto F,
Maffei P, et al: Alstrom syndrome: Mutation spectrum of ALMS1. Hum
Mutat. 36:660–668. 2015. View Article : Google Scholar
|
|
135
|
Ronquillo CC, Bernstein PS and Baehr W:
Senior-Loken syndrome: A syndromic form of retinal dystrophy
associated with nephronophthisis. Vision Res. 75:88–97. 2012.
View Article : Google Scholar
|
|
136
|
Braun DA and Hildebrandt F: Ciliopathies.
Cold Spring Harb Perspect Biol. 9:a0281912017. View Article : Google Scholar
|
|
137
|
Saigusa T and Bell PD: Molecular pathways
and therapies in autosomal-dominant polycystic kidney disease.
Physiology (Bethesda). 30:195–207. 2015.
|
|
138
|
Bergmann C: ARPKD and early manifestations
of ADPKD: The original polycystic kidney disease and phenocopies.
Pediatr Nephrol. 30:15–30. 2015. View Article : Google Scholar
|
|
139
|
Chebib FT and Torres VE: Autosomal
dominant polycystic kidney disease: Core Curriculum 2016. Am J
Kidney Dis. 67:792–810. 2016. View Article : Google Scholar
|
|
140
|
Wolf MT and Hildebrandt F:
Nephronophthisis. Pediatr Nephrol. 26:181–194. 2011. View Article : Google Scholar
|
|
141
|
Konig J, Kranz B, Konig S, Schlingmann KP,
Titieni A, Tönshoff B, Habbig S, Pape L, Häffner K, Hansen M, et
al: Phenotypic spectrum of children with nephronophthisis and
related ciliopathies. Clin J Am Soc Nephrol. 12:1974–1983. 2017.
View Article : Google Scholar
|
|
142
|
Zaghloul NA and Katsanis N: Mechanistic
insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin
Invest. 119:428–437. 2009. View Article : Google Scholar
|
|
143
|
Forsythe E and Beales PL: Bardet-Biedl
syndrome. Eur J Hum Genet. 21:8–13. 2013. View Article : Google Scholar
|
|
144
|
Forsythe R and Gunay-Aygun M: Bardet-Biedl
syndrome overview. Adam MP, Ardinger HH, Pagon RA, et al:
GeneReviews® [Internet]. University of Washington; Seattle, WA:
2003, updated July 23, 2020.
|
|
145
|
Bujakowska KM, Zhang Q, Siemiatkowska AM,
Liu Q, Place E, Falk MJ, Consugar M, Lancelot ME, Antonio A, Lonjou
C, et al: Mutations in IFT172 cause isolated retinal degeneration
and Bardet-Biedl syndrome. Hum Mol Genet. 24:230–242. 2015.
View Article : Google Scholar
|
|
146
|
Alazami AM, Alshammari MJ, Salih MA,
Alzahrani F, Hijazi H, Seidahmed MZ, Abu Safieh L, Aldosary M, Khan
AO and Alkuraya FS: Molecular characterization of Joubert syndrome
in Saudi Arabia. Hum Mutat. 33:1423–1428. 2012. View Article : Google Scholar
|
|
147
|
Stephen J, Vilboux T, Mian L, Kuptanon C,
Sinclair CM, Yildirimli D, Maynard DM, Bryant J, Fischer R,
Vemulapalli M, et al: Mutations in KIAA0753 cause Joubert syndrome
associated with growth hormone deficiency. Hum Genet. 136:399–408.
2017. View Article : Google Scholar
|
|
148
|
Shaheen R, Jiang N, Alzahrani F, Ewida N,
Al-Sheddi T, Alobeid E, Musaev D, Stanley V, Hashem M, Ibrahim N,
et al: Bi-allelic Mutations in FAM149B1 cause abnormal primary
cilium and a range of ciliopathy phenotypes in humans. Am J Hum
Genet. 104:731–737. 2019. View Article : Google Scholar
|
|
149
|
Radha Rama Devi A, Naushad SM and Lingappa
L: Clinical and molecular diagnosis of joubert syndrome and related
disorders. Pediatr Neurol. 106:43–49. 2020. View Article : Google Scholar
|
|
150
|
Oka M, Shimojima K, Yamamoto T, Hanaoka Y,
Sato S, Yasuhara T, Yoshinaga H and Kobayashi K: A novel HYLS1
homozygous mutation in living siblings with Joubert syndrome. Clin
Genet. 89:739–743. 2016. View Article : Google Scholar
|
|
151
|
Beck BB, Phillips JB, Bartram MP, Wegner
J, Thoenes M, Pannes A, Sampson J, Heller R, Göbel H, Koerber F, et
al: Mutation of POC1B in a severe syndromic retinal ciliopathy. Hum
Mutat. 35:1153–1162. 2014. View Article : Google Scholar
|
|
152
|
Bachmann-Gagescu R, Dempsey JC, Phelps IG,
O'Roak BJ, Knutzen DM, Rue TC, Ishak GE, Isabella CR, Gorden N,
Adkins J, et al: Joubert syndrome: A model for untangling recessive
disorders with extreme genetic heterogeneity. J Med Genet.
52:514–522. 2015. View Article : Google Scholar
|
|
153
|
Schmidts M: Jeune syndrome and the ciliary
chondrodysplasias. Ciliopathies. Oxford University Press; Oxford;
pp. 30–63. 2013, View Article : Google Scholar
|
|
154
|
Haycraft CJ, Zhang Q, Song B, Jackson WS,
Detloff PJ, Serra R and Yoder BK: Intraflagellar transport is
essential for endochondral bone formation. Development.
134:307–316. 2007. View Article : Google Scholar
|
|
155
|
St-Jacques B, Hammerschmidt M and McMahon
AP: Indian hedgehog signaling regulates proliferation and
differentiation of chondrocytes and is essential for bone
formation. Genes Dev. 13:2072–2086. 1999. View Article : Google Scholar
|
|
156
|
Schmidts M, Hou Y, Cortes CR, Mans DA,
Huber C, Boldt K, Patel M, van Reeuwijk J, Plaza JM, van Beersum
SE, et al: TCTEX1D2 mutations underlie Jeune asphyxiating thoracic
dystrophy with impaired retrograde intraflagellar transport. Nat
Commun. 6:70742015. View Article : Google Scholar
|
|
157
|
Zhang W, Taylor SP, Ennis HA, Forlenza KN,
Duran I, Li B, Sanchez JAO, Nevarez L, Nickerson DA and Bamshad M:
Expanding the genetic architecture and phenotypic spectrum in the
skeletal ciliopathies. Hum Mutat. 39:152–166. 2018. View Article : Google Scholar
|
|
158
|
Ellis RW and van Creveld S: A syndrome
characterized by ectodermal dysplasia, polydactyly,
Chondro-Dysplasia and congenital Morbus Cordis: Report of three
cases. Arch Dis Child. 15:65–84. 1940. View Article : Google Scholar
|
|
159
|
McKusick VA, Eldridge R, Hostetler JA and
Egeland JA: Dwarfism in the Amish. Trans Assoc Am Physicians.
77:151–168. 1964.
|
|
160
|
Ruiz-Perez VL and Goodship JA: Ellis-van
Creveld syndrome and Weyers acrodental dysostosis are caused by
cilia-mediated diminished response to hedgehog ligands. Am J Med
Genet C Semin Med Genet. 151C:341–351. 2009. View Article : Google Scholar
|
|
161
|
Howard TD, Guttmacher AE, McKinnon W,
Sharma M, McKusick VA and Jabs EW: Autosomal dominant postaxial
polydactyly, nail dystrophy, and dental abnormalities map to
chromosome 4p16, in the region containing the Ellis-van Creveld
syndrome locus. Am J Hum Genet. 61:1405–1412. 1997. View Article : Google Scholar
|
|
162
|
Levin LS, Perrin JC, Ose L, Dorst JP,
Miller JD and McKusick VA: A heritable syndrome of
craniosynostosis, short thin hair, dental abnormalities, and short
limbs: Cranioectodermal dysplasia. J Pediatr. 90:55–61. 1977.
View Article : Google Scholar
|
|
163
|
Lin AE, Traum AZ, Sahai I, Keppler-Noreuil
K, Kukolich MK, Adam MP, Westra SJ and Arts HH: Sensenbrenner
syndrome (Cranioectodermal dysplasia): Clinical and molecular
analyses of 39 patients including two new patients. Am Med Genet A.
161A:2762–2776. 2013. View Article : Google Scholar
|
|
164
|
Arts HH, Bongers EM, Mans DA, van Beersum
SE, Oud MM, Bolat E, Spruijt L, Cornelissen EA, Schuurs-Hoeijmakers
JH, de Leeuw N, et al: C14ORF179 encoding IFT43 is mutated in
Sensenbrenner syndrome. J Med Genet. 48:390–395. 2011. View Article : Google Scholar
|
|
165
|
Huber C and Cormier-Daire V: Ciliary
disorder of the skeleton. Am J Med Genet C Semin Med Genet.
160C:165–174. 2012. View Article : Google Scholar
|
|
166
|
Mainzer F, Saldino RM, Ozonoff MB and
Minagi H: Familial nephropathy associatdd with retinitis
pigmentosa, cerebellar ataxia and skeletal abnormalities. Am J Med.
49:556–562. 1970. View Article : Google Scholar
|
|
167
|
Perrault I, Saunier S, Hanein S, Filhol E,
Bizet AA, Collins F, Salih MA, Gerber S, Delphin N, Bigot K, et al:
Mainzer-Saldino syndrome is a ciliopathy caused by IFT140
mutations. Am J Hum Genet. 90:864–870. 2012. View Article : Google Scholar
|
|
168
|
Beales PL, Bland E, Tobin JL, Bacchelli C,
Tuysuz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J, et al:
IFT80, which encodes a conserved intraflagellar transport protein,
is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet.
39:727–729. 2007. View
Article : Google Scholar
|
|
169
|
Tuysuz B, Baris S, Aksoy F, Madazli R,
Ungur S and Sever L: Clinical variability of asphyxiating thoracic
dystrophy (Jeune) syndrome: Evaluation and classification of 13
patients. Am J Med Genet A. 149A:1727–1733. 2009. View Article : Google Scholar
|
|
170
|
Zhang W, Taylor SP, Nevarez L, Lachman RS,
Nickerson DA and Bamshad M: IFT52 mutations destabilize anterograde
complex assembly, disrupt ciliogenesis and result in short rib
polydactyly syndrome. Hum Mol Genet. 25:4012–4020. 2016. View Article : Google Scholar
|
|
171
|
Duran I, Taylor SP, Zhang W, Martin J,
Qureshi F, Jacques SM, Wallerstein R, Lachman RS, Nickerson DA,
Bamshad M, et al: Mutations in IFT-A satellite core component genes
IFT43 and IFT121 produce short rib polydactyly syndrome with
distinctive campomelia. Cilia. 6:72017. View Article : Google Scholar
|
|
172
|
Roosing S, Romani M, Isrie M, Rosti RO,
Micalizzi A, Musaev D, Mazza T, Al-Gazali L, Altunoglu U,
Boltshauser E, et al: Mutations in CEP120 cause Joubert syndrome as
well as complex ciliopathy phenotypes. J Med Genet. 53:608–615.
2016. View Article : Google Scholar
|
|
173
|
Bruel AL, Franco B, Duffourd Y, Thevenon
J, Jego L, Lopez E, Deleuze JF, Doummar D, Giles RH, Johnson CA, et
al: Fifteen years of research on oral-facial-digital syndromes:
From 1 to 16 causal genes. J Med Genet. 54:371–380. 2017.
View Article : Google Scholar
|
|
174
|
Li C, Jensen VL, Park K, Kennedy J,
Garcia-Gonzalo FR, Romani M, De Mori R, Bruel AL, Gaillard D, Doray
B, et al: MKS5 and CEP290 dependent assembly pathway of the ciliary
transition zone. PLoS Biol. 14:e10024162016. View Article : Google Scholar
|
|
175
|
Al-Qattan MM, Shaheen R and Alkuraya FS:
Expanding the allelic disorders linked to TCTN1 to include Varadi
syndrome (Orofaciodigital syndrome type VI). Am J Med Genet A.
173:2439–2441. 2017. View Article : Google Scholar
|
|
176
|
Franco B and Thauvin-Robinet C: Update on
oral-facial-digital syndromes (OFDS). Cilia. 5:122016. View Article : Google Scholar
|
|
177
|
Toriello HV: Are the oral-facial-digital
syndromes ciliopathies? Am J Med Genet A. 149A:1089–1095. 2009.
View Article : Google Scholar
|
|
178
|
Toriello HV, Franco B, Bruel AL and
Thauvin-Robinet C: Oral-Facial-Digital Syndrome Type I.
GeneReviews((R)). Adam MP, Ardinger HH and Pagon RA: University of
Washington; Seattle, WA: 1993
|
|
179
|
Shamseldin HE, Rajab A, Alhashem A,
Shaheen R, Al-Shidi T, Alamro R, Al Harassi S and Alkuraya FS:
Mutations in DDX59 implicate RNA helicase in the pathogenesis of
orofaciodigital syndrome. Am J Hum Genet. 93:555–560. 2013.
View Article : Google Scholar
|
|
180
|
Toriyama M, Lee C, Taylor SP, Duran I,
Cohn DH, Bruel AL, Tabler JM, Drew K, Kelly MR, Kim S, et al: The
ciliopathy-associated CPLANE proteins direct basal body recruitment
of intraflagellar transport machinery. Nat Genet. 48:648–656. 2016.
View Article : Google Scholar
|
|
181
|
Saari J, Lovell MA, Yu HC and Bellus GA:
Compound heterozygosity for a frame shift mutation and a likely
pathogenic sequence variant in the planar cell
polarity-ciliogenesis gene WDPCP in a girl with polysyndactyly,
coarctation of the aorta, and tongue hamartomas. Am J Med Genet A.
167A:421–427. 2015. View Article : Google Scholar
|
|
182
|
Thevenon J, Duplomb L, Phadke S, Eguether
T, Saunier A, Avila M, Carmignac V, Bruel AL, St-Onge J, Duffourd
Y, et al: Autosomal recessive IFT57 hypomorphic mutation cause
ciliary transport defect in unclassified oral-facial-digital
syndrome with short stature and brachymesophalangia. Clin Genet.
90:509–517. 2016. View Article : Google Scholar
|
|
183
|
Baker K and Beales PL: Making sense of
cilia in disease: The human ciliopathies. Am J Med Genet C Semin
Med Genet. 151C:281–295. 2009. View Article : Google Scholar
|
|
184
|
Laquerriere A, Maluenda J, Camus A,
Fontenas L, Dieterich K, Nolent F, Zhou J, Monnier N, Latour P,
Gentil D, et al: Mutations in CNTNAP1 and ADCY6 are responsible for
severe arthrogryposis multiplex congenita with axoglial defects.
Hum Mol Genet. 23:2279–2289. 2014. View Article : Google Scholar
|
|
185
|
Filges I, Bruder E, Brandal K, Meier S,
Undlien DE, Waage TR, Hoesli I, Schubach M, de Beer T, Sheng Y, et
al: Stromme syndrome is a ciliary disorder caused by mutations in
CENPF. Hum Mutat. 37:359–363. 2016. View Article : Google Scholar
|
|
186
|
Waters AM, Asfahani R, Carroll P, Bicknell
L, Lescai F, Bright A, Chanudet E, Brooks A, Christou-Savina S,
Osman G, et al: The kinetochore protein, CENPF, is mutated in human
ciliopathy and microcephaly phenotypes. J Med Genet. 52. pp.
147–156. 2015, View Article : Google Scholar
|
|
187
|
Luijten MN, Basten SG, Claessens T,
Vernooij M, Scott CL, Janssen R, Easton JA, Kamps MA, Vreeburg M,
Broers JL, et al: Birt-Hogg-Dube syndrome is a novel ciliopathy.
Hum Mol Genet. 22:4383–4397. 2013. View Article : Google Scholar
|
|
188
|
Jenkins D, Seelow D, Jehee FS, Perlyn CA,
Alonso LG, Bueno DF, Donnai D, Josifova D, Mathijssen IM, Morton
JE, et al: RAB23 mutations in Carpenter syndrome imply an
unexpected role for hedgehog signaling in cranial-suture
development and obesity. Am J Hum Genet. 80:1162–1170. 2007.
View Article : Google Scholar
|
|
189
|
Symoens S, Barnes AM, Gistelinck C,
Malfait F, Guillemyn B, Steyaert W, Syx D, D'hondt S, Biervliet M,
De Backer J, et al: Genetic defects in TAPT1 disrupt ciliogenesis
and cause a complex lethal osteochondrodysplasia. Am J Hum Genet.
97:521–534. 2015. View Article : Google Scholar
|
|
190
|
Reijnders MR, Zachariadis V, Latour B,
Jolly L, Mancini GM, Pfundt R, Wu KM, van Ravenswaaij-Arts CM,
Veenstra-Knol HE, Anderlid BM, et al: De Novo Loss-of-Function
mutations in USP9X cause a Female-Specific recognizable syndrome
with developmental delay and congenital malformations. Am J Hum
Genet. 98:373–381. 2016. View Article : Google Scholar
|
|
191
|
Twigg SRF, Hufnagel RB, Miller KA, Zhou Y,
McGowan SJ, Taylor J, Craft J, Taylor JC, Santoro SL, Huang T, et
al: A recurrent Mosaic Mutation in SMO, encoding the Hedgehog
signal transducer smoothened, is the major cause of Curry-Jones
syndrome. Am J Hum Genet. 98:1256–1265. 2016. View Article : Google Scholar
|
|
192
|
Ali BR, Silhavy JL, Akawi NA, Gleeson JG
and Al-Gazali L: A mutation in KIF7 is responsible for the
autosomal recessive syndrome of macrocephaly, multiple epiphyseal
dysplasia and distinctive facial appearance. Orphanet J Rare Dis.
7:272012. View Article : Google Scholar
|
|
193
|
Putoux A, Thomas S, Coene KL, Davis EE,
Alanay Y, Ogur G, Uz E, Buzas D, Gomes C, Patrier S, et al: KIF7
mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat
Genet. 43:601–606. 2011. View
Article : Google Scholar
|
|
194
|
Dammermann A, Pemble H, Mitchell BJ,
McLeod I, Yates JR III, Kintner C, Desai AB and Oegema K: The
hydrolethalus syndrome protein HYLS-1 links core centriole
structure to cilia formation. Genes Dev. 23:2046–2059. 2009.
View Article : Google Scholar
|
|
195
|
Alby C, Piquand K, Huber C, Megarbané A,
Ichkou A, Legendre M, Pelluard F, Encha-Ravazi F, Abi-Tayeh G,
Bessières B, et al: Mutations in KIAA0586 cause lethal ciliopathies
ranging from a hydrolethalus phenotype to Short-Rib Polydactyly
syndrome. Am J Hum Genet. 97:311–318. 2015. View Article : Google Scholar
|
|
196
|
Demurger F, Ichkou A, Mougou-Zerelli S, Le
Merrer M, Goudefroye G, Delezoide AL, Quélin C, Manouvrier S,
Baujat G, Fradin M, et al: New insights into genotype-phenotype
correlation for GLI3 mutations. Eur J Hum Genet. 23:92–102. 2015.
View Article : Google Scholar
|
|
197
|
Oud MM, Bonnard C, Mans DA, Altunoglu U,
Tohari S, Ng AYJ, Eskin A, Lee H, Rupar CA, de Wagenaar NP, et al:
A novel ICK mutation causes ciliary disruption and lethal
endocrine-cerebro-osteodysplasia syndrome. Cilia. 5:82016.
View Article : Google Scholar
|
|
198
|
Karaca E, Buyukkaya R, Pehlivan D, Charng
WL, Yaykasli KO, Bayram Y, Gambin T, Withers M, Atik MM, Arslanoglu
I, et al: Whole-exome sequencing identifies homozygous GPR161
mutation in a family with pituitary stalk interruption syndrome. J
Clin Endocrinol Metab. 100:E140–147. 2015. View Article : Google Scholar
|
|
199
|
Guen VJ, Gamble C, Perez DE, Bourassa S,
Zappel H, Gärtner J, Lees JA and Colas P: STAR syndrome-associated
CDK10/Cyclin M regulates actin network architecture and
ciliogenesis. Cell Cycle. 15:678–688. 2016. View Article : Google Scholar
|
|
200
|
Alessandri JL, Dagoneau N, Laville JM,
Baruteau J, Hébert JC and Cormier-Daire V: RAB23 mutation in a
large family from Comoros Islands with Carpenter syndrome. Am J Med
Genet A. 152A:982–986. 2010. View Article : Google Scholar
|
|
201
|
Jenkins D, Baynam G, De Catte L, Elcioglu
N, Gabbett MT, Hudgins L, Hurst JA, Jehee FS, Oley C and Wilkie AO:
Carpenter syndrome: Extended RAB23 mutation spectrum and analysis
of nonsense-mediated mRNA decay. Hum Mutat. 32:E2069–E2078. 2011.
View Article : Google Scholar
|
|
202
|
Lefroy H, Hurst JA and Shears DJ: STAR
syndrome: A further case and the first report of maternal
mosaicism. Clin Dysmorphol. 26:157–160. 2017. View Article : Google Scholar
|
|
203
|
Ng SB, Bigham AW, Buckingham KJ, Hannibal
MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM,
Mefford HC, et al: Exome sequencing identifies MLL2 mutations as a
cause of Kabuki syndrome. Nat Genet. 42:790–793. 2010. View Article : Google Scholar
|
|
204
|
Miyake N, Koshimizu E, Okamoto N, Mizuno
S, Ogata T, Nagai T, Kosho T, Ohashi H, Kato M, Sasaki G, et al:
MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am J Med
Genet A. 161A:2234–2243. 2013. View Article : Google Scholar
|
|
205
|
Shaheen R, Rahbeeni Z, Alhashem A, Faqeih
E, Zhao Q, Xiong Y, Almoisheer A, Al-Qattan SM, Almadani HA,
Al-Onazi N, et al: Neu-Laxova syndrome, an inborn error of serine
metabolism, is caused by mutations in PHGDH. Am J Hum Genet.
94:898–904. 2014. View Article : Google Scholar
|
|
206
|
Acuna-Hidalgo R, Schanze D, Kariminejad A,
Nordgren A, Kariminejad MH, Conner P, Grigelioniene G, Nilsson D,
Nordenskjöld M, Wedell A, et al: Neu-Laxova syndrome is a
heterogeneous metabolic disorder caused by defects in enzymes of
the L-serine biosynthesis pathway. Am J Hum Genet. 95:285–293.
2014. View Article : Google Scholar
|
|
207
|
Duplomb L, Duvet S, Picot D, Jego G, El
Chehadeh-Djebbar S, Marle N, Gigot N, Aral B, Carmignac V, Thevenon
J, et al: Cohen syndrome is associated with major glycosylation
defects. Hum Mol Genet. 23:2391–2399. 2014. View Article : Google Scholar
|
|
208
|
El Chehadeh S, Aral B, Gigot N,
Thauvin-Robinet C, Donzel A, Delrue MA, Lacombe D, David A, Burglen
L, Philip N, et al: Search for the best indicators for the presence
of a VPS13B gene mutation and confirmation of diagnostic criteria
in a series of 34 patients genotyped for suspected Cohen syndrome.
J Med Genet. 47:549–553. 2010. View Article : Google Scholar
|
|
209
|
Emery NJ, Seed AM, von Bayern AM and
Clayton NS: Cognitive adaptations of social bonding in birds.
Philos Trans R Soc Lond B Biol Sci. 362:489–505. 2007. View Article : Google Scholar
|
|
210
|
Bozal-Basterra L, Martin-Ruiz I, Pirone L,
Liang Y, Sigurðsson JO, Gonzalez-Santamarta M, Giordano I,
Gabicagogeascoa E, de Luca A, Rodríguez JA, et al: Truncated SALL1
impedes primary cilia function in townes-brocks syndrome. Am J Hum
Genet. 102:249–265. 2018. View Article : Google Scholar
|
|
211
|
Devlin LA and Sayer JA: Renal
ciliopathies. Curr Opin Genet Dev. 56:49–60. 2019. View Article : Google Scholar
|