Introduction
Chimeric antigen receptor (CAR)-T cell therapies are
efficient methodologies for the immunotherapy of refractory
malignancies. Anti-CD19-CAR-T cell therapy shows excellent
therapeutic effects against B-cell lymphoblastic leukemia (1,2)
and B-cell lymphoma (3,4). CAR-T cells recognizing B-cell
maturation antigen (BCMA) are used for refractory multiple myeloma
and efficiently eradicate myeloma cells (5). Current CARs have a domain that
recognizes a molecule on tumor cells, a transmembrane domain, a
domain of a co-stimulation molecule such as CD28 or 4-1BB, and a
cytoplasmic domain of CD3-ζ (6).
Since current CARs recognize a single target
molecule on tumor cells, the CAR-T cells also attack normal cells
expressing the antigen. For example, normal B cells in patients who
are treated with anti-CD19-CAR-T cells are attacked since the cells
express CD19. To protect patients from severe infections, those
patients need life-long supplemental immunoglobulin infusions,
which is an anti-infectious product of normal B cells (7). This is called an
'on-target/off-tumor effect'. This adverse effect is one of the
major obstacles to be overcome in this treatment. Improvement of
target-cell-specificity of CAR-T cell therapy could diminish this
'on-target/off-tumor effect', leading to less toxicity of this
therapy. Here, we show a novel regulatory CAR-T cell system with
higher target-cell-specificity by recognizing the expression
patterns of two distinct proteins on target cells.
Materials and methods
Cell lines
A human T-cell acute lymphoblastic leukemia cell
line, Jurkat, a human Burkitt's lymphoma cell line, Raji, and a
human chronic myelogenous leukemia cell line, K562 were purchased
from American Type Culture Collection (ATCC) and maintained in
RPMI-1640 media with 10% fetal bovine serum (FBS). The 293T cell
line and a human breast cancer cell line, SK-BR-3 were purchased
from ATCC and maintained in DMEM supplemented with 10% FBS. All of
the cells were maintained at 37°C with 5% CO2.
Reagents and antibodies
Recombinant human interleukin (IL)-2 was purchased
from R&D Systems. For manipulation of human primary T cells,
the Dynabeads Untouched Human T cell kit and Human T-Activator
CD3/CD28 were purchased from Thermo Fisher Scientific, Inc. For
western blot analysis, anti-RFP/mCherry rabbit polyclonal antibody
(PM005; 1:1,000 dilution) was purchased from Medical and Biological
Laboratories; anti-GFP/YFP rabbit polyclonal antibody (SC-8334;
1:5,000 dilution) and anti-syntaxin 4 mouse mAb (QQ-17; 1:1,000
dilution) were purchased from Santa Cruz Biotechnology, Inc.;
anti-β-actin mouse mAb (A1978; 1:10,000 dilution) was purchased
from Sigma Aldrich; Merck KGaA; anti-rabbit horseradish peroxidase
(HRP)-conjugated antibody (NA934; 1:5,000 dilution) and anti-mouse
HRP conjugated antibody (NA9310; 1:5,000 dilution) were purchased
from GE Healthcare. For flow cytometric analysis, PE anti-human
CD69 antibody (FN50; 1:20 dilution), PerCP/Cy5.5 anti-human HER2
antibody (24D2; 1/50 dilution), and FITC anti-human CD19 antibody
(4G7; 1/50 dilution) were purchased from BioLegend, Inc. Hoechst
33258 was purchased from Dojindo Molecular Technologies, Inc. HIV
protease (HIVPR) inhibitors, saquinavir and nelfinavir, were kindly
provided by Professor Yamaoka at the Department of Virology, TMDU
(Tokyo, Japan).
Preparation and expansion of human
primary T cells
Ten milliliters of peripheral blood collected from
healthy donors was diluted with 10 ml of phosphate-buffered saline
(PBS), and 10 ml of the diluted blood was applied onto 3 ml of
Lymphoprep (Abbott Diagnostics Technologies AS). After
centrifugation at 800 x g (at room temperature) for 20 min, white
blood cells were collected. Human primary T cells were purified
using Dynabeads Untouched Human T cell kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. CAR-T
cells were established as previously reported (8,9).
Briefly, 1.0×105 cells/ml of T cells were expanded in
RPMI-1640 media with 10% FBS, 30 U/ml IL-2, and CD3/CD28 beads (1:3
cell:bead ratio) for 3 days. Expanded T cells were transduced with
lentivirus. In the present study, we collected peripheral blood
from three healthy donors. Donor 1 was a 35-year-old male. His
peripheral blood was collected in November, 2020 and June, 2021.
Donor 2 was a 36-year-old male. His peripheral blood was collected
in December, 2020 and April, 2021. Donor 3 was a
25-year-old-female. Her peripheral blood was collected in January
and August, 2021. All the procedures including peripheral blood
collection, T-cell isolation and manipulation were performed at
TMDU, after informed consents were obtained. All the procedures
involving human cells were approved by the TMDU Ethics Committee
(M2019-294).
Vector construction
Regions encoding scFv domains were amplified from
plasmids, pHR_PGK_antiCD19_synNotch_TetRVP64 (Addgene plasmid
#79126), pHR_PGK_antiHer24D5-3_synNotch_Gal4VP64 (Addgene plasmid
#85422), and pHR_PGK_antiHer24D5-8_synNotch_Gal4VP64 (Addgene
plasmid #85425). The plasmids were gifted by Dr W.A. Lim (10). All the scFv regions were
subcloned and ligated into other plasmids. A plasmid encoding CD28,
pcDNA3.1-PS11-scFvFc-CD28-gp41(706-713) (Addgene plasmid #60606)
and a plasmid encoding HIVPR, pcDNA3/GFP-PR (Addgene plasmid
#20253) were gifted by Dr W.A. Marasco (11) and Dr N.P. Dantuma (12), respectively. To establish
constructs of regulatory CARs, sequences of the scFv fragments, the
CD28 transmembrane and co-stimulatory domains, the HIVPR, and the
fluorescence proteins were connected using PCR. A HIVPR recognition
peptide sequence (SFSFPQIT) was inserted as a cleavage site in the
constructs. A CD3-ζ fragment was amplified from pGEM-human TCR
zeta/2470 (Addgene plasmid #11507) gifted by Dr A.M. Weissman
(13) and inserted in the
constructs. A gag/pol vector, psPAX2 (Addgene plasmid #12260), and
a VZV envelope vector, pMD2.G (Addgene plasmid #12259), for
lentivirus packaging were gifted by Dr D. Trono. Schematic
descriptions of the constructs are shown in Fig. S1.
Transfection and lentiviral
infection
293T cells were transfected using Lipofectamine 3000
reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Twenty-four hours after transfection, the
cells were examined under a microscopy and/or harvested for western
blot analysis. To obtain SK-BR-3 cells with CD19,
pcDNA3-CD19-Hygro, was transfected into the cells using
Lipofectamine 3000 and selected in medium containing 100
μg/ml of hygromycin. To obtain Raji cells with HER2,
pcDNA3-HER2-Hygro was transduced using Nucleofector kit V (Lonza)
according to the manufacturer's protocols and selected in medium
containing 300 μg/ml of hygromycin. Jurkat cells with CARs
were obtained by transduction of pcDNA3-anti-CD19-Signal-CA
R-T2A-YFP using Nucleofector kit V (Lonza) and selected in medium
containing 1 mg/ml of G418.
For lentivirus packaging, vectors encoding CARs,
psPAX2, and pMD2.G were co-transfected into 293T cells using
Lipofectamine 3000 according to the manufacturer's protocols.
Briefly, one day before gene transduction, 2.0×106 of
293T cells were plated on 10-cm collagen-coated plates (Iwaki) in
10 ml of DMEM with 10% FBS. 293T cells were transduced with 10
μg of a vector encoding CARs, 7.5 μg of psPAX2 and
2.5 mg of pMD2.G. Twenty-four hours after the transfection,
supernatants containing lentivirus were collected and 10 ml of
fresh medium was added on the cells. Forty-eight hours after the
transfection, the second viral supernatants were collected. All the
supernatants were filtrated through 0.45-μm syringe filters
(Pall Life Sciences). Then, the viral particles were precipitated
using Lenti-X concentrator (Takara) according to the manufacturer's
protocols. For lentivirus infection, 2 ml of 10-time concentrated
lentiviral supernatants were applied onto 6-well plates coated with
Retronectin (Takara), and the virus particles were captured on the
plates by centrifugation at 1,080 x g (at 32°C) for 120 min. After
removal of the supernatant, 2.0×106 of Jurkat cells or
primary T cells were seeded onto the plates for infection. One day
after the infection, the cells were transferred to new plates for
further experiments.
Western blot analysis
Cells were lysed in a 40 μl of lysis buffer
containing 1% Triton X-100, 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1
mM EDTA, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl
fluoride, 10 μg/ml of aprotinin, and 10 μg/ml of
leupeptin. Following incubation (at 4°C for 15 min) and
centrifugation at 17,400 x g (at 4°C) for 15 min, supernatants were
mixed with equal volume of 2X Laemmli sample buffer and heated at
100°C for 5 min. Separation of membranous and cytosolic fractions
from samples was performed using Trident Membrane Protein
Extraction kit (Genetex) according to the manufacturer's protocols.
Ten microliters of the samples was electrophoresed in 12.5% PAGE
gel (Atto) and transferred onto Immnobilon-P membranes (Merck
Millipore). The membranes were blocked with 5% skim milk at room
temperature for 1 h. Primary antibodies were diluted with Tris-base
buffered saline with Tween (TBS-T) and incubated at 4°C for
overnight. Subsequently, the membranes were incubated with diluted
secondary antibodies at room temperature for 1 h. The transferred
proteins detected by antibodies were visualized by the Western
Lightning Plus-ECL chemiluminescence kit (PerkinElmer, Inc.)
according to the manufacturer's protocols. All the data shown are
representatives of results of at least three independent
experiments.
Microscopic examination
Cells (200,000) were seeded on 35-mm glass-bottom
plates (Matsunami) in 10 ml of DMEM with 10% FBS. One day after
cultivation, plasmids were transfected into the cells using
Lipofectamine 3000 according to the manufacturer's protocols. For
nuclear staining, Hoechst 33258 solution was added into the medium
at 1:500 dilution. The cells were examined with an all-in-one
fluorescence microscopy BZ-X800 (Keyence, Osaka, Japan;
magnification, ×600). Optical sectioning images (14) were captured and analyzed using a
BZ-X800 Analyzer (Keyence). All the data and images shown are
representatives of results of at least three independent
experiments.
Flow cytometric analysis
For flow cytometric analysis, 4.0×105
cells were stained with 50 μl of antibody solution diluted
with PBS with 3% FBS at 4°C for 30 min. The cells were washed with
1 ml of the buffer and resuspended in 500 μl of the buffer.
The cells were analyzed on a FACS Calibur flowcytometer (BD
Bioscience). The cells with YFP and/or mCherry were sorted by a
FACS Aria II (BD Bioscience). The data were analyzed using FlowJo
ver.8.8.7 software (BD Bioscience). All the data shown are
representatives of results of at least three independent
experiments.
Cytotoxicity assays
To assess the cytotoxicity of CAR-T cells, lactate
dehydrogenase (LDH) release assays were carried out using
Cytotoxicity LDH Assay kit-WST (Dojindo) according to the
manufacturer's protocols. CAR-T and target cells were mixed at
indicated effector-to-target (E:T) ratios and co-cultivated. Ten
thousand SK-BR-3 cells or 2.0×104 Raji or K562 cells
were used as target cells in 100 μl of medium in 96-well
plates. Twenty-four hours after co-cultivation, the supernatants
were harvested and levels of LDH released from the target cells
were measured using Multiskan FC (Thermo Fisher Scientific, Inc.).
Specific cytotoxicity (%) was calculated as follows: [(Experimental
release)-(Effector spontaneous release)-(Target spontaneous
release)]/[(Target maximum release)-(Target spontaneous release)]
×100. To measure maximum releases from target cells, the target
cells were lysed and levels of LDH released from the cells were
measured according to the manufacturer's protocols.
Statistical analysis
Differences in the datasets containing two groups
were analyzed with the Student's t-test. Differences in the
datasets containing more than three groups were analyzed with
one-way analysis of variance (ANOVA) with Dunnett's post-hoc test
or Tukey post-hoc test. P<0.05 was regarded as indicative of a
statistically significant difference. Statistical analyses were
performed using EZR software version 1.55 (15).
Results
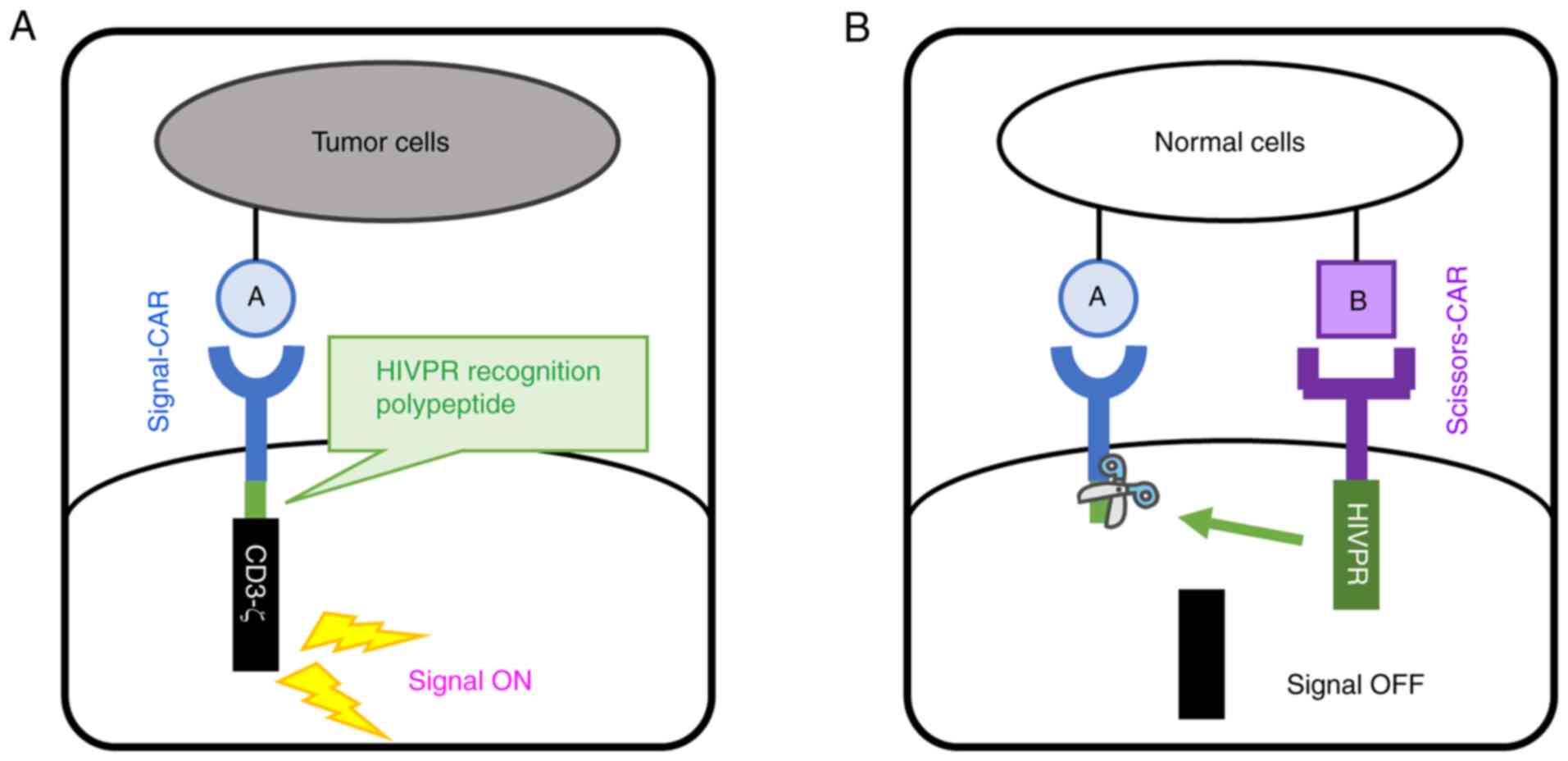
Concepts of a novel CAR system
recognizing two distincT cell-surface proteins on tumor/normal
cells
Current CARs expressed in human T cells are composed
of four elements including i) an extracellular domain recognizing a
cell-surface protein on tumor cells, ii) a trans-membrane
domain, iii) a cytoplasmic co-stimulatory domain, and iv) a
cytoplasmic CD3-ζ. To increase target-cell-specificity of CAR-T
cells, we designed two CARs which recognize two distincT
cell-surface proteins (Proteins A and B in Fig. 1). T cells expressing these two
CARs distinguish target cells depending on expression patterns of
these two proteins. To regulate a functional property of CARs, we
designed two CARs with two distinct cytoplasmic domains. One is CAR
with a protease domain of HIVPR in the cytoplasmic domain
(Scissors-CAR); the other has a HIVPR recognition peptide sequence
between the co-stimulatory domain and the CD3-ζ in the cytoplasmic
domain (Signal-CAR) as shown in Fig.
1. T cells expressing this Signal-CAR transduce the T-cell
activation signal (Fig. 1A). We
hypothesized that T cells expressing both Signal- and Scissors-CARs
were in contact with cells expressing both the proteins A and B,
leading to accumulation of these two types of CARs on the
contacting surface of T cells. Interaction between HIVPR and HIVPR
recognition peptide sequence leads protein cleavage of Signal-CAR
and CD3-ζ in Signal-CAR was released and spread in the cytoplasm.
Multimer formation of CD3-ζ under the plasma membrane is a trigger
for T-cell activation, thus, this Scissors-CAR-mediated protein
cleavage suppresses the CAR-T cell activation (Fig. 1B). To establish this methodology,
we designed and constructed vectors as described above, and
performed experiments to prove this principle.
Establishment of a novel regulatory CAR
construct: Scissors-CAR
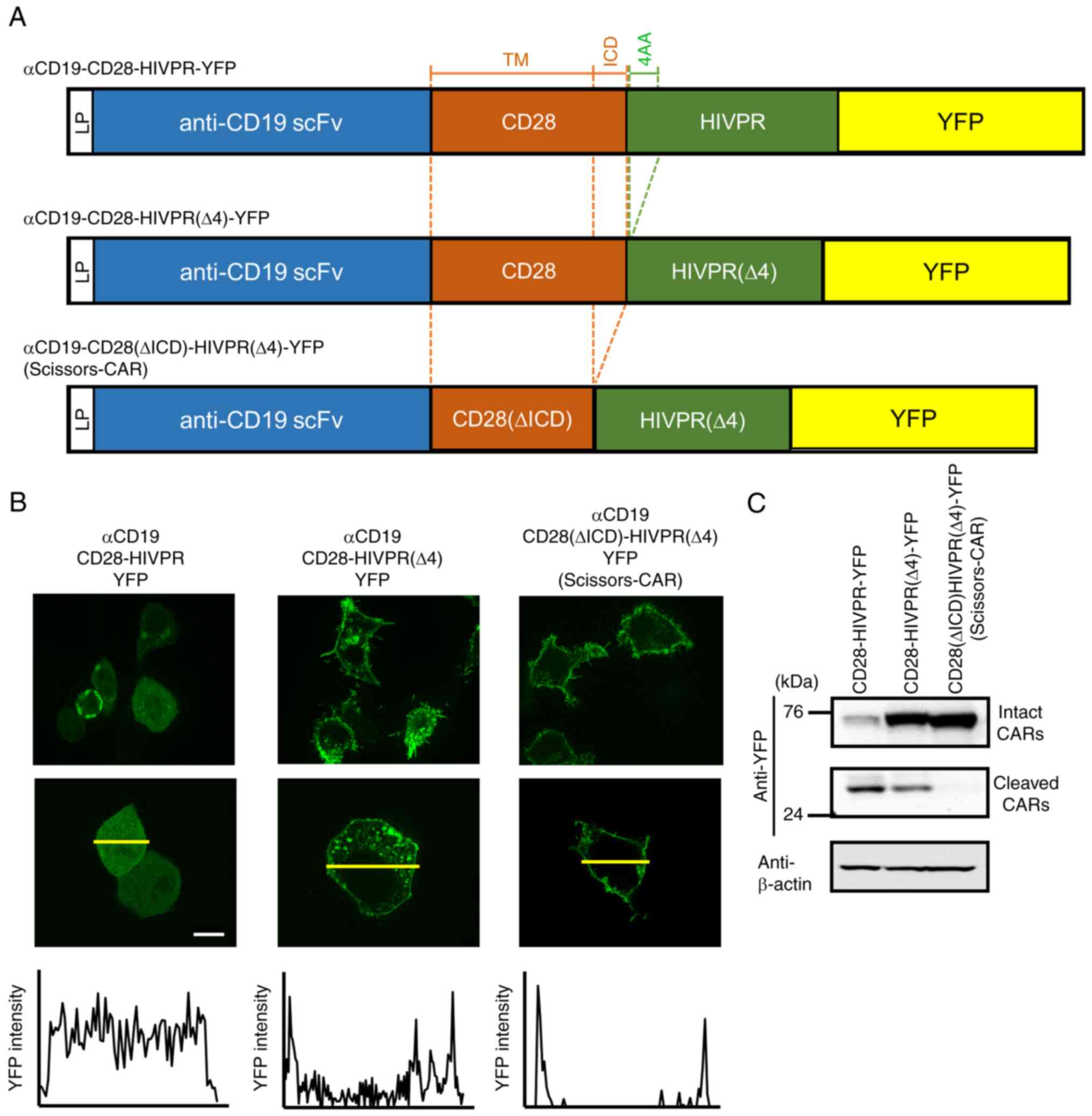
We constructed a vector with anti-CD19 scFv (FMC63),
CD28, and a protease domain of HIVPR. To examine localization of
this CAR, a YFP fluorescence protein was inserted at the C-terminus
(Fig. 2A, top) and fluorescence
optical sectioning imaging by a microscope was performed, which
allowed us to capture sliced images of viable cells (14). This αCD19-CD28-HIVPR-YFP showed
cytoplasmic distribution of YFP (Fig. 2B, left panel). Western blot
analysis using an anti-YFP antibody detected a band of 40 kDa
(Fig. 2C), not an expected size
of anti-CD19-CD28-HIVPR-YFP (76 kDa). These results suggested that
this CAR was auto-cleaved at the N-terminal of the HIVPR domain. We
speculated that this CAR had cleavage site(s) recognized by HIVPR
in its cytoplasmic region. One candidate site was four amino acids
sequence (PQIT) in HIVPR (12,16). We deleted this site and examined
localization of the CAR (Fig.
2A, middle and B, middle). Deletion of the four amino acids
(4AA) in HIVPR enhanced the membranous localization of the CAR
(Fig. 2B, middle) and partially
suppressed the auto-cleavage (Fig.
2C). Furthermore, we found another candidate site in the CD28
inner cellular domain (ICD) as shown in Fig. 2A that was predicted by ExPASy
PeptideCutter tool (https://web.expasy.org/peptide_cutter/) (17). Elimination of both the candidate
sequences of CD28 and HIVPR lead to the membranous localization of
the CAR (Fig. 2B, right) and
strongly suppressed the auto-cleavage (Fig. 2C). We named this construct
Scissors-CAR and was used as a regulatory CAR in further
experiments.
A protease-domain of HIVPR cleaves the
recognition sequence in CAR and induces translocation of the
cytoplasmic domain
To examine whether cleavage of CARs leads to
translocation of a cytoplasmic domain of CARs, we constructed a CAR
with anti-CD19 scFv (FMC63), CD28, and mCherry fluorescence protein
in the cytoplasmic domain (Fig.
3A). The HIVPR recognition peptide sequence was inserted
upstream of mCherry, and we named this construct
anti-CD19-mCherry-CAR (Fig. 3A).
This anti-CD19-mCherry-CAR showed membranous localization after
transfection into 293T cells (Fig.
3B). Since the results shown above suggested that the HIVPR-YFP
protein cleaved from anti-CD19-CD28-HIVPR-YFP was localized in the
cytoplasm (Fig. 2B), we expected
that the HIVPR-YFP in the cytoplasm would cleave the recognition
sequence in anti-CD19-mCherry-CAR. As expected, western blot
analysis using cytoplasmic and plasma membrane fractions of the
transfected cells showed that the mCherry-CAR was cleaved when
anti-CD19-CD28-HIVPR-YFP was co-expressed (Fig. 3C). Furthermore, HIVPR inhibitors,
saquinavir or nelfinavir, suppressed the cleavage in a
dose-dependent manner (Fig. 3D),
suggesting that the cleavage was induced by the HIVPR-YFP. Cells
transduced both anti-CD19-mCherry-CAR and anti-CD19-CD28-HIVPR-YFP
showed cytoplasmic distribution of the mCherry signal (Fig. 3E). These results suggested that
the HIVPR-YFP in the cytoplasm cleaved mCherry-CAR and induced
translocation of the cytoplasmic domain of the CAR. By contrast,
cells expressing both anti-CD19-mCherry-CAR and
anti-CD19-Scissors-CAR showed membranous localization of mCherry
(Fig. 3F). This result suggested
that Scissors-CAR on the membrane was enzymatically inactive
without target cells.
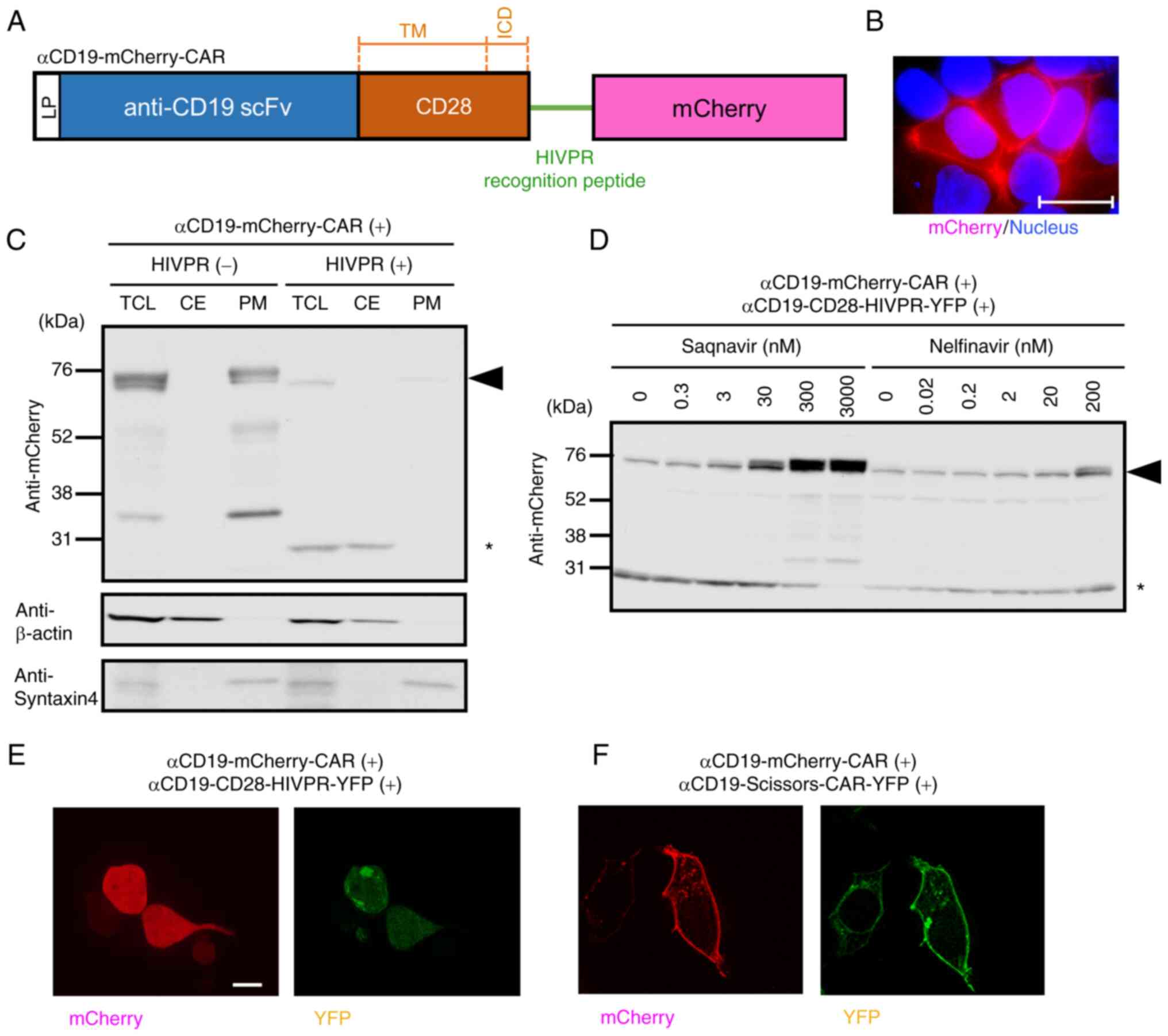 | Figure 3Cleavage of anti-CD19-mCherry-CAR by
the HIVPR protease induces translocation of mCherry from the
membrane to the cytoplasm. (A) A structure of
anti-CD19-mCherry-CAR. The construct has a leader peptide (LP)
sequence (white box), an anti-CD19 scFv (FMC63) domain (blue box),
a CD28 transmembrane (TM) and inner-cellular (ICD) domains (orange
box), the HIVPR recognition peptide sequence (green line) and a
mCherry protein (pink box). (B) Localization of
anti-CD19-mCherry-CAR. The anti-CD19-mCherry-CAR was expressed in
293T cells. Twenty-four hours after gene transduction,
localizations of the mCherry-CAR (red color) were examined. Nuclei
were stained with Hoechst 33258 (blue color). Scale bar, 20
μm. (C) Western blot analysis of fractionated cell lysates
of 293T cells expressing the indicated CAR constructs.
Anti-CD19-CD28-HIVPR-YFP (Fig.
1) was used to express cytoplasmic HIVPR protease (indicated as
HIVPR). Twenty-four hours after gene transduction, the cells were
lysed. Plasma membrane fractions and cytosolic proteins were
separated, and analyzed. Syntaxin4 protein was used as an internal
control of plasma membrane fractions. β-actin was used as an
internal control of cytosolic proteins. An arrowhead on the right
indicates the position of the full-length anti-CD19-mCherry-CAR (76
kDa); an asterisk on the right indicates the position of the
cleaved mCherry (28 kDa). (D) Western blot analysis of cells
treated with HIVPR inhibitors. Both anti-CD19-mCherry-CAR and
anti-CD19-CD28-HIVPR-YFP were transfected into 293T cells.
Twenty-four hours after gene transduction, the cells were treated
with a HIVPR protease inhibitor, saquinavir or nelfinavir, at
indicated concentrations. Twenty-four hours after the treatment,
CARs and mCherry proteins were detected with an anti-mCherry
antibody. A black arrowhead indicates the intact
anti-CD19-mCherry-CARs (76 kDa). A black asterisk indicates the
cleaved mCherry proteins (28 kDa). (E) Fluorescence microscopic
examination of 293T cells with both anti-CD19-mCherry-CAR and
anti-CD19-CD28-HIVPR-YFP. (F) Fluorescence microscopic examination
of 293T cells with anti-CD19-mCherry-CAR and
anti-CD19-Scissors-CAR-YFP. (E and F) Twenty-four hours after gene
transduction of the indicated constructs, localizations of the
mCherry (red color) and YFP (green color) were examined. Scale bar,
20 μm. TCL, total cell lysate; CE, cytosolic extract; PM,
plasma membrane fraction CAR, chimeric antigen receptor. |
Scissors-CAR cleaves
anti-CD19-mCherry-CAR, leading to translocation of mCherry when
target cells express two distinct proteins
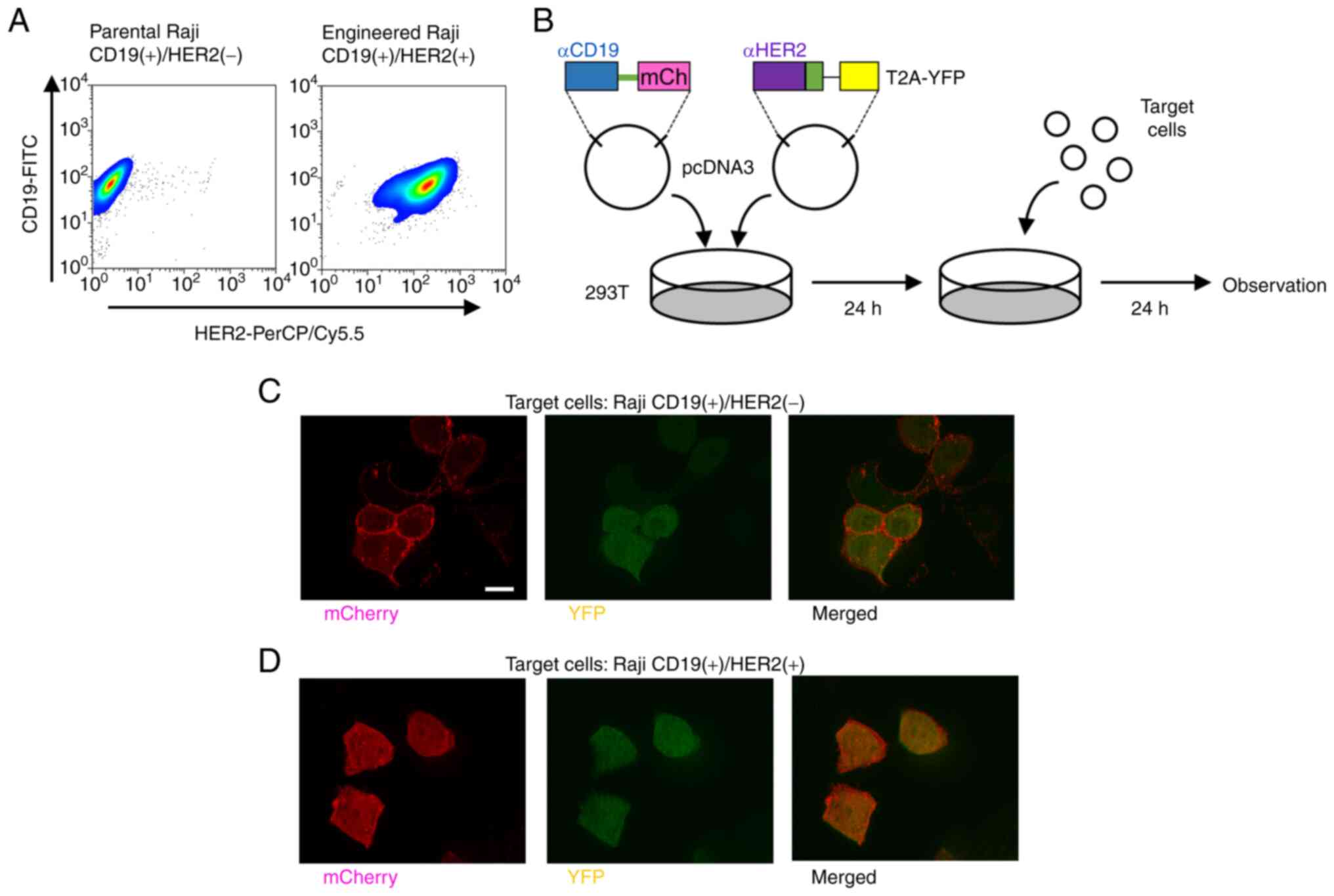
To assess our regulatory CAR system using
Scissors-CAR, we used CD19 and HER2 cell-surface target proteins.
We established Raji cells stably expressing HER2 as target cells.
The parental Raji cells expressed only CD19, not HER2; the
engineered Raji cells expressed both CD19 and HER2 (Fig. 4A). To evaluate
target-cell-dependent cleavage of CAR, 293T cells expressing both
anti-CD19-mCherry-CAR and anti-HER2-Scissors-CAR were co-cultivated
with Raji cells (Fig. 4B). The
293T cells co-cultivated with Raji cells expressing only CD19, the
mCherry CAR signal showed membranous localization (Fig. 4C). By contrast, the 293T cells
co-cultivated with the engineered Raji cells expressing both CD19
and HER2, the mCherry signal was translocated from the membrane to
the cytoplasm (Fig. 4D). Not
only microscopic analysis, but also western blot analysis indicated
that the 293T cells transduced with anti-CD19-mCherry-CAR and
anti-HER2-Scissors-CAR showed mCherry-CAR cleavage, which was
induced by engineered Raji cells expressing both CD19 and HER2
(Fig. S2). Furthermore, the
cleavage was saturated at 24 h after initiation of co-cultivation
as shown in Figs. 4 and S2. On the other hand, in the absence
of target cells, minimal levels of cleaved mCherry-CAR were
detected (left four lanes in each panel in Fig. S2).
The data also showed that ligand-independent
cleavage of mCherry-CAR was associated with the amounts of
transfected Scissors-CAR (left four lanes of each panel in Fig. S2). The experiments showed that
higher numbers of target cells increased the cleavage of
mCherry-CAR (right four lanes of each panel in Fig. S2). However, the higher numbers
of target cells also increased ligand-independent cleavage (left
four lanes of the bottom panel in Fig. S2). These results suggested that
our CAR system using Scissors-CAR cleaved the other CAR when target
cells expressed both the cell-surface target proteins, CD19 and
HER2.
Activation of the novel CAR-T cells is
controlled depending on patterns of proteins expressed on target
cells
To evaluate functional properties of our system, we
constructed the anti-CD19-Signal-CAR with anti-CD19 scFv (FMC63),
CD28, HIVPR recognition peptide sequence, and CD3-ζ. As a marker, a
T2A-YFP fluorescence protein cassette was connected in the
C-terminus (Fig. 5A). Since T2A
site acted as a ribosomal skipping motif (18), this construct expressed both the
full-length CAR protein and YFP protein. Therefore, cells
expressing the Signal-CAR showed the YFP signal in the cytoplasm.
To assess activity of the Signal-CAR, Jurkat cells stably
expressing anti-CD19-Signal-CAR were established (Fig. S3A) and activation of the cells
after co-cultivation with target cells was analyzed by measuring
levels of CD69, a T-cell activation marker, on the cells (Fig. 5B and C). The Jurkat cells were
not activated by co-cultivation with K562 cells, which lacked CD19
(Fig. 5C, left). By contrast,
the Jurkat cells were activated by co-cultivation with the parental
Raji cells expressing CD19 since levels of CD69 on the Jurkat cells
were enhanced (Fig. 5C, right
panels). Furthermore, to assess the effects of Scissors-CAR,
anti-HER2-Scissors-CAR was transduced into the Jurkat cells stably
expressing anti-CD19-Signal-CAR. The Scissors-CAR, in which mCherry
fluorescence protein was connected via the T2A sequence as a
marker, was constructed (Fig.
5A, lower). Jurkat cells expressing both the CARs (Fig. S3B) were co-cultivated with
target cells (Fig. 5D). When the
Jurkat cells were co-cultivated with the parental Raji cells
expressing CD19 alone, expression of CD69 on the CAR-Jurkat cells
was not affected by the expression of Scissors-CAR (Fig. 5E, left). By contrast, when
co-cultivated with the engineered Raji cells expressing both CD19
and HER2, levels of CD69 on the CAR-Jurkat cells were significantly
diminished by expression of Scissors-CAR (Fig. 5E, right; P=0.019, Student's
t-test, and Fig. S3C).
Furthermore, expression of CD69 on the Jurkat cells expressing both
the CARs was diminished not only by the engineered Raji cells
(suspension cells) expressing HER2 but also by the engineered
SK-BR-3 cells (adhesion cells) expressing both the target proteins
(Fig. S3D and E).
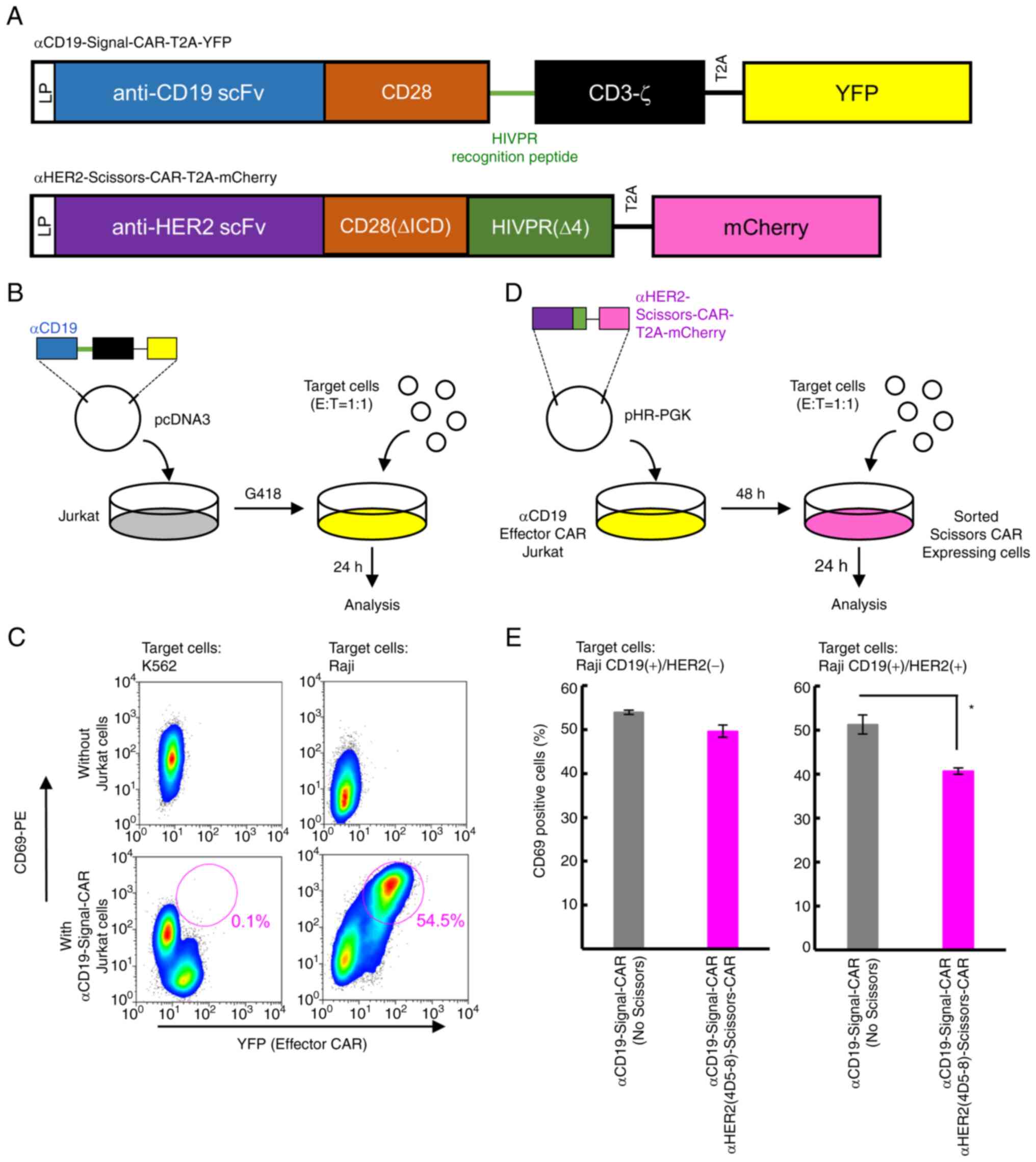 | Figure 5Anti-HER2-Scissors-CAR attenuates
activation of Jurkat cells driven by anti-CD19-Signal-CAR when the
cells are co-cultivated with target cells expressing both CD19 and
HER2. (A) Structures of anti-CD19-Signal-CAR-T2A-YFP (upper) and
anti-HER2-Scissors-CAR-T2A-mCherry (lower). These constructs have a
leader peptide (LP) sequence, an anti-CD19 scFv (FMC63) or
anti-HER2 scFv, a CD28 domain, a HIVPR recognition peptide
sequence, and a CD3-ζ domain or HIVPR. YFP or mCherry protein was
connected with the T2A sequence. Blue box, scFv domain against
CD19; orange boxes, CD28 trans-membrane and co-stimulatory domains
(upper) and CD28 trans-membrane domain lacking ICD (lower); a black
box, CD3-ζ domain; a yellow box, YFP, a pink box, mCherry. A green
horizontal line indicates the HIVPR recognition peptide sequence.
Black horizontal lines indicate the T2A sequences. (B) A schema of
co-cultivation assays using Jurkat cells stably expressing
anti-CD19-Signal-CAR with target cells. The
anti-CD19-Signal-CAR-T2A-YFP vector was transduced into Jurkat
cells. The cells stably expressing the CAR were co-cultivated with
target cells. Blue box, scFv domain against CD19; black box, CD3-ζ
domain; yellow box, YFP. CD3-ζ and YFP are connected with the T2A
sequence. Gray in the dish, culture medium containing parental
Jurkat cells; yellow in the dish, culture media containing Jurkat
cells expressing anti-CD19-Signal-CAR-T2A-YFP. (C) Activation of
Jurkat cells with anti-CD19-Signal-CAR was examined by flow
cytometry. Expression of YFP (x-axis) and CD69 (y-axis) was
examined. Upper two panels show the data of target cells alone.
Lower two panels show the data of Jurkat cells expressing
anti-CD19-Signal-CAR that were co-cultivated with K562 cells (left
panel) and Raji cells (right panel). Red circles indicate
CD69-positive cells (percentages of positive cells are shown under
the circles). (D) A schema of co-cultivation assays using Jurkat
cells stably expressing both anti-CD19-Signal-CAR and
anti-HER2-Scissors-CAR. Anti-HER2-Scissors-CAR-T2A-mCherry was
introduced into Jurkat cells stably expressing
anti-CD19-Signal-CAR-T2A-YFP. Forty-eight hours after infection,
Jurkat cells expressing both YFP and mCherry were sorted and
co-cultivated with target cells. Twenty-four hours after the
co-cultivation, flowcytometric analysis was performed. Purple box,
scFv domain against HER2; green box, CD28(∆ICD) and HIVPR(∆4)
domains; pink box, mCherry. HIVPR(∆4) domain and mCherry are
connected with the T2A sequences. Yellow in the dish, culture
medium containing Jurkat cells expressing
anti-CD19-Signal-CAR-T2A-YFP; pink in the dish, culture media
containing Jurkat cells expressing both
anti-CD19-Signal-CAR-T2A-YFP and
anti-HER2-Scissros-CAR-T2A-mCherry. (E) Levels of CD69 expression
on Jurkat cells. Percentages of CD69-positive Jurkat cells are
plotted. A left panel indicates results of co-cultivation with Raji
cells expressing only CD19. A right panel indicates results of
co-cultivation with the engineered Raji cells expressing both CD19
and HER2. Gray bars indicate Jurkat cells with only
anti-CD19-Signal-CAR. Pink bars indicate Jurkat cells with both the
Signal- and Scissors-CARs. Mean values ± SD of three independent
experiments are plotted. Data were analyzed using the Student's
t-test and values showing significant difference are indicated as
*P<0.05. CAR, chimeric antigen receptor. |
Scissors-CAR attenuates the cytotoxicity
of primary CAR-T cells when the cells are co-cultivated with target
cells expressing two distincT cell-surface proteins
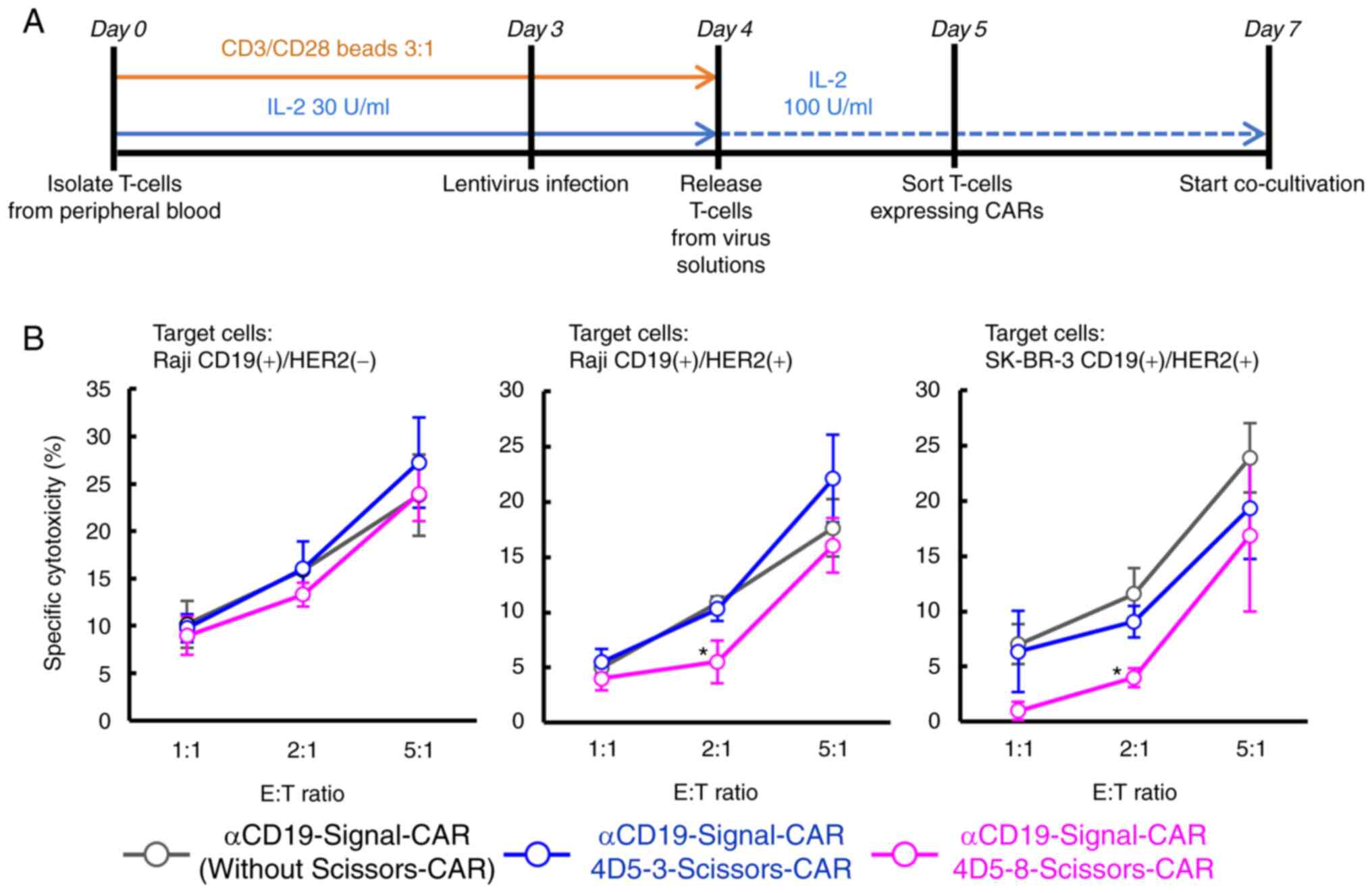
To assess the cytotoxicity of CAR-T cells with
Signal- and Scissors-CARs, the CARs were transduced into primary T
cells, which were collected from healthy donors, using lentivirus.
The manipulated T cells were co-cultivated with target cells
(Fig. 6A). Cytotoxicity of the
CAR-T cells against each the targeT cell was evaluated by measuring
levels of lactate dehydrogenase (LDH) released from the target
cells (Fig. 6B). The CAR-T cells
with anti-CD19-Signal-CAR showed cytotoxicity against Raji cells,
which showed statistical significance, but not K562 cells (Fig. S4A and Table SI). We constructed two types of
Scissors-CARs using anti-HER2 (4D5-8) scFv with a high affinity to
HER2 or anti-HER2 (4D5-3) scFv with a low affinity to HER2
(19). Lentiviruses encoding
anti-CD19-Signal-CAR, in which YFP was connected via T2A sequence,
and/or anti-HER2-Scissors-CAR, in which mCherry was connected via
T2A sequence, were infected into primary T cells (Fig. S4B). CAR-T cells with both the
CARs were sorted and co-cultivated with target cells. The CAR-T
cells with anti-HER2-Scissors-CAR showed comparable cytotoxicity
against the parental Raji cells only expressing CD19 to CAR-T cells
without the Scissors-CAR (Fig.
6B, left). The CAR-T cells with the high affinity
anti-HER2(4D5-8)-Scissors-CAR showed significant attenuation of
cytotoxicity against the engineered Raji cells expressing both CD19
and HER2 (Fig. 6B, middle;
53.0±0.20% in 2:1 mixture, P=0.039, one-way ANOVA with Dunnett's
post-hoc test, control group=anti-CD19-Signal-CAR without
Scissors-CAR). Furthermore, they showed attenuated cytotoxicity
against the engineered SK-BR-3 cells expressing both CD19 and HER2
as well (Fig. 6B, right;
43.2±0.17% in 2:1 mixture, P=0.039, one-way ANOVA with Dunnett's
post-hoc test, control group=anti-CD19-Signal-CAR without
Scissors-CAR).
Discussion
High target-cell-specificity of chimeric antigen
receptor (CAR)-T cells may improve the safety of CAR-T cell
therapy, leading to expansion of treatable diseases by this
immunotherapy. Our novel CAR-T cell system showed different
cytotoxicity toward target cells depending on expression patterns
of proteins on the target cells.
Previously, CAR systems recognizing multi-antigens
have been reported including i) complement CARs, in which a
cytoplasmic domain of CAR was divided into two components on two
distinct CARs (20), ii)
two-step CARs, in which the first CAR of a synthesized Notch
receptor recognizes the first antigen, inducing expression of the
second CAR recognizing the second antigen (21) and iii) inhibitory CARs with
immuno-checkpoint proteins, PD-1 or CTLA-4, controlling activation
of CAR-T cells (22). In the
present study, we established the fourth strategy of CARs
recognizing two distincT cell-surface proteins on target cells
using a protease domain of HIVPR.
HIVPR is an exogenous protease derived from HIV that
has been fully characterized. This enzyme has high specificity for
the recognition peptide sequence. In addition, this enzyme becomes
active only when it forms a homo-dimer (15,23), which probably kept Scissors-CAR
on the membrane inactive without contacting target cells. By
contrast, HIVPR in the cytoplasm was probably automatically
dimerized and cleaved the recognition peptide sequence.
In the present study, as a proof of principle, we
used engineered cells expressing both CD19 and HER2 for our
experiments. We successfully demonstrated that T-cell activation
was controlled by patterns of protein-expression on target cells in
our novel CAR-T cell system. Our system allows cells expressing two
target proteins to escape from attacks by the CAR-T cells while
tumor cells expressing only one protein fail to escape. In the
treatment of B-cell malignancies, small fractions of normal B cells
escaping from attacks by CAR-T cells would produce sufficient
number of immunoglobulin. Therefore, partial attenuation of T-cell
activation by our system would improve the safety of CAR-T cell
therapies for B-cell malignancies.
We need to confirm that our system allows CAR-T
cells to distinguish tumor cells from normal cells in vivo
using immunocompromised animals before initiation of clinical
trials. However, our current in vitro data suggest that it
is too early to perform in vivo experiments since this study
still has limitations to be overcome.
One is to define two distinct surface molecules
expressed on normal cells while primary tumor cells only express
one of the two proteins to apply this method to clinical practice.
Since the herpes virus entry mediator (HVEM) protein has been
reported to be expressed on normal B cells, but not on B-cell
malignancies (24), this protein
is a good candidate as a target protein for Scissors-CAR in B-cell
malignancies. Since CD19 is expressed on both normal and neoplastic
B cells, CD19 and HVEM could be good candidate surface molecules
for development of this novel CAR-T therapy targeting B-cell
malignancies.
The second is improvement of the suppressive effect
of Scissors-CAR. Our study demonstrated that a higher amount of
Scissors-CAR led to more efficient cleavage of Signal-CAR. However,
the high levels of Scissors-CAR caused ligand-independent cleavage
of Signal-CAR. Higher expression of Scissors-CAR may not be an
optimal way to improve the suppressive effect. Therefore,
improvement of binding affinity between Scissors-CAR and its ligand
may increase the suppressive effect of Scissors-CAR.
Another way to increase the suppressive effect could
be enhancement of proteolytic activity of HIVPR or the HIVPR
recognition poly-peptide sequence used in this system. Amino-acid
replacements in the protease domain and/or the recognition sequence
may increase cleavage efficiency, leading to more potent
suppression of Signal-CAR activity. Nevertheless, more
modifications would be needed to improve the quality of our
system.
Since the present study aimed to develop a novel
system regulating CAR-signal based on the expression patterns of
surface proteins on tumor/normal cells, we focused on the
functional analysis of T cells such as CD69 expression or
cytotoxicity. Therefore, we have not fully optimized/characterized
several elements, including scFv binding affinity, cleavage
efficiency and kinetics of the protease and its recognition
sequence. Since our mCherry-CAR system is a beneficial tool with
which to evaluate Scissors-CAR activity, more detailed analysis
using this system could improve the quality of this novel system.
Because of the COVID-19 pandemic, our research was restricted and
several experiments we designed were not allowed to be performed in
our institute. We hope that we will be able to fully
optimize/characterize this system after this pandemic.
Currently, only several types of malignancies are
effectively treated by CAR-T cell therapy. A number of clinicians
and researchers are searching good targeT cell-surface proteins for
CAR-T cell therapy. However, such tumor-specific cell-surface
proteins are very rare and CAR-T cells unexpectedly attack
important normal cells, causing severe adverse events in clinical
trials (25,26). Our system could help such
clinicians and researchers to define good candidate target proteins
for CAR-T cell therapies since only the proteins differentially
expressed between tumor and normal cells need to be identified.
Therefore, our system may expand the number of treatable diseases
by CAR-T cell therapies.
In summary, our novel CAR-T cell system using a
protease and the protease recognition peptide sequence allowed
CAR-T cells to become active depending on the expression patterns
of cell-surface proteins on target cells in vitro. This is
one of the first steps to improve target-cell-specificity of this
immunotherapy.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SA, SY, DW, YU, KO, AN, OM and NK designed the
experiments. SA and HL performed the experiments. SA and NK
confirmed the authenticity of all the raw data and wrote the paper.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
These experiments were reviewed and approved by the
Tokyo Medical and Dental University (TMDU) Ethics Committee
(M2019-294).
Patient consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing
interests.
Acknowledgements
We thank Ms. Okada and Ms. Liu Meiou for the
technical assistance. We also thank Mr. Sasaki and Mr. Ishitani for
assistance with the cell sorting.
Funding
This research was supported by Nagao-Takeshi Research Grant (to
YU), TMDU Young Investigator Research Grant (to YU), Takeda
Research Grant (to KO), and Bristol-Myers-Squibb Research Grant (to
NK).
References
|
1
|
Maude SL, Laetsch TW, Buechner J, Rives S,
Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers
GD, et al: Tisagenlecleucel in children and young adults with
B-Cell lymphoblastic leukemia. N Engl J Med. 378:439–448. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Park JH, Rivière I, Gonen M, Wang X,
Sénéchal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, et
al: Long-term follow-up of CD19 CAR therapy in acute lymphoblastic
leukemia. N Engl J Med. 378:449–459. 2018. View Article : Google Scholar
|
|
3
|
Cappell KM, Sherry RM, Yang JC, Goff SL,
Vanasse DA, McIntyre L, Rosenberg SA and Kochenderfer JN: Long-term
follow-up of Anti-CD19 chimeric antigen receptor T-Cell therapy. J
Clin Oncol. 38:3805–3815. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kochenderfer JN, Dudley ME, Kassim SH,
Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ,
Hughes MS, Sherry RM, et al: Chemotherapy-refractory diffuse large
B-cell lymphoma and indolent B-cell malignancies can be effectively
treated with autologous T cells expressing an anti-CD19 chimeric
antigen receptor. J Clin Oncol. 33:540–549. 2015. View Article : Google Scholar :
|
|
5
|
Raje N, Berdeja J, Lin Y, Siegel D,
Jagannath S, Madduri D, Liedtke M, Rosenblatt J, Maus MV, Turka A,
et al: Anti-BCMA CAR T-Cell therapy bb2121 in relapsed or
refractory multiple myeloma. N Engl J Med. 380:1726–1737. 2019.
View Article : Google Scholar
|
|
6
|
Sterner RC and Sterner RM: CAR-T cell
therapy: Current limitations and potential strategies. Blood Cancer
J. 11:692021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kochenderfer JN, Dudley ME, Feldman SA,
Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes
MS, Sherry RM, et al: B-cell depletion and remissions of malignancy
along with cytokine-associated toxicity in a clinical trial of
anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood.
119:2709–2720. 2012. View Article : Google Scholar
|
|
8
|
Long AH, Haso WM, Shern JF, Wanhainen KM,
Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara
VR, et al: 4-1BB costimulation ameliorates T cell exhaustion
induced by tonic signaling of chimeric antigen receptors. Nat Med.
21:581–590. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lynn RC, Weber EW, Sotillo E, Gennert D,
Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, et al: c-Jun
overexpression in CAR T cells induces exhaustion resistance.
Nature. 576:293–300. 2019. View Article : Google Scholar
|
|
10
|
Roybal KT, Williams JZ, Morsut L, Rupp LJ,
Kolinko I, Choe JH, Walker WJ, McNally KA and Lim WA: Engineering T
cells with customized therapeutic response programs using synthetic
notch receptors. Cell. 167:419–432.e416. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Taube R, Zhu Q, Xu C, Diaz-Griffero F, Sui
J, Kamau E, Dwyer M, Aird D and Marasco WA: Lentivirus display:
Stable expression of human antibodies on the surface of human cells
and virus particles. PLoS One. 3:e31812008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lindsten K, Uhlíková T, Konvalinka J,
Masucci MG and Dantuma NP: Cell-based fluorescence assay for human
immunodeficiency virus type 1 protease activity. Antimicrob Agents
Chemother. 45:2616–2622. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weissman AM, Hou D, Orloff DG, Modi WS,
Seuanez H, O'Brien SJ and Klausner RD: Molecular cloning and
chromosomal localization of the human T-cell receptor zeta chain:
Distinction from the molecular CD3 complex. Proc Natl Acad Sci USA.
85:9709–9713. 1988. View Article : Google Scholar
|
|
14
|
Conchello JA and Lichtman JW: Optical
sectioning microscopy. Nat Methods. 2:920–931. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanda Y: Investigation of the freely
available easy-to-use software 'EZR' for medical statistics. Bone
Marrow Transplant. 48:452–458. 2013. View Article : Google Scholar
|
|
16
|
Navia MA and McKeever BM: A role for the
aspartyl protease from the human immunodeficiency virus type 1
(HIV-1) in the orchestration of virus assembly. Ann NY Acad Sci.
616:73–85. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wilkins MR, Gasteiger E, Bairoch A,
Sanchez JC, Williams KL, Appel RD and Hochstrasser DF: Protein
identification and analysis tools in the ExPASy server. Methods Mol
Biol. 112:531–552. 1999.PubMed/NCBI
|
|
18
|
Liu Z, Chen O, Wall JB, Zheng M, Zhou Y,
Wang L, Vaseghi HR, Qian L and Liu J: Systematic comparison of 2A
peptides for cloning multi-genes in a polycistronic vector. Sci
Rep. 7:21932017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Carter P, Presta L, Gorman CM, Ridgway JB,
Henner D, Wong WL, Rowland AM, Kotts C, Carver ME and Shepard HM:
Humanization of an anti-p185HER2 antibody for human cancer therapy.
Proc Natl Acad Sci USA. 89:4285–4289. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cho JH, Collins JJ and Wong WW: Universal
chimeric antigen receptors for multiplexed and logical control of T
cell responses. Cell. 173:1426–1438.e1411. 2018. View Article : Google Scholar :
|
|
21
|
Choe JH, Watchmaker PB, Simic MS, Gilbert
RD, Li AW, Krasnow NA, Downey KM, Yu W, Carrera DA, Celli A, et al:
SynNotch-CAR T cells overcome challenges of specificity,
heterogeneity, and persistence in treating glioblastoma. Sci Transl
Med. 13:eabe73782021. View Article : Google Scholar :
|
|
22
|
Fedorov VD, Themeli M and Sadelain M:
PD-1- and CTLA-4-based inhibitory chimeric antigen receptors
(iCARs) divert off-target immunotherapy responses. Sci Transl Med.
5:215ra1722013. View Article : Google Scholar
|
|
23
|
Mager PP: The active site of HIV-1
protease. Med Res Rev. 21:348–353. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boice M, Salloum D, Mourcin F, Sanghvi V,
Amin R, Oricchio E, Jiang M, Mottok A, Denis-Lagache N, Ciriello G,
et al: Loss of the HVEM tumor suppressor in lymphoma and
restoration by modified CAR-T cells. Cell. 167:405–418.e413. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lamers CH, Klaver Y, Gratama JW, Sleijfer
S and Debets R: Treatment of metastatic renal cell carcinoma (mRCC)
with CAIX CAR-engineered T-cells-a completed study overview.
Biochem Soc Trans. 44:951–959. 2016. View Article : Google Scholar
|
|
26
|
Morgan RA, Yang JC, Kitano M, Dudley ME,
Laurencot CM and Rosenberg SA: Case report of a serious adverse
event following the administration of T cells transduced with a
chimeric antigen receptor recognizing ERBB2. Mol Ther. 18:843–851.
2010. View Article : Google Scholar : PubMed/NCBI
|