Introduction
The receptor activator of nuclear factor-κB ligand
(RANKL, also known as TNFSF11) is an essential cytokine that
regulates osteoclast differentiation and function (1). RANKL-mediated regulation of
osteoclast proliferation, differentiation and function dictate the
degree of skeletal remodeling, a process that maintains calcium
homeostasis and removes accumulated aged or weakened bone (2). Several pharmaceutical agents are
used for treating osteoporosis, including bone antiresorptive
agents (such as bisphosphonates, estrogen and denosumab) and drugs
that stimulate bone formation (such as parathyroid hormone)
(3-5). However, currently available drugs
that promote bone formation either have side effects; for example,
they can increase the risk of breast cancer and cardiovascular
disease, or do not improve bone quality sufficiently to reduce
fracture susceptibility (6).
Therefore, the development of agents that minimize bone resorption
may be beneficial for treating osteoporosis. Denosumab is a human
monoclonal antibody against RANKL that blocks the binding of RANKL
to its receptor RANK (also known as TNFRSF11A), thereby inhibiting
osteoclast differentiation and activity, leading to suppression of
bone resorption in osteoporosis and other bone-related disorders
(7). The United States Food and
Drug Administration-approved indications for denosumab include the
prevention of skeletal-related events (e.g., bone pain and
fractures) secondary to multiple myeloma or bone metastases from
solid tumors, giant cell tumors of the bone, hypercalcemia related
to malignancy, osteoporosis in postmenopausal women and men at high
risk of fracture, glucocorticoid-induced osteoporosis and bone loss
(8-11). However, its side effects include
hypocalcemia, serious infections, skin reactions, inhibition of
bone turnover and jaw necrosis (12,13). A recently reported side effect of
denosumab is rebound resorption, which occurs when denosumab
treatment is discontinued, especially in patients on long-term
treatment (14).
The leucine-rich repeat-containing G protein-coupled
receptor 4 (LGR4, also known as GPR48) is another potential target
for inhibiting bone resorption, as activation of this receptor
triggers a signaling pathway that inhibits RANK-RANKL signaling
during osteoclastogenesis (15).
Previous studies have indicated that LGR4 competes with RANK for
binding to RANKL and suppresses canonical RANK signaling during
osteoclast differentiation (15,16). LGR4 belongs to the LGR family of
receptors; two other LGR family members, the thyroid-stimulating
hormone receptor and follicle-stimulating hormone receptor, also
regulate osteoclast differentiation and resorption (17,18). The binding of RANKL to LGR4
activates the Gαq-mediated glycogen synthase kinase-3β (GSK-3β)
signaling pathway. When activated by RANKL, this pathway is crucial
for osteoclast differentiation and the subsequent suppression of
the expression and activity of nuclear factor of activated T cells,
cytoplasmic, calcineurin-dependent 1 (NFATc1) during osteoclast
development (15,16,19). In addition, RANKL-RANK-AKT-NFATc1
signaling may directly induce LGR4 expression. Notably, LGR4 is
upregulated in severe pathological bone environments, such as
osteoporosis, suggesting that therapies that target LGR4 would be
ideal for rebalancing bone remodeling (20).
In our previous studies, mutant RANKL (MT RANKL),
based on the wild-type RANKL (WT RANKL) sequence, was developed,
and whether it bound to LGR4 and acted as an agonist was
investigated. In in vitro and in vivo osteoporosis
models, MT RANKL inhibited osteoclast differentiation and
production, suggesting that crosstalk exists between RANKL, RANK,
and LGR4 signaling (21,22). However, it is currently unknown
whether MT RANKL triggers LGR4 downstream signaling pathways, such
as GSK-3β and AKT.
Therefore, the present study aimed to demonstrate
that MT RANKL binds to LGR4, instead of RANK, and that via this
ligand, LGR4 negatively regulates the RANK signaling cascade during
RANKL-induced osteoclast differentiation and bone remodeling. In
addition, by clarifying the effect and mechanism of action of MT
RANKL-LGR4 signaling on osteoclast differentiation and bone
resorption, the study aimed to determine whether an agonist that
binds LGR4 could be considered a pharmacological approach for
treating osteoporosis.
Materials and methods
Chemicals and reagents
Unless otherwise indicated, all of the chemical
reagents used in the present study were purchased from Millipore
Sigma, and cell culture medium was purchased from Thermo Fisher
Scientific, Inc.
Generation and purification of WT and MT
RANKL, RANK and LGR4
WT RANKL and MT RANKL were generated as described
previously (23). The genes
encoding RANK and LGR4 were synthesized and codon-optimized for
Escherichia coli expression by GeneArt Gene Synthesis
(Thermo Fisher Scientific, Inc.), and then subcloned into the
pETM-13 vector (EMBL) between the NcoI and XhoI
restriction sites. This plasmid enables a histidine (His) tag to be
placed at the C-terminus of the mini-protein. The recombinant
protein was successfully overexpressed in E. coli BL21(DE3)
cells (Thermo Fisher Scientific, Inc.), resulting in a yield of 70
mg/l. Briefly, an overnight starting culture of 10 ml was prepared
for growth in 1 l of Luria Bertani medium containing kanamycin (50
μg/l), which was then induced with isopropyl
β-D-1-thiogalactopyranoside (0.8 mM) at 16°C for 16 h. The protein
was purified by ultrasonicating the bacterial cells at 20 KHz, at
5°C for 15 min, which were then resuspended in binding buffer [300
mM NaCl, 50 mM Tris-HCl, 10 mM imidazole, 2.5% (v/v) glycerol, pH
7.8] containing a protease inhibitor cocktail (Roche Diagnostics).
The cell lysate was then centrifuged for 30 min at 14,000 × g and
5°C to remove the cell debris and inclusion bodies. The supernatant
was centrifuged again for 30 min at 40,000 × g and 5°C after which,
the resulting membrane pellet was resuspended in PBS lysis buffer
[PBS, pH 8.0; 10% (w/v) glycerol, 1 mM DTT, 0.002% (w/v)
phenylmethylsulfonyl fluoride (PMSF) and 10 mg/l DNase I (PanReac
AppliChem); 5 ml PBS lysis buffer per 1 g cells] and the cell
membranes were isolated by centrifugation at 34,000 × g and 5°C for
30 min. The membrane pellets were flash-frozen in liquid nitrogen
and stored at −80°C. To solubilize the protein, the membranes were
resuspended in buffer S [50 mM Tris/HCl, pH 7.8; 200 mM NaCl; 1.2%
(w/v) FosCholine-16; 2.5 mM DTT; 0.002% (w/v) PMSF] and stirred at
700 rpm for 1 h. The cell lysate was ultracentrifuged at 230,000 ×
g and 5°C for 60 min. The resulting supernatant was loaded onto a 5
ml Ni-NTA HisTrap HP column (Cytiva) equilibrated in buffer C-P
[buffer C (50 mM HEPES/NaOH, pH 7.6; 300 mM NaCl, 5% (w/v)
glycerol; 5 ml lysis buffer C per 1 g cells) with 0.002% (w/v)
PMSF]. The column was washed with 10 column volumes (CV) of buffer
C-P, followed by 20 CV of buffer C-ATP [buffer C with an additional
50 mM KCl, 20 mM MgCl2, 10 mM ATP, and 0.002% (w/v)
PMSF]. The column was further washed with 10 CV of buffer C-P
containing 50 mM imidazole and 100 mM imidazole. The RANK and LGR4
proteins were eluted with 500 mM imidazole in the C-P buffer. The
purified proteins were concentrated in a 50 kDa Amicon Ultra-15
concentrator (MilliporeSigma). The buffer was changed by desalting
on a PD-10 column (Cytiva) for binding affinity measurement.
Binding affinity measurement
The protein-binding affinity was measured using
microscale thermophoresis (MST) (24). The MST experiments were performed
using Monolith NT.115 systems (NanoTemper Technologies GmbH) and a
red filter. Briefly, all dilutions were prepared to ensure that no
other gradient (salt, glycerol, DMSO, etc.) was created during the
buffer mixing. To minimize the adsorption of the sample to the
material, 0.05% Tween 20 was added to PBS, which was used to dilute
all of the receptors and ligands. To measure protein-protein
binding, the receptor protein RANK or LGR4 and the ligand WT RANKL
or MT RANKL were mixed with an equal volume of the fluorescent
ligand spiperone-Cy5 (NanoTemper Technologies GmbH) to obtain final
ligand concentrations of 0.125, 5, 7.5 and 12 nM. After incubation
at 20°C for 1 h, the samples were loaded onto capillaries and the
LED was set to 20% for 0.125 nM samples, and 1% for 5, 7.5 and 12
nM samples, using medium MST power. For the receptor titration
assay, various concentrations of protein (10 mg/ml-5 μg/ml
total protein) were mixed with a specific concentration of the
fluorescent ligand, namely, spiperone-Cy5 was added at a final
concentration of 5, 7.5 or 12 nM to each protein dilution point.
The sample was incubated at 20°C for 1 h before capillary loading.
The LED power was set to 1% and the MST power was set to medium.
The intersection points in the binding curves were determined
according to the manufacturer's protocol and equilibrium
dissociation constant (Kd) values were obtained using Frobenius
normalization (%) from the mean of three replicates.
Primary cell culture and in vitro
osteoclast differentiation
A total of 20 female C57BL/6 mice (age, 5 weeks;
weight, ~25 g; Orient Bio, Inc.) were housed under controlled 12-h
light/dark cycle conditions, at a constant room temperature of
20±1°C and humidity of 40-60%. The mice were fed ad libitum
for 1 week prior to euthanasia with CO2 at a
displacement rate of 50%/min for use in the in vitro
studies. Bone marrow cells were obtained from the tibiae and femurs
of mice by flushing the bones with α-MEM (Gibco; Thermo Fisher
Scientific, Inc.) using previously described methods (25). After the removal of red blood
cells, bone marrow cells were resuspended in complete α-MEM
containing 10% (v/v) fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 μg/ml
streptomycin, and incubated at 37°C for 24 h in the presence of 10
ng/ml macrophage colony-stimulating factor (M-CSF; Cell Guidance
Systems Ltd.). Non-adherent cells were collected and cultured with
30 ng/ml M-CSF for 3 days to generate bone marrow-derived
macrophages (BMDMs). To generate pre-osteoclast lineage cells and
osteoclast lineage cells, BMDMs (50,000/cm2) were
cultured in complete α-MEM containing 30 ng/ml M-CSF and 75 ng/ml
WT RANKL at 37°C for 72 h.
RNA interference
SMART pool mixtures of small interfering RNA (siRNA)
targeting mouse LGR4 (cat. no. E-047291-00-0050, lot no. 210719)
and Accell non-targeting mouse RNA (cat. no. D-001910-10-50, lot
no. 3026176) were designed and synthesized by GE Healthcare
Dharmacon, Inc. For siRNA transfection, BMDMs were seeded on a
24-well plate at a density of 100,000 cells/well, and after 24 h,
the cells were transfected with each siRNA (20 pmol) at 37°C for 1
day using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. After
transfection, the BMDMs were cultured with M-CSF (30 ng/ml) and
RANKL (75 ng/ml) for 3 days to induce differentiation into
osteoclasts.
Tartrate-resistant acid phosphatase
(TRAP) assay
After the BMDMs were cultured with WT RANKL or MT
RANKL, the cells were washed once with PBS and fixed in 10%
formalin (in PBS) at 5°C for 5 min. After three washes with
distilled water, TRAP staining was performed for 30-40 min
according to the manufacturer's instructions (Kamiya Biomedical
Co.). The stained cells were examined under an ECLIPSE Ts2R
inverted light microscope (Nikon Corporation) and images were
captured using a digital camera (Nikon Corporation) with
NIS-Elements imaging software (Nikon Corporation). The number of
multinucleated cells was counted manually.
Reverse transcription-quantitative PCR
(RT-PCR) analysis
The BMDMs were incubated with 30 ng/ml M-CSF and 75
ng/ml WT RANKL, or 75 ng/ml WT RANKL plus 75 ng/ml MT RANKL at
37.5°C for the indicated times (0, 1 and 2 days) in 6-well plates.
Total RNA was extracted from the cells using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The cDNA was
then obtained from 2 μg total RNA using ReverTra Ace qPCR RT
master mix (Toyobo Life Science) according to the manufacturer's
protocol. The mRNA expression levels were measured using qPCR and
GAPDH was used as a control. qPCR was performed with a CFX Connect
real-time PCR detection system (Bio-Rad Laboratories, Inc.) using a
20 μl reaction mixture containing 10 μl IQ SYBR Green
supermix (Bio-Rad Laboratories, Inc.), 10 pmol forward primer, 10
pmol reverse primer and 1 μg cDNA. The sequences of the
primers used to target TRAP, OSCAR, NFATc1 and GAPDH are presented
as follows: TRAP, 5′-TAC CTG TGT GGA CAT GAC C-3′ (forward) and
5′-CAG ATC CAT AGT GAA ACC GC-3′ (reverse); OSCAR, 5′-CTG CTG GTA
ACG GAT CAG CTC CCC AGA-3′ (forward) and 5′-CCA AGG AGC CAG AAC CTT
CGA AAC T-3′ (reverse); NFATc1, 5′-GAG TAC ACC TTC CAG CAC CTT-3′
(forward) and 5′-TAT GAT GTC GGG GAA AGA GA-3′ (reverse) and GAPDH,
5′-TCA AGA AGG TGG TGA AGC AG-3′ (forward) and 5′-AGT GGG AGT TGC
TGT TGA AGT-3′ (reverse). The amplification parameters consisted of
an initial denaturation step at 95.8°C for 5 min followed by 40
cycles of three-step PCR (denaturation at 95.8°C for 1 min,
annealing at 60.8°C for 30 sec and extension at 72.8°C for 1 min).
The fluorescence resulting from the incorporation of SYBR Green dye
into the double-stranded DNA produced during PCR was quantified
using the 2−ΔΔCq method (26).
Western blot analysis
The BMDMs were serum-starved for 8 h and treated
with 2 μg/ml WT RANKL or MT RANKL at 0, 5, 10 and 15 min
intervals at 37°C. The cells were lysed in lysis buffer (50 mM
Tris-HCl, pH 7.5; 1% NP-40; 150 mM NaCl; 0.02% sodium azide; 1
mg/ml pepstatin A; 2 mg/ml aprotinin; 20 mg/ml leupeptin; 150 mg/ml
PMSF). After protein quantification using the BCA protein assay kit
(Pierce; Thermo Fisher Scientific, Inc.), ~30 μg cell lysate
was separated by SDS-PAGE 10% on gels and transferred onto a
polyvinylidene difluoride membrane (Amersham; Cytiva). Each
membrane was blocked for 30 min with a blocking solution containing
5% skim milk in Tris-buffered saline containing Tween-20 (TBST;
2.42 g/l Tris-HCl, 8 g/l NaCl, 0.1% Tween-20, pH 7.6) and rinsed
with TBST. The membrane was then incubated overnight at 4°C with
the following primary antibodies: Phosphorylated (p)-AKT (1:1,000;
cat. no. 9271S; Cell Signaling Technology, Inc.), AKT (1:1,000;
cat. no. 9272S; Cell Signaling Technology, Inc.), p-GSK-3β (Ser9,
1:1,000; cat. no. 9336S; Cell Signaling Technology, Inc.), GSK-3β
(1:1,000; cat. no. 9315S; Cell Signaling Technology, Inc.), p-Src
(1:1,000; cat. no. 2105; Cell Signaling Technology, Inc.), Src
(1:1,000; cat. no. 2108; Cell Signaling Technology, Inc.), His Tag
(1:1,000; cat. no. SAB1306082; MilliporeSigma), NFATc1 (1:1,000;
cat. no. 8032; Cell Signaling Technology, Inc.), Histone H1
(1:1,000; cat. no. sc-393358; Santa Cruz Biotechnology, Inc.),
β-Actin (1:1,000; cat. no. sc-47778; Santa Cruz Biotechnology,
Inc.), RANK (1:1,000; cat. no. 4845S; Cell Signaling Technology,
Inc.) and LGR4 (1:500; cat. no. MBS468030; MyBioSource, Inc.). A
mouse monoclonal immunoglobulin G antibody specific for GAPDH
(1:1,000; cat. no. 97166; Cell Signaling Technology, Inc.) was used
as a control. After rinsing with TBST, the membrane was incubated
at 4°C for 1 h with anti-rabbit (1:2,000; cat no. sc-2357; Santa
Cruz Biotechnology, Inc.) or anti-mouse (1:2,000; cat. no.
sc-525409; Santa Cruz Biotechnology, Inc.) horseradish
peroxidase-conjugated secondary antibodies. The membrane was then
rinsed with TBST, and the protein immunoreactivity was detected
using an enhanced chemiluminescence detection kit (Amersham;
Cytiva). Finally, the blot images were acquired using a
chemiluminescence imaging system (Vilber Lourmat) and the
densitometric semi-quantification of the detected bands was
performed using Image J 1.52a (National Institutes of Health) after
normalization to GAPDH.
Cell viability assay
Cell viability was evaluated using the MTT assay
(MilliporeSigma). The BMDMs were seeded into 96-well plates at
5×103 cells/well in 200 μl medium and cultured
for 1 day with MK2206 (0, 0.1, 0.2, 0.5, 1, 2, 5, and 10 μM;
MilliporeSigma) or LiCl (0, 2, 5, 10, 15, 20, 25 and 30 mM;
MilliporeSigma) at 37°C. For each experiment, the medium was
removed and 20 μl MTT (50 μg/ml) was added to the
wells. The cells were then incubated at 37°C for 4.5 h to allow the
color to develop, and the formazan was solubilized with the
addition of 50 μl dimethyl sulfoxide (Calbiochem; Merck
KGaA). The optical density was measured at 570 nm using a
microtiter plate reader (Bio-Rad Laboratories, Inc.).
Separation of nuclear and cytoplasmic
fractions
After siRNA transfection for 24 h or treatment with
LiCl for 8 h, the BMDMs were incubated with 75 ng/ml WT RANKL or 75
ng/ml MT RANKL for 24 h in 6-well plates. The cells were rinsed in
PBS and collected in microtubes (Eppendorf). A 0.5-ml volume of
Solution A (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM
DTT, 0.05% NP40, pH 7.9) was then added, after which the cells were
centrifuged at 805 × g for 10 min at 4°C. The supernatant,
containing mostly cytoplasmic constituents, was then removed and
transferred to another tube. To yield a nuclear pellet, 0.4 ml
solution B [5 mM HEPES, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5
mM DTT, 26% glycerol (v/v), 300 mM NaCl, pH 7.9] was added and the
contents of the tube were mixed thoroughly and placed on a small
rotator shaker for 15 min. Finally, the mixture was centrifuged at
24,000 × g for 20 min at 4°C. The supernatant containing proteins
from the nuclear extract was removed and transferred carefully into
a fresh tube. The nuclear and cytosolic extracts were frozen at
−80°C in aliquots prior to western blot analysis. The protein
concentration of each sample was determined using the BCA protein
assay kit (Thermo Fisher Scientific, Inc.), and the cytosolic and
nuclear fractions were subjected to western blot analysis.
Histone-H1 and β-actin were used as loading controls for the
nuclear and cytoplasmic fractions, respectively.
Animal study
A total of 20 female C57BL/6 mice (age, 5 weeks;
weight, ~25 g; Orient Bio, Inc.) were housed under a controlled
12-h light/dark cycle, at a constant room temperature of 20±1°C)
and humidity of 40-60%. The mice were fed ad libitum for 1
week prior to being randomly divided into four groups (n=5/group).
The control group was intraperitoneally injected with PBS; the WT
group was injected with WT RANKL (2 mg/kg) in PBS; and the WT + MT
group was injected with WT RANKL (2 mg/kg) and MT RANKL (2 mg/kg)
in PBS at 24-h intervals for 2 days. The MT group was also injected
with MT RANKL (2 mg/kg) in PBS. The total injections were performed
twice in each group. On day 3 after injection, all animals were
euthanized with 50% CO2 and femur bone samples were
collected for further study.
Micro-computed tomography (CT) imaging
and data acquisition
Micro-CT scanning of the distal femurs of mice was
initiated at the level of the growth plate using a Quantum GX
micro-CT imaging system (PerkinElmer, Inc.) located at the Korea
Basic Science Institute (Gwangju, Republic of Korea), according to
the methods described in a previous study (23). The bone volume/tissue volume
(BV/TV), trabecular separation (Tb. Sp.) and bone mineral density
(BMD) of the femurs were calculated using the region of interest
tool. Parameter values are shown as mean ± standard deviation
(SD).
Histological and immunohistochemical
analysis of bone specimens
Mouse femur tissues were fixed for 1 week in cold 4%
formalin at 5°C. The bone tissue was then decalcified using 0.5 M
EDTA, cut into 3-mm sections at the midpoint and embedded in
paraffin. The paraffin-embedded tissue sections were stained with
hematoxylin and eosin (H&E) using a staining kit (cat. no.
ab245880; Abcam) according to the manufacturer's protocol, and
images were acquired using an ECLIPSE Ts2R inverted light
microscope (Nikon Corporation).
For the immunohistochemical study, bone sections
were deparaffinized using three changes of xylene and then
rehydrated using graded concentrations of ethanol solutions, ending
with distilled water. For antigen retrieval, the slides were placed
in 0.01 M citrate buffer (pH 6.0) and heated in a steamer for 30
min. The endogenous peroxidases were quenched by incubating the
sections with 3% hydrogen peroxide for 20 min at room temperature
and blockading was performed with 3% BSA (in PBS; cat. no.
9048-46-8; VWR; Avantor) at 4°C for 1 h. The sections were then
incubated overnight at 4°C with a 1:50 dilution of the following
appropriate primary antibodies: Anti-LGR4 (1:100; cat. no.
PA5-67868; Invitrogen; Thermo Fisher Scientific, Inc.), ani-RANK
(1:100; cat. no. PA5-88904; Invitrogen; Thermo Fisher Scientific,
Inc.) or NFATc1 (1:100; cat. no. MA5-32686; Invitrogen; Thermo
Fisher Scientific, Inc.). Subsequently, the sections were incubated
at 20°C for 30 min with a biotin-labeled mouse anti-rabbit
secondary antibody (1:200; cat. no. 31824; Invitrogen; Thermo
Fisher Scientific, Inc.), washed in PBS, and incubated at 20°C for
30 min with a streptavidin-peroxidase conjugate (Dako; Agilent
Technologies, Inc.). The reaction was developed for 5 min using
3,30-diaminobenzidine tetrahydrochloride (MilliporeSigma). The
slides were counterstained with hematoxylin, dehydrated and a
coverslip was added. Finally, immune-stained slides were imaged
using an ECLIPSE Ts2R inverted light microscope (Nikon Corporation)
and the immune-positive area was analyzed using ImageJ
software.
Immunofluorescence analysis of bone
specimens
Paraffin-embedded bone sections were prepared
according to the aforementioned protocol and incubated overnight at
4°C with the following primary antibodies; Anti-LGR4 (1:100; cat.
no. PA5-67868; Invitrogen; Thermo Fisher Scientific, Inc.),
ani-RANK (1:100; cat. no. PA5-88904; Invitrogen; Thermo Fisher
Scientific, Inc.) and anti-glutathione S-transferase (GST; 1:100;
cat. no. 13-6700; Invitrogen; Thermo Fisher Scientific, Inc.).
Subsequently, sections were stained at 4°C for 1 h using Alexa
Fluor 594 goat anti-rabbit (1:500; cat. no. A11037; Invitrogen;
Thermo Fisher Scientific, Inc.) and Alexa Fluor 594 goat anti–mouse
secondary antibodies (1:500; cat. no. A11032; Invitrogen; Thermo
Fisher Scientific, Inc.). After washing with PBST, immunolabeled
cells were counter-stained with DAPI in Pro-Long Gold mounting
solution (Invitrogen; Thermo Fisher Scientific, Inc.). Digital
images were acquired using a TCS SP5 AOBS laser-scanning confocal
microscope (Leica Microsystems GmbH) and co-localization of RANKL
and LGR4, or RANKL and RANK was analyzed comparing the Pearson
correlation coefficient between WT RANKL- and MT RANKL-treated
groups.
Statistical analysis
All in vitro and in vivo studies were
conducted at least in triplicate. All quantitative results are
presented as the mean ± SD. The primary comparisons of cell-based
data and data from all the animal studies were analyzed using a
one-way or two-way analysis of variance with a Bonferroni
multiple-comparisons test, or unpaired Student's t-test. All of the
reported P-values are two-sided, and P<0.05 was considered to
indicate a statistically significant difference. All statistical
analyses were performed using GraphPad Prism version 7
(Dotmatics).
Results
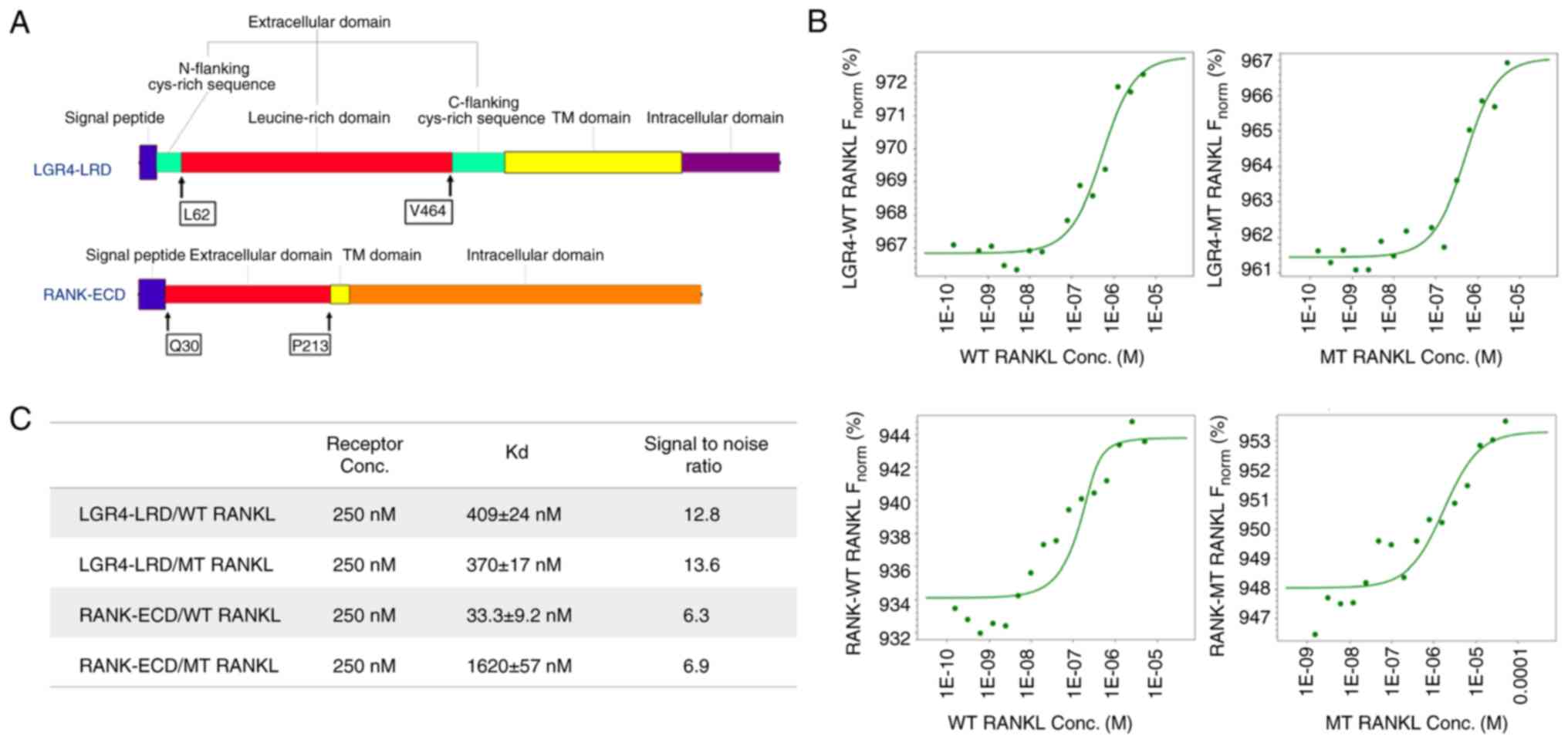
Binding affinity of WT RANKL and MT RANKL
for RANK and LGR4
The extracellular domains of RANK and LGR4 used in
the ligand-binding assays are shown in Fig. 1A. RANK and LGR4 were detected as
His-tagged proteins of 24 kDa for RANK and 47 kDa for LGR4 (the
impure form of LGR4 was detected above 47 kDa), respectively, by
western blot analysis (Fig.
S1).
MST assays were carried out to determine the binding
affinities of WT RANKL and MT RANKL for RANK and LGR4 (Fig. 1B and C). The MST measurements
showed that the Kd for the binding of WT RANKL to RANK was 33.3±9.2
nM and that for binding of MT RANKL to RANK was 1.62±0.057
μM, indicating that the affinity of MT RANKL for RANK was
~48.7-fold lower than the affinity of WT RANKL for RANK. The Kd
values for binding of WT RANKL to LGR4 (409±24 nM) and MT RANKL for
LGR4 (370±17 nM) did not show much difference.
These results showed that MT RANKL bound strongly to
LGR4 but not to RANK, whereas WT RANKL bound with equal affinity to
LGR4 and RANK. This finding suggested that MT RANKL may stimulate
the LGR4 signaling cascade without stimulating the canonical RANK
signaling cascade.
Effect of MT RANKL on the RANK-RANKL and
LGR4-RANKL signaling cascades
To investigate the effect of MT RANKL on the
RANKL-induced LGR4 signaling cascade on osteoclastogenesis in
vitro, BMDMs were transfected with LGR4 siRNA (Fig. 2A). The knockdown of LGR4
expression in LGR4 siRNA-transfected BMDMs was confirmed using
western blotting, which also showed that RANK expression was
unaffected (Fig. 2B).
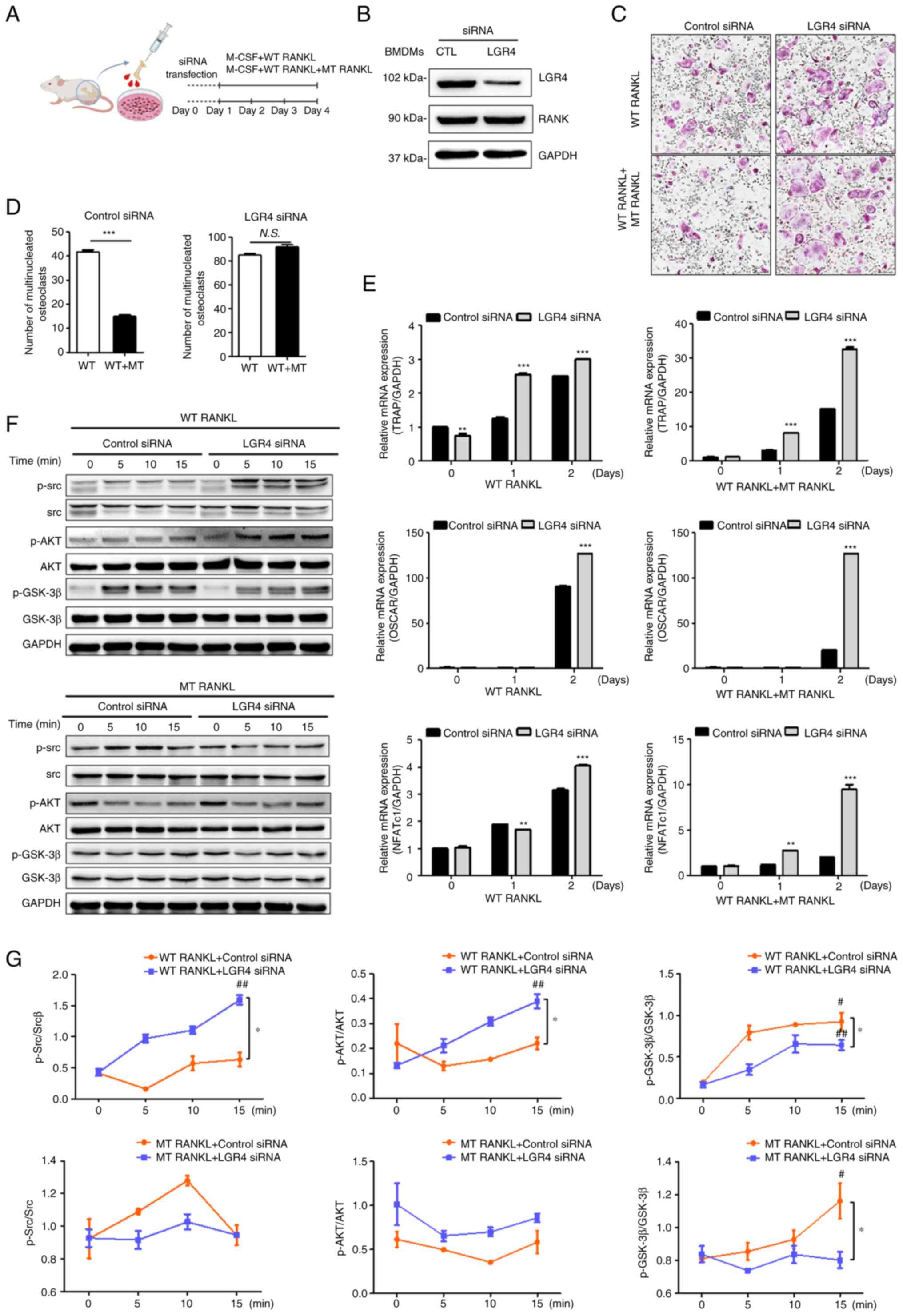 | Figure 2Effect of MT RANKL on osteoclast
differentiation in vitro. (A) Schedule for treating control
or LGR4 siRNA-transfected BMDMs with WT RANKL or WT RANKL + MT
RANKL. (B) Western blotting of LGR4 and RANK expression in LGR4
siRNA-transfected BMDMs. LGR4 expression was markedly lower in LGR4
siRNA-treated BMDMs. (C) A representative image of BMDMs stained
for TRAP (red) following treatment of control siRNA- or LGR4
siRNA-transfected BMDMs with WT RANKL (75 ng/ml) or WT RANKL (75
ng/ml) + MT RANKL (75 ng/ml). Magnification, ×100; scale bar, 20
μm. (D) Number of multinucleated TRAP-positive cells (≥3
nuclei). ***P<0.001. (E) Osteoclast-related gene
expression in control siRNA- or LGR4 siRNA-transfected BMDMs. BMDMs
were exposed to WT RANKL (75 ng/ml) or WT RANKL (75 ng/ml) + MT
RANKL (75 ng/ml) for 2 days. Gene expression was determined by
reverse transcription-quantitative PCR and normalized to the
expression of GAPDH. **P<0.01 and
***P<0.001 vs. Control siRNA. (F) Western blot
analysis of RANK and LGR4 signaling cascades in control siRNA- or
LGR4 siRNA-transfected BMDMs in the presence of WT RANKL (2
μg/ml) or MT RANKL (2 μg/ml). GAPDH was used as a
loading control. BMDM, bone marrow-derived macrophage. (G)
Densitometric value of p-Src/Src, p-AKT/AKT and p-GSK-3β/GSK-3β, as
determined by western blot analysis. Results are representative of
three separate experiments that had comparable results.
*P<0.05 Control siRNA vs.LGR4 siRNA at 15 min;
#P<0.05 vs. WT RANKL + Control siRNA at 0 min,
##P<0.05 vs. MT RANKL+ Control siRNA at 0 min.
GSK-3β, glycogen synthase kinase-3β; LGR4, leucine-rich
repeat-containing G-protein-coupled receptor 4; M-CSF, macrophage
colony-stimulating factor; MT, mutant; NFATc1, nuclear factor of
activated T cells, cytoplasmic, calcineurin-dependent 1; N.S., not
significant; p-, phosphorylated; RANK, receptor-activated nuclear
factor-κB; RANKL, RANK ligand; siRNA, small interfering RNA; TRAP,
tartrate-resistant acid phosphatase; WT, wild-type. |
The numbers of BMDMs that differentiated into
mature, TRAP-positive, multinucleated osteoclasts were subsequently
counted (Fig. 2C). In the
presence of WT RANKL, MT RANKL decreased the number of
TRAP-positive cells in control siRNA-transfected BMDMs, but not in
LGR4 siRNA-transfected BMDMs. In addition, the number of osteoclast
cells was significantly decreased in the WT RANKL- and MT
RANKL-treated BMDMs in the presence of control siRNA (Fig. 2D) but not in the presence of LGR4
siRNA.
To evaluate the effect of MT RANKL signaling via the
LGR4-dependent pathway on the mRNA expression levels of
osteoclastogenesis-related genes, RT-qPCR was used to investigate
the expression of several osteoclast-specific genes in BMDMs
treated with either WT RANKL, or both WT RANKL and MT RANKL
(Fig. 2E). The results showed
that on day 2 post-treatment, there was a significant increase in
the mRNA expression levels of TRAP, OSCAR and NFATc1 in the LGR4
siRNA-transfected BMDMs treated with either WT RANKL, or both WT
RANKL and MT RANKL, compared with those in the control
siRNA-transfected BMDMs; these genes are markers of osteoclast
differentiation and activity.
To evaluate the effect of MT RANKL on the LGR4-RANKL
signaling cascade, the present study investigated whether treatment
of BMDMs with WT RANKL or MT RANKL induced the phosphorylation of
AKT, Src, and GSK-3β via RANK and LGR4 signaling cascades (Fig. 2F and G). In WT RANKL-treated
BMDMs, transfection with LGR4 siRNA induced the obvious increase in
Src and AKT phosphorylation, and a decrease in GSK-3β
phosphorylation compared with the control siRNA. However, LGR4
siRNA in MT RANKL-treated BMDMs did not affect AKT phosphorylation
compared with control siRNA. In addition, MT RANKL alone
significantly increased the phosphorylation of GSK-3β compared with
untreated BMDMs at 0 min, and LGR4 siRNA transfection decreased
GSK-3β phosphorylation in MT RANKL-treated BMDMs compared with
control siRNA.
Overall, these results demonstrated that in
RANKL-treated BMDMs, MT RANKL may inhibit osteoclast
differentiation and activity via the LGR4-dependent signaling
pathway rather than via the RANK-mediated signaling.
Effect of MT RANKL on the AKT signaling
cascade
To evaluate the effects of WT RANKL and MT RANKL on
the LGR4-mediated AKT signaling cascade, BMDMs were pretreated with
the AKT inhibitor MK2206. Cell viability assays were performed to
determine the optimal concentration of MK2206 (Fig. S2). Cell viability decreased with
increases in MK2206 concentration; 0.2 μM MK2206 was the
maximum concentration that did not affect BMDM cell viability;
therefore, this concentration was used in subsequent
experiments.
Treatment of BMDMs with WT RANKL in the presence of
MK2206 significantly decreased the number of TRAP-positive
multinuclear cells (Fig. 3A and
B), but treatment with MT RANKL in the presence or absence of
MK2206 did not affect the number of TRAP-positive multinuclear
cells.
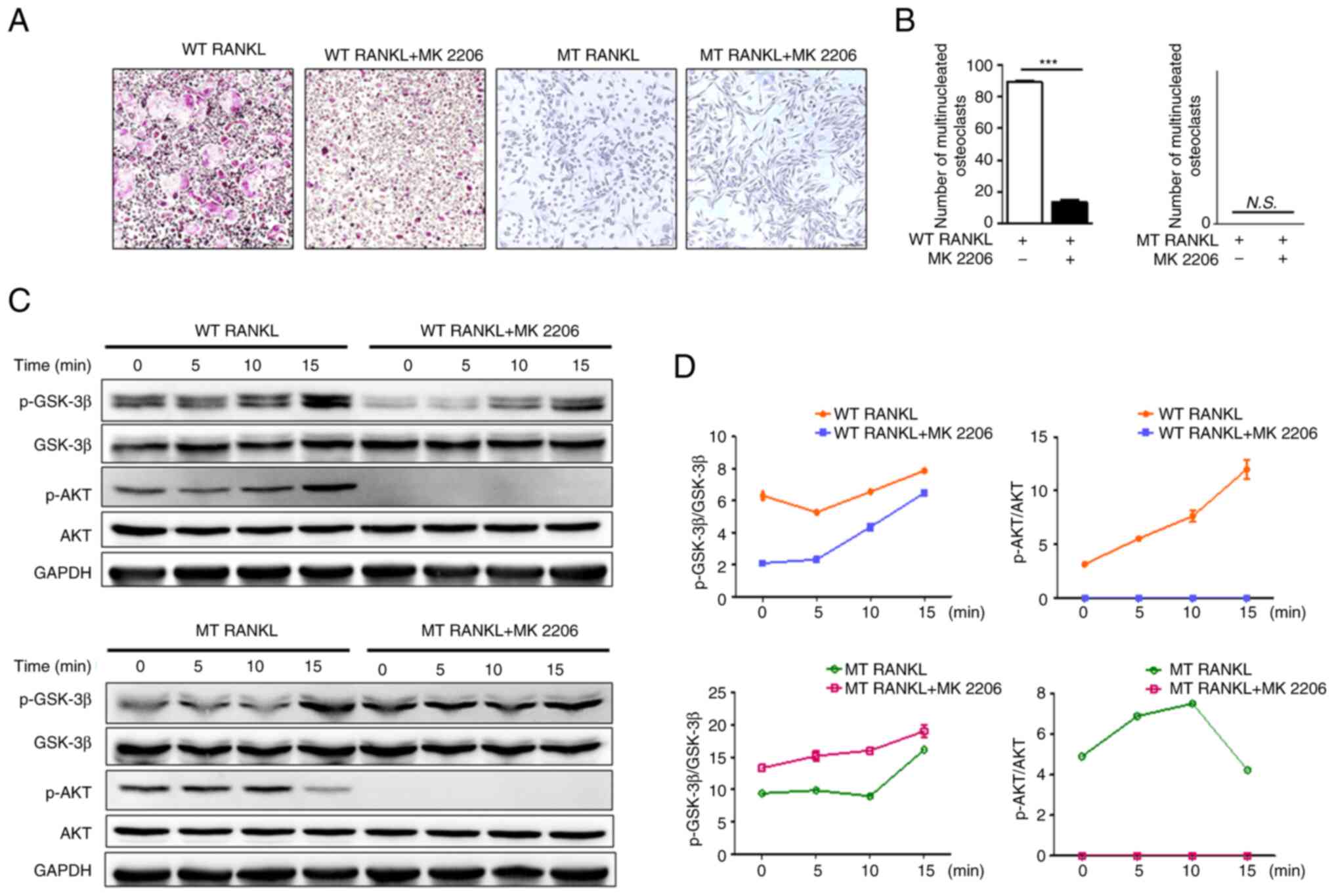 | Figure 3Effect of MT RANKL on the AKT
signaling cascade. (A) TRAP staining in the presence of WT RANKL
(75 ng/ml) or MT RANKL (75 ng/ml) in BMDMs pretreated with MK2206
for 8 h before RANKL treatment. Magnification, ×100; scale bar, 20
μm. (B) Number of multinucleated TRAP-positive cells (≥3
nuclei). ***P<0.001. (C) Western blot analysis of
RANK and LGR4 signaling cascades. BMDMs were exposed to WT RANKL (2
μg/ml) or MT RANKL (2 μg/ml) for 0, 5, 10 and 15 min
after 8 h with or without MK2206 pretreatment. GAPDH was used as a
loading control. (D) Densitometric value of p-GSK-3β/GSK-3β and
p-AKT/AKT determined by western blot analysis. Results are
representative of three separate experiments that had comparable
results. BMDM, bone marrow-derived macrophage; GSK-3β, glycogen
synthase kinase-3β; LGR4, leucine-rich repeat-containing
G-protein-coupled receptor 4; MT, mutant; N.S., not significant;
p-, phosphorylated; RANKL, receptor-activated nuclear factor-κB
ligand; siRNA, small interfering RNA; TRAP, tartrate-resistant acid
phosphatase; WT, wild-type. |
Although WT RANKL slightly decreased GSK-3β
phosphorylation in the presence of MK2206 (Fig. 3C and D), MT RANKL in
MK2206-pretreated BMDMs exhibited increased levels of p-GSK-3β in
compared with MT RANKL alone. In addition, MK2206 completely
blocked phosphorylation of AKT in WT RANKL- and MT RANKL-treated
BMDMs. These results suggested that the phosphorylation of GSK-3β
by MT RANKL may be independent of AKT, since phosphorylation of
GSK-3β still occurred in MT RANKL-treated BMDMS for which AKT
phosphorylation was blocked, and MT RANKL alone did not have an
effect on the osteoclastogenesis progress.
Effect of MT RANKL on the GSK-3β
signaling cascade
To investigate the LGR4-mediated GSK-3β signaling
cascade, WT RANKL- or MT RANKL-treated BMDMs were pretreated with
lithium chloride (LiCl), a powerful GSK-3β inhibitor (27,28). Cell viability assays were
performed to determine the appropriate LiCl concentration (Fig. S3). LiCl
concentration-dependently decreased cell viability; since 5 mM LiCl
did not affect BMDM cell viability, subsequent experiments were
performed using this concentration.
Treatment of LiCl-pretreated BMDMs with WT RANKL
significantly increased the number of TRAP-positive multinuclear
cell (Fig. 4A and B). However,
MT RANKL did not affect the number of TRAP-positive multinuclear
cells in the presence or absence of LiCl.
 | Figure 4Effect of MT RANKL on the GSK-3β
signaling cascade. (A) TRAP staining results in the presence of WT
RANKL (75 ng/ml) or MT RANKL (75 ng/ml) in BMDMs pretreated with
LiCl for 8 h before RANKL treatment. Magnification, ×100; scale
bar, 20 μm. (B) Number of multinucleated TRAP-positive cells
(≥3 nuclei). ***P<0.001. (C) Western blot analysis of
the RANK and LGR4 signaling cascades. BMDMs were exposed to WT
RANKL (2 μg/ml) or MT RANKL (2 μg/ml) for 0, 5, 10
and 15 min after 8 h with or without LiCl pretreatment. GAPDH was
used as a loading control. (D) Densitometric value of
p-GSK-3β/GSK-3β and p-AKT/AKT was determined by western blot
analysis. The results are representative of three separate
experiments that had comparable results. BMDM, bone marrow-derived
macrophage; GSK-3β, glycogen synthase kinase-3β; LGR4, leucine-rich
repeat-containing G-protein-coupled receptor 4; LiCl, lithium
chloride; MT, mutant; N.S., not significant; p-, phosphorylated;
RANKL, receptor-activated nuclear factor-κB ligand; TRAP,
tartrate-resistant acid phosphatase; WT, wild-type. |
In addition, WT RANKL or MT RANKL slightly increased
GSK-3β phosphorylation in a time-dependent manner in cells
pretreated with LiCl, which indicated the successful inhibition of
GSK-3β, although the level of AKT was not altered (Fig. 4C and D). These results suggested
that the inhibition of GSK-3β may lead to an increase in
osteoclastogenesis and MT RANKL could stimulate the GSK-3β
phosphorylation.
Effect of MT RANKL on NFATc1
translocation to the nucleus
To evaluate the effect of MT RANKL on LGR4-dependent
inhibition of NFATc1 nuclear translocation, the present study
examined the nuclear and cytosolic localization of NFATc1 using
western blotting and densitometric analysis of control siRNA- and
LGR4 siRNA-transfected BMDMs (Fig.
5A and B). Nuclear NFATc1 was not detected in MT RANKL-treated
BMDMs in the presence of control siRNA but was detected in BMDMs
treated with WT RANKL or MT RANKL in the presence of LGR4
siRNA.
 | Figure 5Effect of MT RANKL on NFATc1 nuclear
translocation. (A) NFATc1 nuclear translocation in LGR4
siRNA-transfected BMDMs was analyzed in the cytoplasmic and nuclear
fractions. Histone-H1 and β-actin were used as loading controls for
the nuclear and cytoplasmic fractions, respectively. (B)
Densitometric analysis of NFATc1 expression in the cytoplasmic and
nuclear fractions of LGR4 siRNA-transfected BMDMs is presented as
the mean ± standard deviation of three separate experiments. (C)
NFATc1 nuclear translocation in BMDMs pretreated with LiCl for 8 h
before RANKL treatment. (D) Densitometric analysis of NFATc1
expression in BMDMs pretreated with LiCl. The results are
representative of three separate experiments that had comparable
data. Data are presented as the mean ± standard deviation.
*P<0.01 vs. control group; #P<0.01 vs.
WT RANKL. BMDM, bone marrow-derived macrophage; CTL, control; LGR4,
leucine-rich repeat-containing G-protein-coupled receptor 4; LiCl,
lithium chloride; MT, mutant; NFATc1, nuclear factor of activated T
cells, cytoplasmic, calcineurin-dependent 1; N.S., not significant;
RANKL, receptor-activated nuclear factor-κB ligand; si, small
interfering; WT, wild-type. |
In addition, the expression levels of NFATc1 were
detected in LiCl-treated BMDMs to investigate the effect of MT
RANKL on GSK-3β-mediated inhibition of NFATc1 nuclear translocation
(Fig. 5C and D). Nuclear NFATc1
was not detected in untreated BMDMs or MT RANKL-treated BMDMs, but
was detected in BMDMs treated with WT RANKL or MT RANKL in the
presence of LiCl, and in BMDMs treated with LiCl alone.
These results further support the hypothesis that
LGR4 signaling serves a critical role in the GSK-3β-mediated
inhibition of NFATc1 nuclear translocation in RANKL-treated BMDMs,
suggesting that MT RANKL compensates for the inhibition of WT
RANKL-RANK- NFATc1 signaling pathway via MT RANKL-LGR4-GSK-3β.
Effect of MT RANKL on bone loss in
mice
To investigate the effect of MT RANKL on bone lysis,
healthy mice were treated with MT RANKL in the presence or absence
of WT RANKL and their femur bones were examined using micro-CT
(Fig. 6A). Mice in the WT
RANKL-treated group exhibited marked bone loss compared with the
control (untreated) group, whereas those treated with WT RANKL + MT
RANKL exhibited little bone loss. Bone loss in the MT RANKL-treated
group was similar to that in the control group. The BV/TV, Tb. Sp
and BMD scores were assessed using quantitative micro-CT (Fig. 6B). As expected, the BV/TV and BMD
scores were lower in WT RANKL-treated mice than control mice, and
significantly increased after MT RANKL treatment. The BV/TV, Tb.Sp
and BMD scores in mice treated only with MT RANKL were similar to
those in control mice. These results demonstrated the therapeutic
effects of MT RANKL in a model of RANKL-induced bone loss.
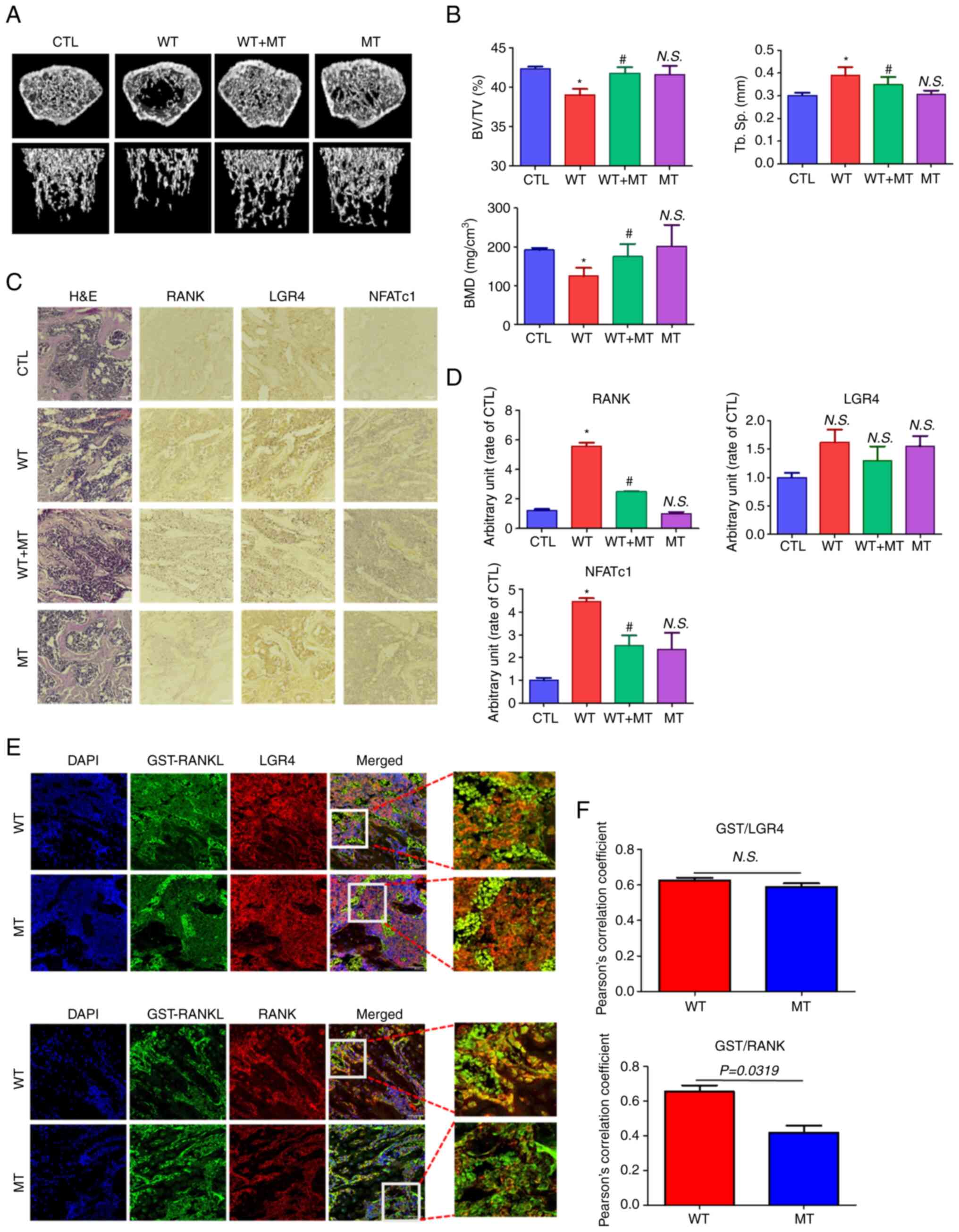 | Figure 6Effect of MT RANKL on RANKL-induced
bone loss in a mouse model. (A) Representative micro-computed
tomography images of the distal femurs of mice. (B) Measurements of
BV/TV, Tb. Sp and BMD. Data are presented as the mean ± SD.
*P<0.01 vs. control group; #P<0.01 vs.
WT RANKL. (C) Immunohistochemistry staining of RANK, LGR4 and
NFATc1 in femurs. Magnification, ×200; scale bar, 10 μm. (D)
Densitometric analysis of immunohistochemistry. (E) Confocal
microscopic images of the co-localization of GST-RANKL with LGR4
and RANK in WT RANKL- and MT RANKL-treated mice. Magnification,
×200; scale bar, 10 μm. (F) Pearson's correlation
coefficient was calculated from the merged images of GST/LGR4 and
GST/RANK. Data are presented as the mean ± SD of three independent
measurements. BMD, bone mineral density; BV/TV, bone volume/tissue
volume; CTL, control; GST, glutathione S-transferase; H&E,
hematoxylin and eosin; LGR4, leucine-rich repeat-containing
G-protein-coupled receptor 4; MT, mutant; NFATc1, nuclear factor of
activated T cells, cytoplasmic, calcineurin-dependent 1; N.S., not
significant; RANK, receptor-activated nuclear factor-κB; RANKL,
RANK ligand; SD, standard deviation; Tb. Sp, trabecular
spacing. |
In addition, the immunopositive expression of RANK,
LGR4 and NFATc1 was detected in H&E-stained histological
sections of the femur (Fig. 6C and
D). RANK and NFATc1 showed decreased immunopositive expression
in mice treated with WT RANKL + MT RANKL compared with in WT
RANKL-treated mice.
Finally, the co-localization of WT RANKL/MT RANKL as
a GST-tagged RANKL with LGR4/RANK in WT RANKL- or MT RANKL-treated
mice was assessed using confocal microscopy (Fig. 6E and F). GST-RANKL and LGR4
colocalized in mice treated with WT RANKL or MT RANKL, whereas
GST-RANKL and RANK only colocalized in mice treated with WT RANKL.
The correlation coefficient for GST-RANKL and LGR4 colocalization
was not significantly different between WT RANK- or MT
RANKL-treated mice, whereas the correlation coefficient of
GST-RANKL and RANK colocalization was significantly higher in WT
RANKL-treated mice than in WT RANKL-treated mice.
Taken together, these results demonstrated that MT
RANKL could inhibit RANKL-induced bone lysis in a mouse model via
the LGR4-dependent compensatory signaling pathway, and as such, MT
RANKL may be a useful pharmaceutical agent in severe
osteoporosis.
Discussion
Increasing evidence has indicated that the binding
of RANKL to its receptor RANK, which drives the development of
osteoclasts, is a key target for drugs that treat osteolytic bone
diseases, including osteoporosis (29). Given the role of the RANKL-RANK
signaling cascade in osteoclast development, the human monoclonal
antibody denosumab, which targets RANKL, has been used as a
clinical therapy for osteoporosis; furthermore, the use of
denosumab validates RANKL as a therapeutic target (30-32). However, concerns have arisen
regarding rebound bone resorption associated with hypercalcemia,
parathyroid hyperplasia, severe BMD loss and multiple fractures
after discontinuation of denosumab treatment (33-35). Scavenging of RANKL by denosumab
in patients with osteoporosis leads to repeated regeneration of
RANKL, resulting in rebound bone resorption that is more severe
than that observed before treatment (14).
Because of the side effects associated with
denosumab, the development of a competitive inhibitor of the
RANKL-RANK signaling cascade may be an alternative approach for
treating osteoporosis. LGR4 is another receptor for RANKL, and
RANKL has a similar binding affinity for both RANK and LGR4,
resulting in the negative regulation of RANKL-RANK signaling during
osteoclastogenesis (36).
Moreover, our previous studies showed that a novel MT RANKL acts as
an agonist of LGR4, and inhibits the differentiation and activation
of osteoclasts in RANKL-induced osteoclastogenesis in in
vitro and in vivo models (15,22,23). Thus, agonist activation of
LGR4-mediated signaling inhibits NFATc1 signaling through the
intracellular LGR4-GSK-3β signaling pathway in osteoclast
progenitor cells, and this pathway predominates over the canonical
RANKL-RANK pathway, resulting in a block in osteoclast development
(15,37).
The present study investigated the effects of the MT
RANKL protein, in which the RANK binding site was modified using
minimal amino acid changes, resulting in a protein that had a high
binding affinity for LGR4 but a 500-fold lower binding affinity for
RANK (22). The minimal change
in the RANK-binding domain in MT RANKL means that if this protein
is used in humans, RANKL homeostasis will be maintained without
causing additional RANKL release, meaning that rebound resorption
should not occur. Furthermore, the extracellular domain of LGR4 is
known to have a higher binding affinity for MT RANKL than for RANK,
implying that LGR4 -MT RANKL binding could have fewer physiological
side effects than MT RANKL-RANK binding on osteoclast
differentiation and activation. The potential reduction in side
effects may be because MT RANKL competes with endogenous RANKL for
binding to LGR4 and RANK.
In the mouse model, MT RANKL colocalized with only
LGR4, whereas WT RANKL colocalized with RANK and LGR4. This finding
suggested that in RANKL-induced osteoclastogenesis, WT RANKL
interacts with RANK and LGR4, but MT RANKL interacts only with
LGR4. Additional stimulation of LGR4 in RANKL-induced osteoclast
precursor cells may trigger a negative regulatory signal that
predominates over RANKL-RANK signaling and reduces NFATc1-related
signaling. Luo et al (15) reported that the LGR4
extracellular domain acts as a molecular decoy receptor for RANKL
binding both in vitro and in vivo, and inhibits
RANKL-induced osteoclast activation. Another report showed that
NFATc1 drives early osteoclast differentiation and causes
osteoclast precursors to commit to the osteoclast lineage (38). The suppression of RANK signaling
may lead to the upregulation of NFATc1 in differentiating
osteoclast cells, as it has been suggested that targeting AKT
signaling promotes IκBα degradation, resulting in the nuclear
translocation of NFATc1. Moreover, LGR4 signaling prevents the
inactivation of GSK-3β; active GSK-3β prevents the activation and
nuclear translocation of NFATc1 (39,40). Thus, in the current study,
signaling from LGR4-MT RANKL could inhibit AKT phosphorylation and
stimulate GSK-3β phosphorylation, resulting in the inhibition of
NFATc1 nuclear translocation that was independent of the RANK
signaling cascade. In particular, MT RANKL signaling did not
stimulate AKT phosphorylation and osteoclast development in BMDMs
treated with an AKT inhibitor. The present study showed that MT
RANKL, but not WT RANKL, acted as an LGR4 agonist and triggered
LGR4-GSK-3β signaling (Fig. 7).
A previous report showed that LGR4 expression is induced during
osteoclast differentiation, resulting in RANKL-NFATc1 signaling
(41). LGR4 expression is
elevated in mature osteoclasts, and the LGR4-mediated signaling
pathway is activated at a maximum level in mature osteoclasts,
resulting in decreased RANKL-RANK signaling (15). This mechanism suggests that LGR4
could be used as a target for the development of novel osteoporosis
therapies. Notably, mature osteoclasts ultimately undergo apoptosis
in a RANKL-containing environment, suggesting that the existence of
a RANKL-induced signaling pathway limits the maintenance of mature
osteoclasts (42). The present
study indicated that LGR4 may be a pivotal player in the
negative-feedback mechanism that controls the activity of
osteoclasts. The LGR4 signaling cascade activated by MT RANKL
inactivated AKT and activated the GSK-3β signaling pathway, which
resulted the inhibition of the activity of NFATc1 during osteoclast
differentiation.
In conclusion, MT RANKL, a novel agonist of LGR4,
may activate an inhibitory signaling pathway during
osteoclastogenesis that is different from the pathway activated by
RANK-RANKL. MT RANKL could modulate the RANKL-AKT-NFATc1 signaling
cascade through LGR4-induced GSK-3β phosphorylation and provides a
negative-feedback mechanism to control osteoclast activity. The
critical role of the RANK-RANKL protein interaction during
osteoclast development means that it is an important target for
drugs that treat osteoporosis. The results of the present study
showed that in in vitro or in vivo experimental
models of bone loss induced by RANKL, MT RANKL induced GSK-3β
phosphorylation, and inhibited NFATc1 nuclear translocation and
bone resorption. Furthermore, the results of this study suggested
that MT RANKL, by activating a pathway that inhibits the effects of
the RANKL-RANK signaling cascade, has the potential for treating
osteoporosis.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YJ and HL conducted the majority of the experiments,
and YJ, HL and WL wrote the original manuscript. YC, SJ and EC
performed the statistical analysis. YJ, HL and WL contributed to
study conception and design. YJ and HL contributed to acquisition
of data, and YC, SJ and HMS performed analysis and interpretation
of data. HMS and WL reviewed and edited the original manuscript.
BCK and YJK provided research materials and developed the
methodology. YC and SJ confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All of the experimental procedures involving animals
were performed in compliance with institutional and governmental
requirements, and were approved by the Institutional Animal Care
and Use Committee (approval no. CIACUC2018-S0012-1) of Chosun
University (Gwangju, Republic of Korea).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by research funding from Chosun
University (awarded in 2021).
References
|
1
|
Hofbauer L, Kuhne C and Viereck V: The
OPG/RANKL/RANK system in metabolic bone diseases. J Musculoskel
Neuro Inter. 4:268–275. 2004.
|
|
2
|
Wang L, You X, Zhang L, Zhang C and Zou W:
Mechanical regulation of bone remodeling. Bone Res. 10:162022.
|
|
3
|
Hanley DA, Adachi JD, Bell A and Brown V:
Denosumab: Mechanism of action and clinical outcomes. Int J Clin
Pract. 66:1139–1146. 2012.
|
|
4
|
Gowen M, Stroup GB, Dodds RA, James IE,
Votta BJ, Smith BR, Bhatnagar PK, Lago AM, Callahan JF, DelMar EG,
et al: Antagonizing the parathyroid calcium receptor stimulates
parathyroid hormone secretion and bone formation in osteopenic
rats. J Clin Invest. 105:1595–1604. 2000.
|
|
5
|
Martin TJ: Bone biology and anabolic
therapies for bone: Current status and future prospects. J Bone
Metab. 21:8–20. 2014.
|
|
6
|
Sozen T, Ozisik L and Basaran NC: An
overview and management of osteoporosis. Eur J Rheumatol. 4:46–56.
2017.
|
|
7
|
Miller PD: Denosumab: Anti-RANKL antibody.
Curr Osteoporos Rep. 7:18–22. 2009.
|
|
8
|
Cadieux B, Coleman R, Jafarinasabian P,
Lipton A, Orlowski RZ, Saad F, Scagliotti GV, Shimizu K and Stopeck
A: Experience with denosumab (XGEVA(R)) for prevention of
skeletal-related events in the 10 years after approval. J Bone
Oncol. 33:1004162022.
|
|
9
|
Lei MM, Tavares E, Buzgo E, Lou U, Raje N
and Yee AJ: Denosumab versus intravenous bisphosphonate use for
hypercalcemia in multiple myeloma. Leuk Lymphoma. 63:1–4. 2022.
|
|
10
|
Terpos E, Jamotte A, Christodoulopoulou A,
Campioni M, Bhowmik D, Kennedy L and Willenbacher W: A
cost-effectiveness analysis of denosumab for the prevention of
skeletal-related events in patients with multiple myeloma in four
European countries: Austria, Belgium, Greece, and Italy. J Med
Econ. 22:766–776. 2019.
|
|
11
|
Benlidayi IC: Denosumab in the treatment
of glucocorticoid-induced osteoporosis. Rheumatol Int.
38:1975–1984. 2018.
|
|
12
|
Pittman K, Antill YC, Goldrick A, Goh J
and de Boer RH: Denosumab: Prevention and management of
hypocalcemia, osteonecrosis of the jaw and atypical fractures. Asia
Pac J Clin Oncol. 13:266–276. 2017.
|
|
13
|
Gkoufa A, Angelousi A, Neonaki A,
Athanasouli F and Cholongitas E: Severe symptomatic hypocalcemia
associated with denosumab administration in a patient with
decompensated cirrhosis and renal dysfunction. Ann Pharmacother.
56:853–855. 2022.
|
|
14
|
Anastasilakis AD, Makras P, Yavropoulou
MP, Tabacco G, Naciu AM and Palermo A: Denosumab discontinuation
and the rebound phenomenon: A narrative review. J Clin Med.
10:1522021.
|
|
15
|
Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G,
Huang J, Dai W, Li C, Zheng C, et al: LGR4 is a receptor for RANKL
and negatively regulates osteoclast differentiation and bone
resorption. Nat Med. 22:539–546. 2016.
|
|
16
|
Takegahara N, Kim H and Choi Y: RANKL
biology. Bone. 159:1163532022.
|
|
17
|
Yue Z, Niu X, Yuan Z, Qin Q, Jiang W, He
L, Gao J, Ding Y, Liu Y, Xu Z, et al: RSPO2 and RANKL signal
through LGR4 to regulate osteoclastic premetastatic niche formation
and bone metastasis. J Clin Invest. 132:e1445792022.
|
|
18
|
Jin Y and Yang Y: LGR4: A new receptor for
a stronger bone. Sci China Life Sci. 59:735–736. 2016.
|
|
19
|
Luo W, Tan P, Rodriguez M, He L, Tan K,
Zeng L, Siwko S and Liu M: Leucine-rich repeat-containing G
protein-coupled receptor 4 (Lgr4) is necessary for prostate cancer
metastasis via epithelial-mesenchymal transition. J Biol Chem.
292:15525–15537. 2017.
|
|
20
|
Elango J, Bao B and Wu W: The hidden
secrets of soluble RANKL in bone biology. Cytokine.
144:1555592021.
|
|
21
|
Ko Y, Lee G, Kim B, Park M, Jang Y and Lim
W: Modification of the RANKL-RANK-binding site for the
immunotherapeutic treatment of osteoporosis. Osteoporos Int.
31:983–993. 2020.
|
|
22
|
Ko YJ, Sohn HM, Jang Y, Park M, Kim B, Kim
B, Park JI, Hyun H, Jeong B, Hong C and Lim W: A novel modified
RANKL variant can prevent osteoporosis by acting as a vaccine and
an inhibitor. Clin Transl Med. 11:e3682021.
|
|
23
|
Jang Y, Sohn HM, Ko YJ, Hyun H and Lim W:
Inhibition of RANKL-induced osteoclastogenesis by novel mutant
RANKL. Int J Mol Sci. 22:4342021.
|
|
24
|
Romain M, Thiroux B, Tardy M, Quesnel B
and Thuru X: Measurement of protein-protein interactions through
microscale thermophoresis (MST). Bio Protoc. 10:e35742020.
|
|
25
|
Bartell SM, Kim HN, Ambrogini E, Han L,
Iyer S, Ucer SS, Rabinovitch P, Jilka RL, Weinstein RS, Zhao H, et
al: FoxO proteins restrain osteoclastogenesis and bone resorption
by attenuating H2O2 accumulation. Nat Commun. 5:37732014.
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
27
|
Law SM and Zheng KK: Premise and peril of
Wnt signaling activation through GSK-3β inhibition. iScience.
25:1041592022.
|
|
28
|
Fan X, Xiong H, Wei J, Gao X, Feng Y, Liu
X, Zhang G, He QY, Xu J and Liu L: Cytoplasmic hnRNPK interacts
with GSK3β and is essential for the osteoclast differentiation. Sci
Rep. 5:177322015.
|
|
29
|
Cao X: RANKL-RANK signaling regulates
osteoblast differentiation and bone formation. Bone Res.
6:352018.
|
|
30
|
Tokuyama N and Tanaka S: Updates of
denosumab, anti-RANKL antibody for osteoporosis. Clin Calcium.
24:85–91. 2014.In Japanese.
|
|
31
|
Cipriani C, Piemonte S, Colangelo L, De
Martino V, Diacinti D, Ferrone F, Piazzolla V, Fassino V, Nieddu L,
Minisola S and Pepe J: Inhibition of the RANKL with denosumab has
no effect on circulating markers of atherosclerosis in women with
postmenopausal osteoporosis: A pilot study. Endocrine. 71:199–207.
2021.
|
|
32
|
Lasco A, Morabito N, Basile G, Atteritano
M, Gaudio A, Giorgianni GM, Morini E, Faraci B, Bellone F and
Catalano A: Denosumab inhibition of RANKL and insulin resistance in
postmenopausal women with osteoporosis. Calcif Tissue Int.
98:123–128. 2016.
|
|
33
|
Kim AS, Girgis CM and McDonald MM:
Osteoclast recycling and the rebound phenomenon following denosumab
discontinuation. Curr Osteoporos Rep. 20:505–515. 2022.
|
|
34
|
Anastasilakis AD, Evangelatos G, Makras P
and Iliopoulos A: Rebound-associated vertebral fractures may occur
in sequential time points following denosumab discontinuation: Need
for prompt treatment re-initiation. Bone Rep. 12:1002672020.
|
|
35
|
Anastasilakis AD, Trovas G, Balanika A,
Polyzos SA, Makras P and Tournis S: Progression of
rebound-associated vertebral fractures following denosumab
discontinuation despite reinstitution of treatment: Suppressing
increased bone turnover may not be enough. J Clin Densitom.
24:338–340. 2021.
|
|
36
|
Luo J, Zhou W, Zhou X, Li D, Weng J, Yi Z,
Cho SG, Li C, Yi T, Wu X, et al: Regulation of bone formation and
remodeling by G-protein-coupled receptor 48. Development.
136:2747–2756. 2009.
|
|
37
|
Shi GX, Zheng XF, Zhu C, Li B, Wang YR,
Jiang SD and Jiang LS: Evidence of the role of R-spondin 1 and its
receptor lgr4 in the transmission of mechanical stimuli to
biological signals for bone formation. Int J Mol Sci.
18:5642017.
|
|
38
|
Lee Y, Kim HJ, Park CK, Kim YG, Lee HJ,
Kim JY and Kim HH: MicroRNA-124 regulates osteoclast
differentiation. Bone. 56:383–389. 2013.
|
|
39
|
Cong F, Wu N, Tian X, Fan J, Liu J, Song T
and Fu H: MicroRNA-34c promotes osteoclast differentiation through
targeting LGR4. Gene. 610:1–8. 2017.
|
|
40
|
Moon JB, Kim JH, Kim K, Youn BU, Ko A, Lee
SY and Kim N: Akt induces osteoclast differentiation through
regulating the GSK3beta/NFATc1 signaling cascade. J Immunol.
188:163–169. 2012.
|
|
41
|
Wang M, Liu J, Zhu G and Chen X: Low
levels of cadmium exposure affect bone by inhibiting Lgr4
expression in osteoblasts and osteoclasts. J Trace Elem Med Biol.
73:1270252022.
|
|
42
|
Manolagas SC and Parfitt AM: What old
means to bone. Trends Endocrinol Metab. 21:369–374. 2010.
|