Introduction
Acute pancreatitis (AP) is a widely observed and
severe clinical condition characterised by the abnormal activation
of enzymes within the pancreas, leading to a series of inflammatory
responses including autodigestion, edema, hemorrhage and
potentially necrosis (1-3). Despite advancements in our
understanding of the pathogenesis of AP pathogenesis, the
prevalence of the disease continues to increase each year, with
~20% of patients progressing to severe AP (SAP). SAP can
subsequently evolve into a systemic inflammatory response and a
multiple organ dysfunction syndrome, ultimately resulting in
considerably high mortality rates with a range of 20-40% (4-7).
At present, the primary emphasis in the management of AP is on
providing supportive care, including fluid resuscitation, pain
management and parenteral nutrition support (4,8,9).
The etiologies underlying the development and progression of AP are
complex, encompassing various apoptotic and necrotic signaling
pathways. Nevertheless, the therapeutic options for targeting the
underlying causes are severely limited. The currently available
medications, such as somatostatin and gabexate, solely serve to
inhibit excessive pancreatic enzyme activity, without effectively
intercepting the premature activation of zymogens, oxidative
stress-induced injuries and inflammatory responses during the
course of AP (10,11). AP is characterised by the abrupt
and extensive destruction of pancreatic acinar cells, encompassing
various apoptotic signaling pathways. Merely targeting a single
pathway is insufficient for effectively treating patients with AP,
particularly those with SAP.
The accumulation of Ca2+ within
pancreatic acinar cells plays a pivotal role in the pathological
progression of AP (12). Various
conditions, including biliary tract obstruction, hyperlipidemia,
hyperparathyroidism, alcoholism and abdominal infection among
others, can facilitate the influx and release of Ca2+
from the endoplasmic reticulum, leading to subsequent
Ca2+-related consequences (13-15). The shared mechanism among the
aforementioned diseases involves excessive accumulation of
cytoplasmic Ca2+, which subsequently triggers the
activation of Ca2+-dependent proteases and lipases.
These enzymes then proceed to hydrolyze the cytoskeleton, leading
to cellular collapse and compromised membrane integrity (16). Consequently, this exacerbates
Ca2+ overload, ultimately amplifying cell death
(17,18). Additionally, the sustained
Ca2+ overload in the mitochondria can induce excessive
production of reactive oxygen species (ROS), resulting in lipid
peroxidation of cell membranes which further enhances the
permeability of the cell membranes and exacerbates the
Ca2+ overload. The interplay between Ca2+
overload and ROS results in a reciprocal reinforcement, leading to
a cascading feedback loop that ultimately culminates in cellular
collapse. Additionally, the occurrence of Ca2+ overload
at an early stage in the process of apoptosis and necrosis
stimulates the expression and release of proinflammatory cytokines,
such as TNF-α and IL-6, by inflammatory cells. It is worth noting
that TNF-α not only initiates inflammatory signaling pathways but
also facilitates exogenous cell apoptosis through its binding to
TNF-receptor 1 (TNF-R1). The activation of TNF-R1 triggers
premature zymogen activation and excessive secretion of cathepsin
B, further exacerbating the cellular response (19,20). The excessive production and
activation of zymogens exacerbate the autodigestion of pancreatic
acinar cells. The increased levels of cathepsin B also contribute
to apoptosis, pyroptosis, ferroptosis, necroptosis and autophagic
cell death (21). Specifically,
an overload of cytoplasmic Ca2+ prompts the fusion of
lysosomes and zymogen granules, which is essential for the
aforementioned pathological processes (20,22,23). Therefore, the fluctuation of
intracellular Ca2+ [Ca2+]i plays a crucial
role in the regulation of TNF-α/cathepsin B-mediated necroptotic
and apoptotic signaling pathways, as well as the modulation of
inflammatory responses (23-26). Moreover, an excessive increase in
[Ca2+]i leads to the proliferation of ROS, which
disrupts the functioning of both the cytoplasmic and mitochondrial
membranes, ultimately resulting in cell apoptosis or necrosis
(27,28). Therefore, promptly restoring
[Ca2+]i levels from an overloaded state
(10−5-10−4 mol/l) to a resting state
(~10−7 mol/l) presents a viable approach to mitigate
cellular damage and rescue endangered cells.
In order to achieve this goal,
1,2-bis(2-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid tetrakis
(acetoxymethyl ester) (BAPTA-AM), a cell-permeant Ca2+
chelator, has emerged as a suitable candidate for the management of
AP, as opposed to Ca2+ channel blockers (CCBs) (16). CCBs have limited efficacy in
mitigating Ca2+ overload and do not impact
Ca2+ release from intracellular stores (29). The pathogenesis of acinar cell
death in AP is intricate and interconnected. Targeting a single
aspect of the pathogenesis of AP is insufficient to effectively
halt its progression. Calcium overload serves as a pivotal factor
in this intricate pathogenic process (Fig. S1). BAPTA-AM, a compound capable
of penetrating acinar cells, undergoes hydrolysis by esterase
enzymes upon cellular entry, transforming into the active chelator
BAPTA, which can then selectively and rapidly pair with
Ca2+, thus diminishing the intracellular Ca2+
overload (Fig. S2) (30). Nevertheless, the hydrophobic
nature of BAPTA-AM poses a notable obstacle to its direct use for
the preservation of endangered cells. To address this issue,
liposomes, particularly nanoscale liposomes, have been employed to
enhance the solubility of hydrophobic drugs, modify drug
absorption, prolong biological half-life and imbue more specific
targeting and biocompatible characteristics. Additionally, the
lipid bilayer of liposomes serves as a protective barrier,
preventing extracellular nonspecific hydrolysis of ester compounds
such as BAPTA-AM. Consequently, the ability of BAPTA to chelate
Ca2+ in the extracellular fluid is rendered ineffective.
Given the advantages of liposomes and the good solubility of
BAPTA-AM in lipids, BAPTA-AM liposome nanoparticles (BLNs) were
prepared in the present study.
Our previous studies demonstrated the promising
therapeutic potential of BAPTA-AM in mitigating ischemia
reperfusion-induced acute kidney injury and
D-GalN/lipopolysaccharide (LPS) induced fulminant hepatic failure
through the reduction of overloaded [Ca2+]i (31,32). In the present study, the rescuing
effects of BLN both in vitro and in vivo were
assessed. The evaluation of survival rates, pancreatic function
indicators and pancreas microcirculation were examined.
Additionally, the impact of BLN on the pathways of pancreatic
necrosis and apoptosis was performed to elucidate the underlying
protective mechanism.
Materials and methods
Materials
BAPTA-AM (>95% pure), L-α-phosphatidylcholine,
cholesterol, sodium oleate, sodium cholate, glucose and ulinastatin
were purchased from Shanghai Aladdin Biochemical Technology Co.,
Ltd. Thiazolyl blue and lipopolysaccharide (LPS) were obtained from
Sigma-Aldrich; Merck KGaA. Biochemical test kits used for α-amylase
(AMS), triglycerides (TGs), lactate dehydrogenase (LDH),
malondialdehyde (MDA) and superoxide dismutase (SOD) were obtained
from the Nanjing Jiancheng Bioengineering Institute. ELISA
quantification kits for TNF-α (cat. no. ek0526) and IL-6 (cat. no.
ek0412) were obtained from Wuhan Boster Biological Technology, Ltd.
Annexin V-FITC/PI kits, Kaighn's modified Ham's F-12 Medium (F12K),
Dulbecco's modification of Eagle's medium (DMEM) and D-Hanks buffer
were obtained from Procell Life Science & Technology Co., Ltd.
Fetal bovine serum (FBS) was purchased from Thermo Fisher
Scientific, Inc. Fura-3/AM was purchased from Shanghai Beyotime
Biotech Co., Ltd. Primary antibodies against TNF-α (cat. no.
ab6671), cathepsin B (cat. no. ab214428) and goat anti-rabbit IgG
H&L (HRP; cat. no. ab6721) were purchased from Abcam;
TRIzol® reagent was purchased from Thermo Fisher
Scientific; BioRT cDNA First Strand Synthesis kit was purchased
from Hangzhou Bioer Co., Ltd. and primers targeting cathepsin B,
TNF-α and β-actin mRNA were synthesized by Shanghai Sangon Biotech
Co., Ltd.
Preparation and characterization of
BLN
BLN was prepared using a thin-film hydration method
followed by an extrusion technique (31). Briefly, a mixture of 100 mg
L-α-phosphatidylcholine, 5 mg BAPTA-AM, 10 mg cholesterol and 15 mg
Tween 80 was dissolved in 10 ml chloroform in an eggplant flask.
The chloroform was then evaporated slowly under reduced pressure at
50°C using rotary evaporation, resulting in a uniform lipid film.
Subsequently, the lipid film was hydrated by adding 10 ml PBS and
agitating at 40°C for 1 h, leading to the formation of a primary
suspension. The primary suspension was further processed by
extruding it through a 200-nm polycarbonate membrane for three
cycles to obtain nanoscale liposomes. The mean particle size,
polydispersity index (PDI) and ζ potential of BLN in a 5% glucose
injection were measured using a Malvern Zetasizer NanoZS90 (Malvern
Instruments Ltd.). The morphology of BLN was analyzed using
scanning electron microscopy (SEM; JSM-6510LV, JEOL, Ltd.). BLN was
diluted to a concentration of 0.01 mg/ml, and ~10 μl of the
dilution (equivalent to ~one drop) was carefully dripped onto the
substrate. Subsequently, the solution was allowed to slowly
disperse and dry at room temperature under a nitrogen atmosphere in
order to facilitate uniform spreading. Finally, gold coating was
applied on BLN using vacuum-assisted spraying technique. SEM
detection parameters included: Voltage set at 5 kV, horizontal
field width of 25.4 μm, working distance maintained at 9.4
mm, SEM mode used for imaging and pressure maintained at 3.00e-6
Torr. The encapsulation efficiency of BAPTA-AM in liposome
nanoparticles (LNs) was assessed using microcolumn centrifugation.
Specifically, BLN was added and allowed to seep into a microcolumn
containing Sephadex G-50. The entire assembly was then centrifuged
at room temperature (600 × g for 3 min), with distilled water
serving as the eluent. These procedures were conducted in
triplicate. The unencapsulated free BAPTA-AM was separated from BLN
by passing it through a microcolumn and determined with
high-performance liquid chromatography (HPLC; 1260, Agilent
Technologies, Inc.). The HPLC analysis was performed on a C18
Column (150×4.6 mm i.d., 3.5 μm) with a detection wavelength
of 250 nm. Elution consisted of 65:35 (v/v) methanol/water with
ammonium acetate (50 mmol/l) as the mobile phase initiated at a
flow rate of 1 ml/min at 30°C. The volume of each injection was 20
μl. Encapsulation efficiency (EE) was calculated using the
equation: EE
(%)=(BAPTA-AMtotal-BAPTA-AMunencapsulated)/(BAPTA-AMtotal)
×100.
Liposomes with 7.5% (w/v) trehalose as a
lyoprotectant were freeze-dried on a scheduled procedure using an
Alpha 1-2 LD Plus lyophilizer (Martin Christ Inc.). Size
distribution and drug leakage were analyzed after reconstitution of
the lyophilized BLN in PBS (pH 7.4) solution.
Cell culture
The rat pancreatic exocrine cell line AR42J and the
murine macrophage cell line RAW264.7 were obtained from Shanghai
Institutes for Biological Sciences, The Cell Bank of Type Culture
Collection (The Chinese Academy of Sciences), and were used in the
experiments to evaluate the protective effects of BLN in the AP
cell model. AR42J and RAW264.7 cells were plated in 25
cm2 culture flasks at 37°C in a humidified incubator
supplied with 5% CO2. AR42J cells were cultured in F12K
supplemented with 20% FBS, while RAW264.7 cells were cultured in
DMEM supplemented with 10% FBS. AR42J cells were passaged every 5-7
days, and RAW264.7 cells were passaged every 3-4 days. AR42J and
RAW264.7 cells were subcultured at a ratio of 1:3 and 1:6,
respectively, when they reached 80% confluency.
Establishment of the pancreatitis cell
model
An AP cell model based on a patent (CN114250194A;
http://epub.cnipa.gov.cn) was constructed. AR42J
cells were exposed to F12K containing the toxins of HGO (25-50
mmol/l high glucose plus 100-400 μmol/l sodium oleate) at
37°C for a duration of 24 h. MTT assays were used to evaluate the
extent of cell damage. MTT solution (20 μl; 5 mg/ml) was
added to each well. Following a 4-h incubation at 37°C, the medium
was replaced with 150 μl dimethyl sulfoxide to dissolve
formazan crystals. Optical densities were measured at 490 nm with
microplate reader (SpectraMax M2e; Molecular Devices).
Protective effect of BLN against
HGO-induced cell damage
AR42J cells in the logarithmic phase were seeded in
a 96-well plate (5×104 cells/well) and divided into
seven groups after 24 h incubation at 37°C. In the present study,
five treatment groups were respectively pretreated with 1,000 U/ml
ulinastatin (UTI), blank NPs (nanoparticles without BAPTA-AM), or
10, 50 or 250 nmol/l BLN for 0.5 h before HGO treatment. AR42J
cells without any drug or HGO treatment were used as the normal
control group, while cells exposed to HGO but not to BLN were
considered the model group. A total of 24 h after HGO treatment,
the protective effects of BLN against HGO-induced cell damage were
evaluated using an MTT assay.
Inhibitory effect of BLN on HGO-induced
cell apoptosis and necrosis
For collecting a sufficient quantity of cells, the
AR42J cells were incubated in 6-well plates (1×106
cells/well) and divided into seven groups as aforementioned. After
24 h of exposure to HGO, the cells underwent gentle digestion (at
37°C for 5 min) and centrifugation (300 × g for 4 min) at room
temperature. Subsequently, the cells were rinsed twice with cold
PBS before being resuspended in 500 μl binding buffer
(2×105 cells). Following this, the cells were stained
with 5 μl of Annexin V and 5 μl of PI from the kit at
a temperature of 4°C for 15 min in the dark. The Flow cytometer
(FCM; FACSCanto II; BD Biosciences) and Flowjo V10 software (FlowJo
LLC) were then used to determine the percentage of cells in the
apoptotic stage.
Effect of BLN on intracellular
Ca2+ overload
To investigate the Ca2+ scavenging effect
of BLN on intracellular Ca2+ overload, intracellular
Ca2+ levels were detected in AR42J cells following
HGO-induced damage. Briefly, AR42J cells were seeded in 24-well
plates (1×105 cells/well) and grouped as aforementioned.
After 4 h HGO exposure, the culture medium was removed, cells were
gently washed with D-Hanks buffer once and Fura-3/AM was added to a
final concentration of 5 μmol/l. After incubated with
Fura-3/AM for 2, 5, 10, 15 or 20 min, the cell images were
respectively captured using an inverted fluorescence microscope
(TE2000-U; Nikon Corporation). Image J (version 1.48; National
Institutes of Health) was used to analyze the relative fluorescent
intensity (RFI). The changes in RFI in the cells represented the
fluctuation of intracellular Ca2+ concentration.
Inhibitory effect of BLN on HGO-induced
secretion of AMS from AR42J cells
AMS is primarily derived from pancreatic acinar
cells and its excessive release is one of the characteristic
indicators in the diagnosis of AP. The cells were divided and
treated as described earlier, and the culture media was collected
at 9 h after the HGO exposure. The detection of AMS in the culture
media was performed in accordance with the protocol provided by the
manufacturer.
Effect of BLN on HGO-induced ROS
overproduction in AR42J cells
ROS are involved in numerous physiological
processes. However, the overproduction of ROS also activates
apoptotic pathways. After 9 h of HGO exposure, the ROS indicator
2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA) (1:1,000) was
introduced to the AR42J cells following the guidelines provided by
the manufacturer. When the cells were incubated with the
fluorescent probe for another 30 min, the images of
dichlorodihydrofluorescein (DCF) labeled cells were captured. Image
J was used for quantitative analysis of the DCF fluorescence
intensity. The areas with RFI values >15 and >35 were
respectively regarded as the cell total and the region of ROS
strongly positive cells.
Effect of BLN on the activation of
inflammatory cells and overexpression of inflammatory
cytokines
The RAW264.7 macrophage cell line is frequently used
to explore inflammation. Upon being stimulated with LPS or cell
debris, the macrophage cells were rapidly activated, and the
release of various inflammatory cytokines, including IL-6 and
TNF-α, amplifies the inflammatory response and further exacerbates
existing cell damage. In the present study, the inhibitory effect
of BLN on inflammatory responses was explored. The RAW264.7 cells
were seeded in a 24-well plate and divided into seven groups. In
addition to the normal group, the model, UTI, blank NP and BLN
treatment groups were exposed to 100 ng/ml LPS or AR42J cell debris
(~5,000 apoptotic or necrotic cells/ml) to induce inflammatory
responses. The model group was exposed to the stimuli but without
any pretreatment, while the normal group consisted of cells without
the stimuli and BLN intervention. After a further 9 h of
incubation, the culture media was collected. ELISA was performed to
detect the expression of IL-6 and TNF-α according to the
manufacturer's protocol. The remaining RAW264.7 cells were fixed
with 4% paraformaldehyde at 4°C for 15 min. After removing the
fixative, a solution of Wright-Giemsa stain (0.5 ml) was added at
room temperature incubated for 5 min. Subsequently, the staining
solution was discarded, and the cells were washed three times with
cold PBS before being observed under a light microscope (Nikon
Eclipse Ti-S; Nikon Corporation) at a magnification of ×200 to
assess morphological changes.
Effect of BLN on inflammatory
microenvironment induced AR42J cell apoptosis
The RAW264.7 cells were seeded in a 6-well plate at
a density of 1×106 cells/well. After incubating for 24
h, the cells were exposed to either LPS or AR42J cell debris. Then,
the culture media were collected and centrifuged at room
temperature (900 × g for 5 min). The supernatant, which was used to
mimic an inflammatory microenvironment, was used for culturing the
AR42J cells for a further 24 h. The AR42J cells cultured in
supplemented DMEM were used as the normal control group, while the
cells exposed to the aforementioned supernatant with or without BLN
(250 nmol/l) were respectively defined as the model and BLN groups.
FCM was used to determine the proportion of apoptotic cells.
Animal studies
Healthy Sprague-Dawley (SD) rats weighing 250±20 g
and aged 8-10 weeks, with an equal distribution of males and
females were purchased from the Zhejiang Center of Laboratory
Animals (Hangzhou, China), and the experiments reported in the
present study were performed in accordance with The Health Guide
for Care and Use of Laboratory Animals, Zhejiang Center of
Laboratory Animals. All animals were acclimated to the laboratory
for ≥1 week before the experiments were performed and housed in a
light-controlled room (12 h light/dark cycle) at an ambient
temperature of 25°C with ad libitum access to water and
standard food.
Establishment of the sodium taurocholate
(TC)-induced AP rat model
The in vivo retrograde injection of TC into
the common biliopancreatic duct is a widely accepted experimental
model for mimicking AP (33,34). This model involves a sequential
process characterized by excessive activation and release of
pancreatic enzymes, inflammatory cascades and eventually,
pancreatic necrosis. The AP model in the present study was
established as described by Aho et al (35) and Lange et al (36). A laparotomy in the midline was
performed, using intraperitoneal injection of 2% sodium
pentobarbital anesthesia at a dosage of 40 mg/kg (the anesthetic
took effect within 15 min), and an injection needle was then
inserted into the common biliopancreatic duct. The hepatic duct was
closed using small hemostatic forceps and tightened with sutures
around the needle and the wall of the duct. Steady manual pressure
was applied to inject 0.2 ml saline or TC diluted in saline at a
concentration of 4% into the pancreatic duct system for a period of
>60 sec. Following a 5-min dwell time, the needle was removed
after which the abdomen was sutured layer by layer.
Effect of BLN on the survival rate of the
rats in the animal model of AP
A total of 64 rats were randomly allocated into the
following eight groups, each consisting of eight animals: Normal,
sham, model, UTI (20,000 U/kg), blank NP (NPs without BAPTA-AM) and
three BLN treatment groups (75, 150 and 300 μg/kg). UTI is a
trypsin inhibitor for treating AP and is included in the present
study for comparison with BLN (37,38). All animals, except those in the
normal group, underwent laparotomy and received either saline or TC
solution injected into the common biliopancreatic duct, which
connects the liver and pancreas. Mortality rates for each group
were monitored at 0, 6, 12, 18, 24, 30, 36, 42 and 48 h after TC
administration.
Effect of BLN on the biochemical
indices
After being exposed to TC for 12 h or before
sacrificing, 1.2-1.4 ml blood samples were collected and placed in
an EP tube containing sodium citrate anticoagulant via tail-tip
amputation. The expression levels of several indicators of
pancreatic function, including AMS, LDH, TGs, TNF-α and IL-6 in the
serum were measured according to the manufacturer's protocol. At
the experimental endpoint or prior to sacrifice, the rats were
transferred to a CO2 euthanasia chamber within their
respective home cages and were euthanized using a CO2
flow displacement rate of 30% per min. This process was continued
until every rodent exhibited lack of respiration and faded eye
color. Subsequently, the pancreas tissues were isolated and
weighed. A portion of the tissues was used to prepare tissue
homogenate to measure oxidative and anti-oxidative indicators, such
as MDA, glutathione (GSH) and SOD.
Histopathological evaluation of the
rescuing effect of BLN on AP
Histopathological changes in the pancreatic tissue
can indicate the extent of necrosis. The pancreatic tissues were
fixed in 10% buffering formalin at 4°C for 24 h, embedded in
paraffin, sliced into 5-μm-thick sections. The slices were
subjected to a sequential washing procedure, commencing with
xylene, followed by a series of ethanol solutions and distilled
water. Subsequently, the sections were subjected to hematoxylin
staining for a duration of 5 min at room temperature, followed by
rinsing with tap water, and then stained with eosin staining
solution for another 2 min. The slices were subsequently dehydrated
and rendered transparent by sequential immersion in alcohol and
xylene, and finally sealed with neutral gum. All pancreatic
sections were observed using light microscopy (Nikon Eclipse Ti-S;
Nikon Corporation), and images were captured in a blinded manner.
Edema, inflammatory cell infiltration, hemorrhage, necrosis and
other degenerative changes were used to assess pancreatic
histology. The extent of pancreatic damage was graded on a scale
from 0-III: 0, normal tissue; I, swelling can be observed in part
of the pancreatic tissue but without obvious necrosis in the visual
field; II, more swelling can be observed in the pancreatic tissue
with visible spotty necrosis and inflammatory cell infiltration in
the portal area, and necrosis in <1/2 of the area; and III,
massive necrosis and swelling of the pancreatic tissue, and
necrosis in >1/2 of the area (31,39,40).
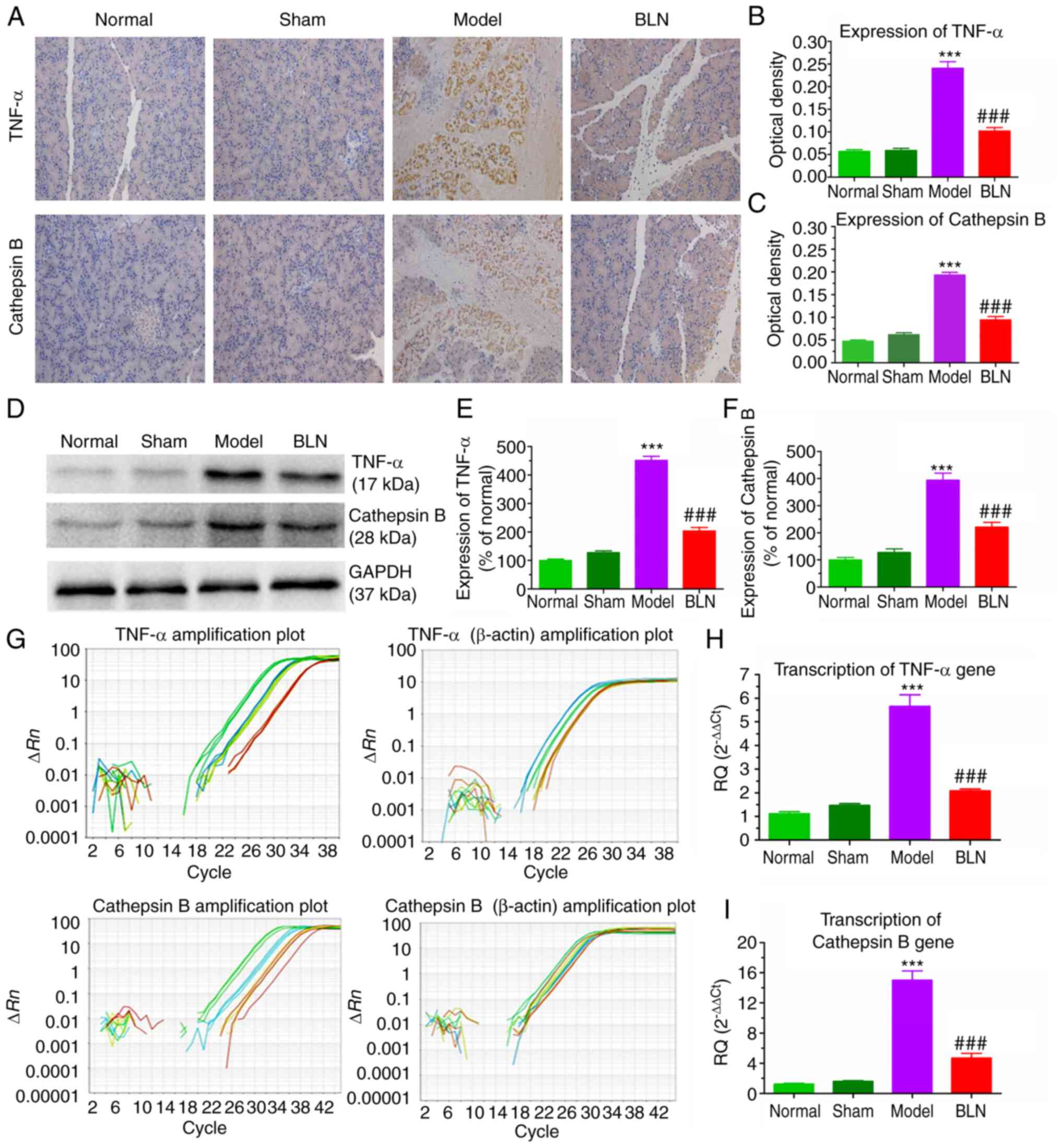
Immunohistochemical analysis for TNF-α
and cathepsin B protein expression
The premature activation of trypsinogen in
pancreatic acinar cells, followed by the activation of other
pancreatic zymogens, such as chymosinogen and prophospholipase, is
considered to cause autodigestion of the gland, which is a crucial
pathological change in AP (41,42). The mechanisms underlying
trypsinogen activation in AP are not entirely clear. However,
hypercalcemia, TNF-α and cathepsin B are hypothesized to play
notable roles (43). TNF-α and
cathepsin B can trigger both necrotic and apoptotic processes,
which are mediated by receptor-interacting protein kinases.
Meanwhile, TNF-α and cathepsin B are closely involved in
inflammatory responses, which consist another type of pathological
processes in AP. The pancreatic tissues were fixed with 10%
buffered formalin, embedded in paraffin, and then sliced into 5
μm-thick sections. Immunohistochemical staining was
performed using a standard peroxidase anti-peroxidase method, with
diaminobenzidine (DAB) as the chromogen. After dewaxing and
rehydration, sections were subjected to a citrate water bath at a
temperature range of 96-98°C for a duration of 15 min to recover
antigens. To inhibit endogenous peroxidase activity, the sections
were exposed to a 3% H2O2 solution at room
temperature for 10 min. Additionally, nonspecific binding was
prevented by treating the sections with 2% bovine serum albumin
(BSA) at room temperature for 30 min. Subsequently, the sections
were incubated with the primary antibody (dilution, 1:500)
overnight at 4°C, followed by incubation with the secondary
antibody (dilution, 1:2,000) at room temperature for 10 min.
Finally, the sections were stained with DAB. All slides were
observed under a light microscope (Nikon Eclipse Ti-S; Nikon
Corporation) and images were obtained for further analysis. The
percentage of TNF-α and cathepsin B-stained areas (brown colored)
was calculated using Image J. The proportion of positive area to
total area was calculated and used as the relative expression
level. The mean values from 10 fields of each section were taken as
the individual rat score (31).
RNA isolation, cDNA synthesis and reverse
transcriptionquantitative (RT-q)PCR
The total RNA was extracted from pancreatic tissues
using TRIzol® reagent (Thermo Fisher Scientific, Inc.),
following the manufacturer's protocol. High-purity RNAs (2.0≥
A260/280 ≥1.8) were used for cDNA synthesis, with ≤0.8 μg
RNA converted to cDNA based on the BioRT cDNA First Strand
Synthesis kit (Hangzhou Bioer Technology Co., Ltd.) protocol, using
RT temperature of 42°C for 60 min. Gene transcription was
quantified through qPCR using a Step One™ Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.), along with
template cDNA, forward/reverse primers and Maxima SYBR Green qPCR
MasterMix reagent. The sequences of the primer are listed in
Table SI. Thermocycling was
carried out following the programmed procedure: Uracil DNA
glycosylase pretreatment for 2 min at 50°C, 5 min at 95°C, followed
by 40 cycles of 95°C for 15 sec, 60°C for 30 sec and 72°C for 30
sec, with a final dissociation stage at 95°C. After running SYBR
Green PCR, data collection and analysis were performed with the
fluorescence intensity (Rn) detected at the end of every cycle. The
threshold cycle (Cq) values are inversely associated
with the number of target cDNA copies. Quantification was
calculated using the 2−ΔΔCq value (44). Cq values of the target
genes were compared with the Cq value of the endogenetic
gene (EG) as follows:
∆Cq=Cq(TG)-Cq(EG) and
∆∆Cq=∆Cq(BLN)-Cq(Normal).
Statistical analysis
All data are presented as mean ± SD. One-way ANOVA
followed by Dunnett's post hoc multiple comparison tests, two-way
ANOVA followed by Tukey's post hoc multiple comparison tests and
log-rank test were used to determine the significance of
differences between experimental groups in GraphPad Prism (version
5.0; Dotmatics). P<0.05 was considered to indicate a
statistically significant difference.
Results
Characterization of BLN
The morphology of BLN was visualized using SEM, as
shown in Fig. 1A, revealing BLN
to be spherical vesicles with a mean size of 113 nm. The size
distribution was narrow, as confirmed by dynamic light scattering
(Fig. 1B), with a mean diameter
of 121.0±7.9 nm and a PDI of 0.17±0.01, consistent with SEM
measurements. The ζ potential of the BLN utilized in this
experiment was -26.61±0.52 mV (Fig.
1C), a moderate potential for liposome stabilization. The
encapsulation efficiency (EE) of BAPTA-AM was 95.3±2.1%. It was
found that 7.5% trehalose (w/v) was an effective cryoprotectant for
BLN. Notably, the lyophilization process did not significantly
affect the size or the surface charge of BLN, and drug leakage was
negligible upon reconstitution, facilitating long-term storage of
this formulation (Table
SII).
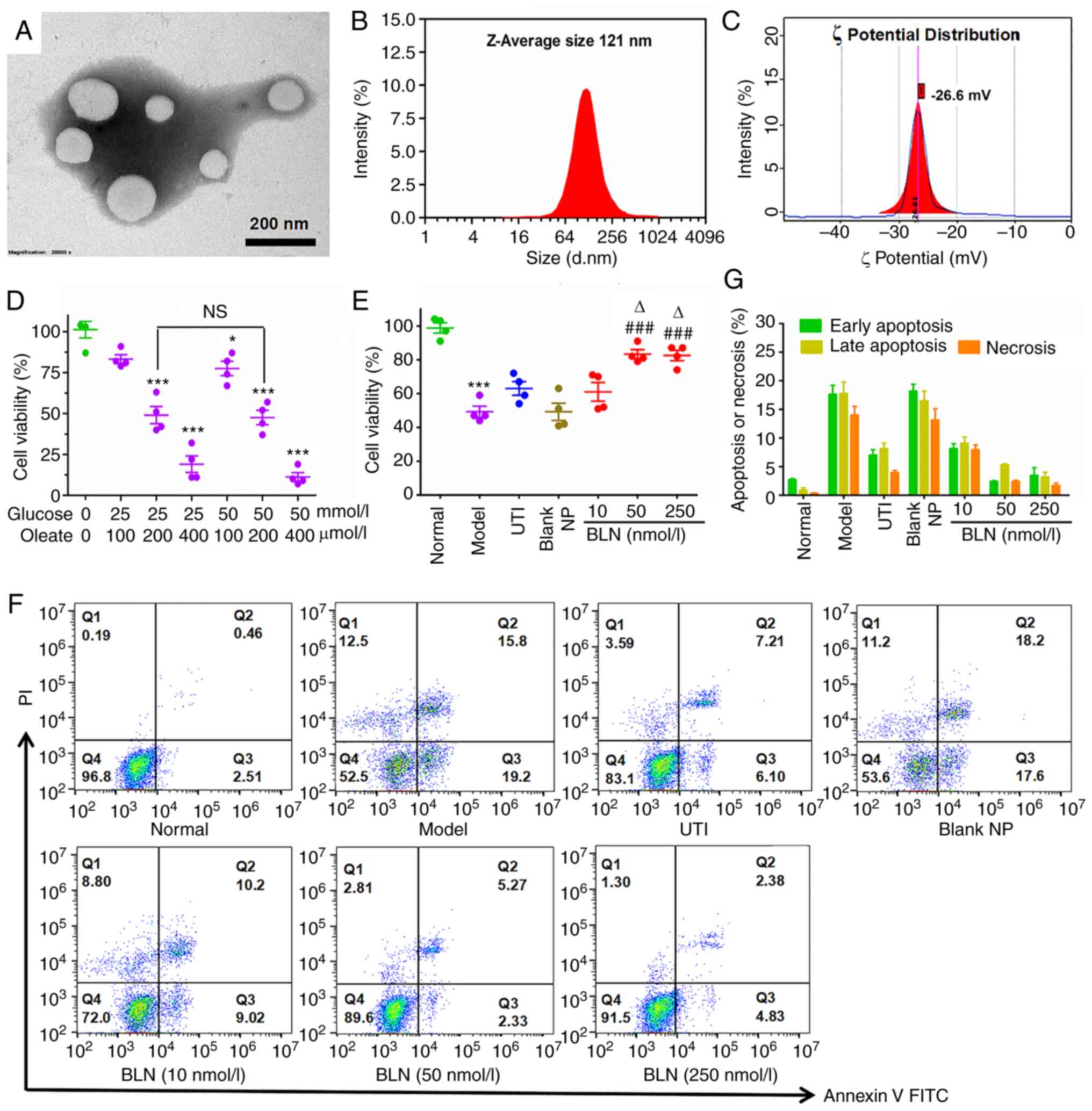 | Figure 1Characterization of BLN and its
protective effect on HGO-induced AR42J cell damage. (A) Morphology
of BLN using scanning electron microscopy. Scale bar, 200 nm. (B)
Size distribution of BLN measured by DLS. (C) Surface charge of
BLN. (D) Cytotoxicity screening of HGO in AR42J cells. (E) Cell
viability of HGO-treated AR42J cells following BLN treatment
measured using an MTT assay. (F) Cell apoptotic stages classified
by flow cytometry. Early and late apoptotic cells are respectively
presented in Q3 and Q2, necrotic cells in Q1 and normal cells in
Q4. (G) Quantitative analysis for necrosis or apoptosis based on
flow cytometry. *P<0.05, ***P<0.001 vs.
normal group; ###P<0.001 vs. model group,
∆P<0.05 vs. UTI group. Data are presented as the mean
± SEM of four repeats. BLN, BAPTA-AM loaded liposome nanoparticles;
HGO, high glucose 25-50 mmol/l plus sodium oleate 100-400
μmol/l; DLS, dynamic light scattering; UTI, ulinastatin; NP,
nanoparticle; BAPTA-AM,
1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid tetrakis
(acetoxymethyl ester). |
BLN prevents HGO-induced pancreatic
acinar cell damage
As shown in Fig.
1D, HGO (25 mmol/l high glucose + 200 μmol/l sodium
oleate) exposure resulted in a modest amount of cell damage, which
was used to establish the AP cell model. Compared with the model
group, the cell viability was significantly increased in the groups
pretreated with BLN at a dose range of 10-250 nmol/l, particularly
at a dose range of 50-250 nmol/l (Fig. 1E). FCM was also used to assess
the viability of cells following BLN pretreatment. As shown in
Fig. 1F and G, BLN pretreatment
resulted in a significant reduction in the proportion of apoptotic
or necrotic cells in a dose-dependent manner, which was consistent
with the trend observed in the MTT assay.
BLN reduces Ca2+ overload in
HGO-induced pancreatic acinar cell damage
The increase in Ca2+ levels in acinar
cells is one of the initiating factors in the premature activation
of zymogens and overproduction of ROS following HGO exposure
(45,46). RFI was used to indicate the
intracellular Ca2+ concentration. In the model group, a
sudden increase in RFI was observed compared with the normal group,
which suggested that the cells had become overloaded with
Ca2+ in the model cells. Following BLN treatment, the
RFI showed a dose-dependent decrease (Fig. 2A). Treatment with 50-100 nmol/l
BLN reversed this Ca2+ overload back to Ca2+
levels observed in the resting state. However, UTI did not result
in the inhibition of Ca2+ influx. This result shows the
potent Ca2+ scavenging ability of BLN, which is a
prerequisite for the treatment of damaged cells.
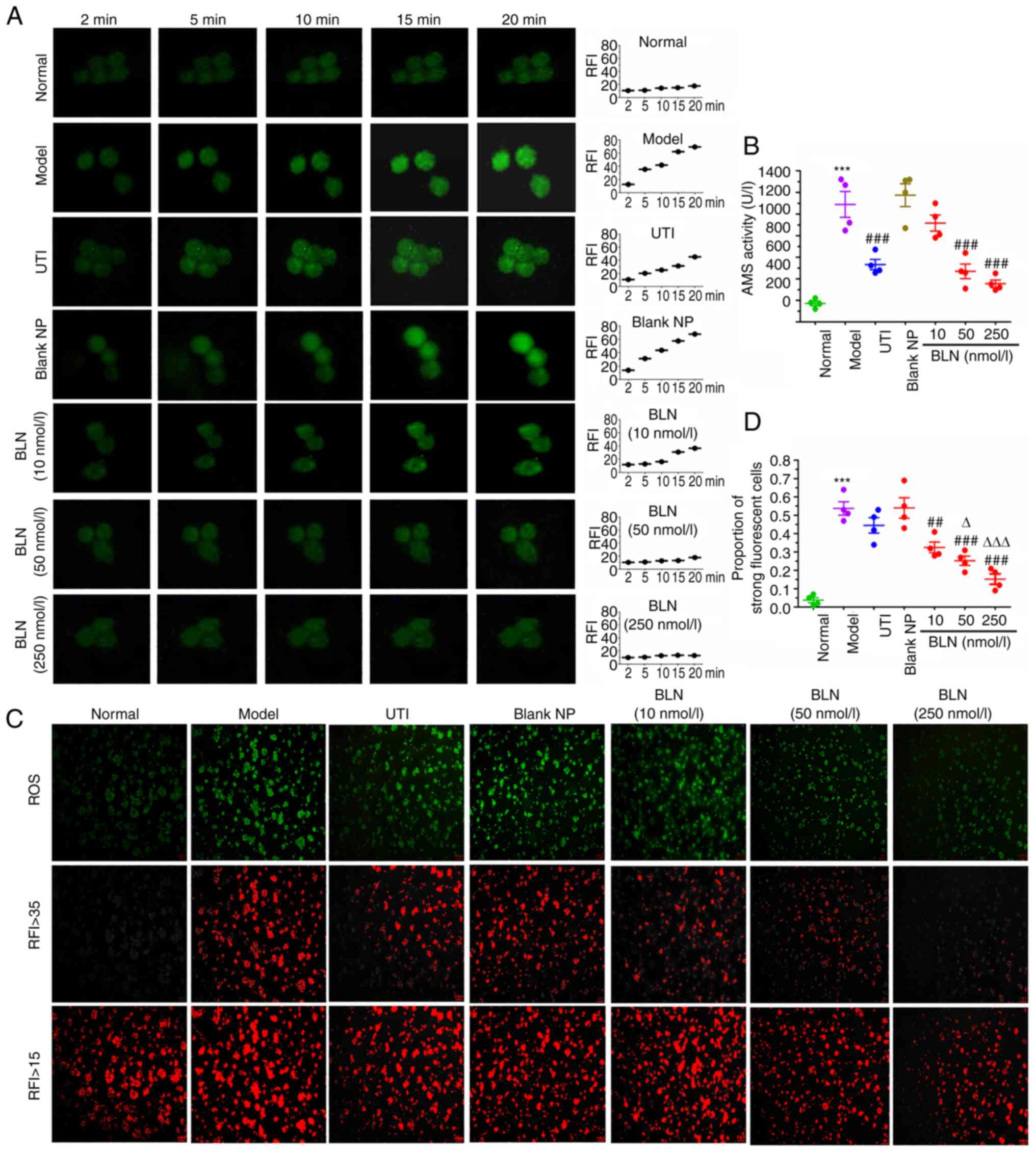 | Figure 2Inhibition of HGO-induced calcium
overload, amylase over secretion, and ROS overproduction in AR42J
cells. (A) Changes in intracellular [Ca2+] in
HGO-treated AR42J cells following BLN treatment. Scale bar, 5
μm. (B) BLN exhibited an inhibitory effect on HGO-induced
amylase over secretion. (C) Representative images of ROS-induced
fluorescence response; RFI values >15 and >35 were
respectively regarded as the cell total and the region of ROS
strongly positive cells. Scale bar, 100 μm. (D) Quantitative
analysis of DCF relative fluorescence intensity.
***P<0.001 vs. normal group; ##P<0.01,
###P<0.001 vs. model group; ∆P<0.05,
∆∆∆P<0.001 vs. UTI group. Data are presented as the
mean ± SEM of four repeats. BLN, BAPTA-AM loaded liposome
nanoparticles; HGO, high glucose 25-50 mmol/l plus sodium oleate
100-400 μmol/l; UTI, ulinastatin; RFI, relative fluorescence
intensity; ROS, reactive oxygen species; BAPTA-AM,
1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid tetrakis
(acetoxymethyl ester); NP nanoparticle; DCF, dichlorofluorescein;
AMS, amylase. |
BLN inhibits HGO-induced AMS
oversecretion from AR42J cells
The abnormal elevation of plasma pancreatic AMS is
an important indicator of pancreatitis (47,48). Excessive secretion and premature
activation of pancreatin can lead to the autodigestion of
pancreatic tissue, resulting in tissue bleeding and necrosis. The
effects of BLN on AMS expression and release are shown in Fig. 2B. Compared with the normal group,
AMS secretion in the culture media of the model group increased
nearly 6-fold after 9 h of HGO co-incubation. However, pretreatment
with BLN at dose range of 50-250 nmol/l significantly inhibited AMS
expression by 57-70%.
BLN alleviates oxidative stress
Oxidative stress is regarded as a major causative
factor in AP. To determine the effect of BLN on ROS production,
intracellular ROS levels were assessed using DCF as a fluorescent
indicator. Intracellular ROS levels were increased after 9 h of HGO
exposure (Fig. 2C), with the
majority of cells presenting strong yellow-green fluorescence. The
proportion of area with RFI values ≥35 in the model group was
48-71%. BLN significantly suppressed ROS overproduction in a
dose-dependent manner by 46-80% (Fig. 2D). These results indicated that
BLN could reduce the cytotoxicity caused by ROS.
BLN inhibits LPS-induced inflammatory
responses in RAW 264.7 macrophages
As shown in Fig. 3A
and B, RAW264.7 cells exposed to LPS or AR42J cell debris for 9
h, exhibit a notable morphological change, characterized by mass
pseudopodia protrusions, indicating activation of the macrophages
compared with the resting state observed in the cells in the normal
group. This activation of inflammatory cells is a prerequisite for
the release of inflammatory factors. Intervention with BLN (10-250
nmol/l) was found to have a notable anti-inflammatory effect and
maintained the cells at rest in a dose-dependent manner.
Inflammatory cytokines, such as TNF-α and IL-6, play a crucial role
in AP initiation. As shown in Fig.
3C and D, TNF-α and IL-6 levels in RAW264.7 cells exposed to
100 ng/ml LPS or AR42J cell debris were significantly increased up
to 3-8-fold the normal level. However, BLN treatment significantly
inhibited the inflammatory responses, especially when the cells
were treated with BLN at concentrations ≥50 nmol/l. UTI did not
alleviate the inflammatory responses. Furthermore, the inflammatory
microenvironment aggravated apoptosis. The supernatant rich in
TNF-α and IL-6 from activated RAW264.7 cell culture medium
significantly promoted the occurrence of apoptosis. BLN (250
nmol/l) blocked this apoptotic process (Fig. 3E and F). These results suggest
that BLN treatment can effectively prevent macrophage activation
and block LPS or cell debris-stimulated inflammatory responses.
These findings highlight the protective effect of BLN against cell
apoptosis or necrosis.
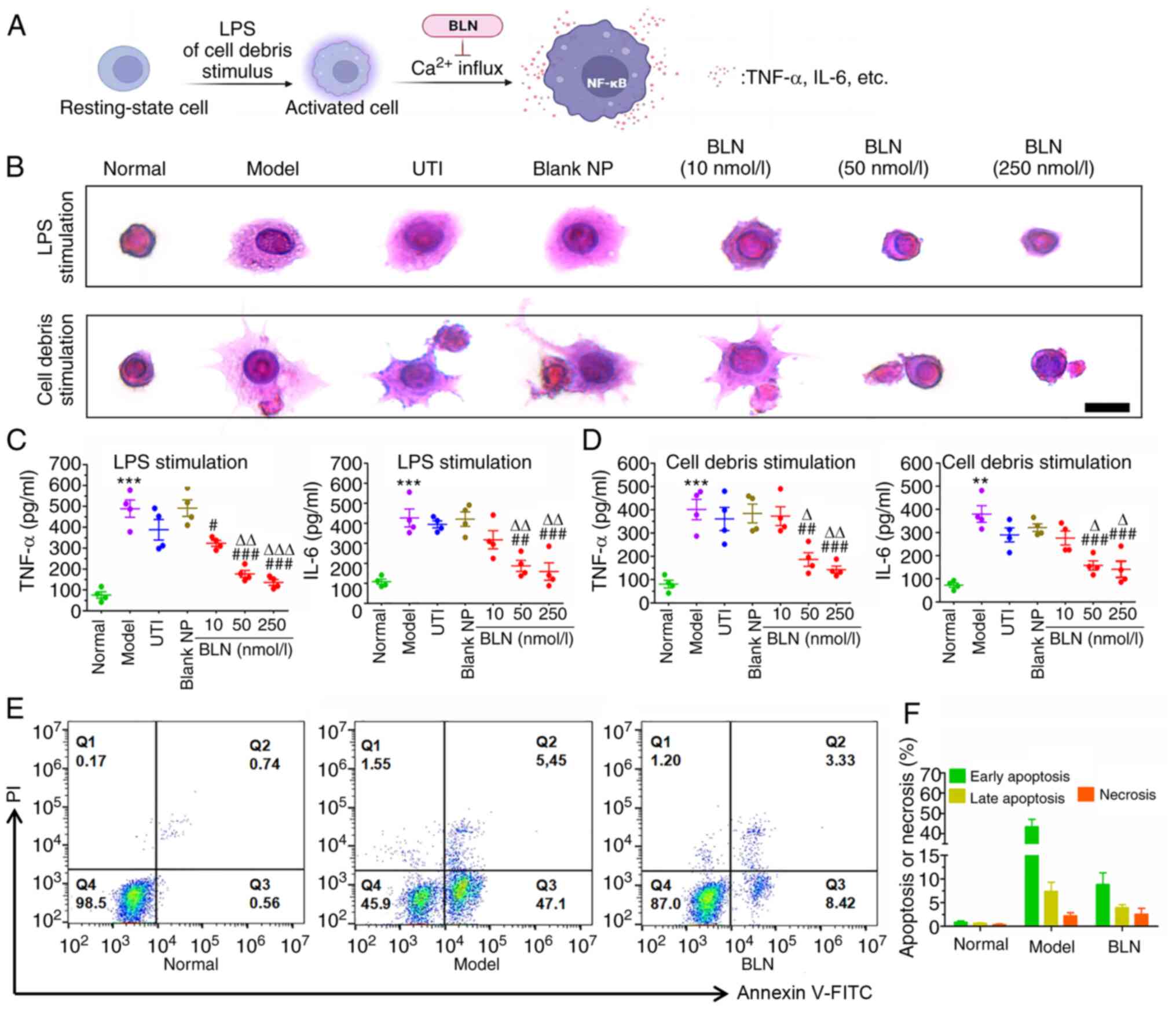 | Figure 3Inhibition of LPS and AR42J cell
debris-stimulated inflammatory responses in RAW264.7 macrophages
following BLN treatment. (A) Schematic diagram of the massive
calcium influx promoting inflammatory cytokine release from
activated monocytes. (B) LPS and cell debris-induced RAW264.7 cells
morphological changes. Scale bar, 10 μm. (C) LPS and (D)
cell debris exposure induced TNF-α and IL-6 expression in RAW264.7
cells. (E) Cell apoptotic stages classified by flow cytometry. (F)
Quantitative analyses of necrosis or apoptosis based on flow
cytometry. Data are presented as the mean ± SEM of four repeats.
**P<0.01, ***P<0.001 vs. normal group,
#P<0.05, ##P<0.01,
###P<0.001 vs. model group, ∆P<0.05,
∆∆P<0.01, ∆∆∆P<0.001 vs. UTI group.
BLN, BAPTA-AM loaded liposome nanoparticles; UTI, ulinastatin; LPS,
lipopolysaccharide; NP, nanoparticle; BAPTA-AM,
1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid tetrakis
(acetoxymethyl ester). |
Effect of BLN on the survival rate in the
animal model of AP
AP is characterized by a rapid deterioration in
health and a high mortality rate. As shown in Fig. 4A, the survival rate of the model
group decreased sharply after the first death occurred 12 h post-TC
assault via retrograde injection of TC into the pancreatic duct.
The survival rate plummeted to 50% after 24 h and to 37.5% after 48
h. However, BLN (75-300 μg/kg) treatment significantly
extended the survival time, and no TC-treated rats died in the
first 24 h, which is a critical period for rescuing patients with
AP. Treatment with BLN at a dose of 150 μg/kg resulted in a
terminal survival rate of 75% at the experimental endpoint, a
significant improvement compared with the model group's rate of
only 37.5%. Furthermore, BLN (150 μg/kg) had a superior
rescuing effect compared with that of UTI, a commonly used
pancreatic protective agent in clinical settings.
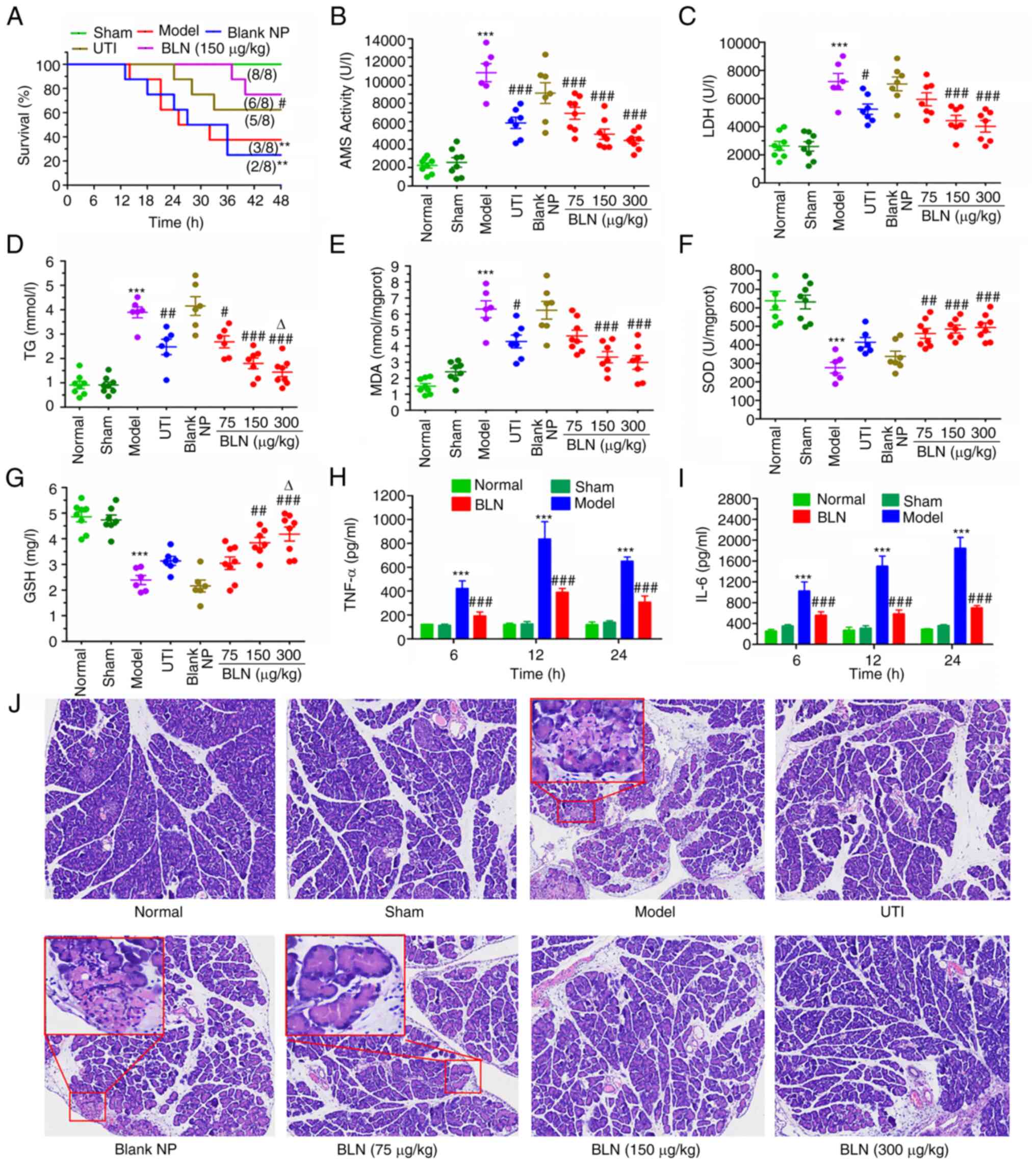 | Figure 4Effects of BLN on the survival rate,
biomarker levels, and inflammatory cytokine levels. (A) The change
in survival rates in different groups was monitored for 48 h
following BLN treatment. Levels of (B) AMS, (C) LDH, and (D) TG in
serum and (E) MDA, (F) SOD and (G) GSH levels in the pancreas
homogenates from different groups after 12 h of BLN treatment.
Levels of (H) TNF-α and (I) IL-6 levels in serum. (J) Pancreatic
histological observation of the different experimental groups.
Original magnification, ×100. Data are presented as the mean ± SEM
of eight repeats. **P<0.01, ***P<0.001
vs. normal group, #P<0.05, ##P<0.01,
###P 0.001 vs. model group, ∆P<0.05 vs.
UTI group. BLN, BAPTA-AM loaded liposome nanoparticles; UTI,
ulinastatin; LPS, lipopolysaccharide; NP, nanoparticle; BAPTA-AM,
1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid tetrakis
(acetoxymethyl ester); AMS, amylase; LDH, lactate dehydrogenase;
TG, triglyceride; MDA, malondialdehyde; SOD, superoxide dismutase;
GSH, glutathione. |
Measurement of serum and homogenate
biochemical indices
Increases in serum AMS, LDH, and TG serve as
important diagnostic indicators of AP, while MDA, GSH and SOD in
homogenates are the most commonly used indices to inspect oxidative
injury (48,49). All biomarker levels in the sham
and normal groups did not differ notably, indicating normal
pancreatic function in these groups. By contrast, the AP model
group showed notably increased levels of serum AMS, LDH, TG and
homogenate MDA by 3-6-fold after 24 h, displaying a significant
reduction in antioxidant levels of homogenate GSH and SOD, when
compared with the normal group, suggesting a notable decrease in
pancreatic function. Treatment with BLN resulted in a
dose-dependent recovery in pancreatic function compared with the
model group (Fig. 4B-G). Taken
together, these results suggest that BLN exhibits excellent
therapeutic effects in the rat model of AP.
Improvement of serum inflammatory
cytokines
ELISA was used to quantitatively analyze the levels
of TNF-α and IL-6 in serum (Fig. 4H
and I). As early contributors to AP events, TNF-α and IL-6 play
a crucial role in the process of the inflammatory cascade,
apoptosis and necrosis. After 12 h of modeling, a significant
increase in TNF-α and IL-6 expression was observed, which was
4-6-fold higher than that in the normal group. By contrast, the
TNF-α and IL-6 levels in the BLN treatment groups were
significantly lower; <30% of those in the model group. These
findings suggest that BLN has the potential to protect the pancreas
by exhibiting anti-inflammatory and anti-apoptotic properties.
Alleviation of the pancreatic
destruction
Based on the aforementioned experimental results,
pancreatic tissues were chosen for H&E staining to explore the
pathological changes (Fig. 4J
and Table I). Both the normal
group and the sham group exhibited no histological abnormalities,
as evidenced by the presence of an intact pancreatic structure and
well-preserved cytoplasm (grade 0). However, the histopathological
results for the AP model group revealed a notable amount of
destruction, including a considerable amount of necrosis,
inflammatory infiltrates, cellular swelling and vacuolization
(grade II and III). Despite exhibiting mild swelling, the
histological structure of the pancreatic tissue in the BLN
treatment group did not show visibly abnormal regions. After BLN
treatment, the injury degree was significantly decreased in a
dose-dependent manner to mainly grade 0 and I.
 | Table IEffects of BLN on the pathological
grade of TC-assaulted rats (n=8). |
Table I
Effects of BLN on the pathological
grade of TC-assaulted rats (n=8).
| Group | Grade
| P-value |
|---|
| 0 | I | II | III |
|---|
| Normal | 8 | 0 | 0 | 0 | |
| Sham | 8 | 0 | 0 | 0 | NS |
| Model | 0 | 0 | 2 | 6 | a |
| UTI | 2 | 5 | 1 | 0 | b |
| Blank NP | 0 | 1 | 3 | 4 | NSa |
| BLN (75
μg/kg) | 1 | 5 | 2 | 0 | b |
| BLN (150
μg/kg) | 3 | 5 | 0 | 0 | b |
| BLN (300
μg/kg) | 5 | 3 | 0 | 0 | b |
Modulation of the expression of
pro-apoptotic and pro-necrotic cytokines
The inflammatory response associated with AP
promotes an increase in the expression of two important cytokines,
namely TNF-α and cathepsin B, leading to the initiation of
apoptotic or necrotic signaling pathways (50-53). The binding of TNF-α to its
receptor TNFR-1 stimulates the release of cathepsin B from
lysosomes, which then promotes the activation of the apoptotic or
necrotic pathways. Thus, lower levels of these cytokines indicate a
reduction in pancreatic acinar cell damage by inhibiting these
signaling pathways. The levels of TNF-α and cathepsin B were
assessed using immunohistochemistry and western blotting. As shown
in Fig. 5A, there was a sharp
increase in TNF-α and cathepsin B expression in the pancreatic
tissues of the model group, suggesting the onset of apoptosis or
necrosis. However, treatment with BLN effectively and rapidly
reduced the expression of these cytokines, leading to a 50-70%
decrease in TNF-α and cathepsin B levels (Fig. 5B and C). Western blotting was
also used to verify the anti-apoptotic or anti-necrotic effects of
BLN, and the results of western blotting were consistent with those
of immunohistochemistry (Fig.
5D-F). Therefore, BLN may exert its therapeutic effect by
inhibiting a TNF-α/cathepsin B signaling pathway to reduce
pancreatic acinar cell damage in AP.
Inhibition of pro-apoptotic and
pro-necrotic gene transcription
As shown in Fig.
5G, the association between the cycles of PCR and fluorescence
intensity followed an ideal amplification curve, with an S-shaped
pattern. The genes of TNF-α, cathepsin B and β-actin, displayed
clear phases of exponential amplification and plateauing. The qPCR
data of this experiment were sufficient for the quantitative
analysis of TNF-α, cathepsin B and β-actin genes, and a wide linear
range of association was detectable in the 14th-32nd cycles.
Amplification of a specific vs. non-specific product could be
differentiated through dissociation curve analysis. The
dissociation curves of TNF-α and cathepsin B gene amplification
products with a single peak indicated specific amplification for
TNF-α and cathepsin B genes, free from primer-dimers or other
impurities. TNF-α and cathepsin B gene melting temperatures were
81.86°C and 80.21°C, respectively (Fig. S3). Compared with the normal
group, the transcription of TNF-α and cathepsin B mRNA in the
pancreas tissue of the model group increased by a factor of 5-10.
However, TNF-α and cathepsin B mRNA were significantly reduced by
57-73% in the BLN treatment group (150 μg/kg; Fig. 5H and I). In conjunction with the
aforementioned data, it was postulated that the rapid suppression
of the TNF-α/cathepsin B signaling pathway subsequent to BLN
treatment significantly contributed to the therapeutic efficacy for
AP.
Discussion
AP is an acute inflammatory process localized to the
pancreas in clinical practice with a high mortality rate
characterized by the premature activation of zymogens, causing
pancreatic tissue edema, hemorrhage and necrosis (54,55). Intracellular Ca2+
overload is generally accepted as a key trigger initiating
pancreatic acinar cell damage (27,56,57). The aberrant Ca2+
elevation in acinar cells can not only provoke the fusion of
zymogen granules with lysosomes to activate the cleavage of
trypsinogen to trypsin causing pancreatic autodigestion (58), but also induces the
overproduction of oxygen radicals that further target and damage
the cytoplasmic and mitochondrial membrane (27,59). The results of the present study
also showed that inhibiting intracellular Ca2+ overload
could simultaneously reduce pancreatic enzyme premature activation
and mitigate oxidative stress injury. However, it is not clear
whether Ca2+ overload directly promotes the activation
and release of pancreatic enzymes, the exact mode of the BAPTA
action must be interpreted with caution. The conclusions of Wang
et al (60) and those of
the current study suggest that Ca2+ chelators can reduce
acute pancreatic cell injury by inhibiting the expression of
inflammatory factors which could induce the activation of
protrypsin. In addition, the activation of
Ca2+-dependent proteases, such as calpain, due to the
Ca2+ elevation, is known to have a substantial impact on
the development of cell damage induced by oxidative stress.
Chelation of intracellular Ca2+ leads to complete
inhibition of H2O2-induced proteolytic
activity (16). ROS and
Ca2+ overload mutually reinforce each other, forming a
feedback loop (61). The pair
can further activate macrophages to release inflammatory cytokines,
such as TNF-α and IL-6, which amplifies the inflammatory cascade,
and eventually leads to further deterioration of the local tissue
and aggravate AP (62,63). These cytokines increase the
capillary permeability and promote inflammatory infiltration. The
binding of TNF-α to its receptor TNF-R1 contributes to the
maturation and release of cathepsin B from lysosomes, which
subsequently triggers the cathepsin B-mediated apoptotic or
necroptotic pathway (50,64).
Thus, intracellular Ca2+ overload plays a pivotal role
in the occurrence and development of AP. Rapidly restoring
Ca2+ to the resting state is useful for lessening the
excessive activation of zymogens, halting the inflammatory
progression and ameliorating pathological acinar cell apoptosis and
necrosis (65).
To mimic hyperlipidemia or gallstone-related
pancreatitis, two well-characterized models were used in the
present study: An in vitro model of HGO-induced AR42J cell
damage and an in vivo model of intra-ductal taurocholate
infusion-induced AP (66). These
models involve stepwise processes characterized by an excessive
release of pancreatic enzymes, severe inflammatory responses and
fulminant pancreatic necrosis, which are consistent with the
clinical manifestations of severe pancreatitis. Of note,
inflammatory cells, such as macrophages and neutrophils, are
intricately involved in the process of AP development. The simple
restriction of zymogen activation is insufficient to rescue the
endangered acinar cells due to the release/induction of pancreatic
toxins (ROS, LPS and Ca2+ overload) provoking
inflammatory cytokine overproduction, which thus exacerbates cell
damage. Since the absence of anti-inflammatory and anti-apoptotic
functions, the rescuing functions for severe AP of the available
anti-pancreatitis drugs (such as somatostatin and gabexate) are
less than satisfactory. Following intervention with somatostatin or
gabexate, ~20% of patients with AP still progress to SAP, which is
characterized by a systemic inflammatory response. In clinical
practice, there is a high mortality of 20-40% for patients with SAP
with currently available anti-pancreatitis regimens.
Clinically, the therapeutic regimen for AP
typically involves symptomatic therapy, such as fluid
resuscitation, low-fat diet, antibiotic therapy and biliary
drainage, rather than etiological treatment (67,68). Reversing the Ca2+
overload in the endangered acinar cells while simultaneously
inhibiting monocyte activation may be a more feasible scheme for
curing AP. Compared to the Ca2+ channel blockers,
BAPTA-AM can more effectively and rapidly reverse the overload in
intracellular Ca2+ concentration to a resting state
(69,70). BAPTA-AM is hydrolyzed by lipases
into BAPTA within acinar cells, and in this hydrolyzed form, it
decreases the degree of Ca2+ overload. In an
ischemia/reperfusion-induced acute kidney injury model, the use of
BAPTA-AM loaded NPs effectively impedes Ca2+ overload
and hinders the activation of the endoplasmic reticulum stress
cascade response while interrupting the feedback loop between
Ca2+ overload and ROS, thereby mitigating oxidative
stress-induced damage (32,28,61). In our previous study, it was also
validated that BLN exhibited a beneficial protective impact on
fulminant hepatic failure induced by D-GalN/LPS via a comparable
mechanism of action (31).
BAPTA-AM prevented various types of fulminant necrosis by
protecting against oxidative damage, maintaining the mitochondrial
membrane potential and suppressing both endogenous and exogenous
apoptotic pathways (71-74). AP is also considered a fulminant
and necrotizing disease (75,76). The results of the current study
suggest that BLN could not only disrupt the reciprocal relationship
between Ca2+ overload and ROS, but also delay the
macrophage activation to block the inflammatory cascades. These
potential mechanisms can correspondingly fight against the
pathological process of AP.
In the present study, the rescuing functions of BLN
were further demonstrated in vitro and in vivo AP
models. HGO exposure results in a pathological rise in cytosolic
Ca2+ in AR42J cells, which triggers the excessive
release of AMS, which is a specific indicator for the diagnosis of
AP (77,78). Furthermore, intracellular
Ca2+ overload can promote excessive ROS production in
pancreatic acinar cells, leading to oxidative stress.
Ca2+ influx and oxidative stress self-amplify and
potentiate each other until mass cell death is observed (31). In addition to the aforementioned
pathological changes, the inflammatory cascade is another
characteristic of SAP. There is sufficient evidence that
inflammatory mediators, such as IL-6 and TNF-α, are closely
involved in the progression of AP (79,80). The endotoxin and acinar cellular
contents from collapsed cells recruit inflammatory cells to
infiltrate the pancreas and augment the inflammatory responses. The
RAW264.7 macrophage cell line, which is frequently employed to
explore inflammation, releases mass quantities of inflammatory
cytokines following endotoxin stimulation. In the present study, it
was found that BLN not only inhibited the premature activation of
zymogens and oxidative stress caused by Ca2+ overload,
but also relieved inflammatory damage. These results illustrated
that Ca2+ overload was a key trigger in the progression
of AP. Rapid elimination of Ca2+ overload is beneficial
to the rescue of the pancreas. The results of the present study
suggested that BLN exerted a promising protective effect on a rat
model of AP, increasing the survival rate from 37.5 to 75%. BLN
exhibited considerably better therapeutic outcomes compared with
those of UTI, a currently available pancreatic protective agent
(81). BLN recovered pancreatic
microcirculation and the function of the pancreas by improving
blood biochemistry indices, alleviating oxidative damage,
restricting the activation of trypsinogen and downregulating the
expression of TNF-α and IL-6. Pancreatic histopathology scores
showed that BLN significantly relieved pancreatic damage. According
to the immunohistochemistry and RT-qPCR results, it was found that
the substantial increase in the levels of cathepsin B was also
involved in the rapid development of AP, boosting the progression
of pancreatic acinar cell necroptosis. In addition to the ability
of BLN to block TNF-α-mediated endogenous apoptotic pathways, BLN
also notably reduced the cathepsin B burden. The multifaceted
interventional mechanisms of BLN for the prevention of cellular
death may underlie its rescuing effects.
In conclusion, BLN exhibits significant promise as
an innovative therapeutic approach for managing AP. Its ability to
promptly mitigate intracellular Ca2+ overload can
greatly enhance survival rates and restore pancreatic function. The
underlying pancreas protective mechanism of BLN is primarily
attributed to its potent anti-necrotic and anti-apoptotic
properties for pancreatic acinar cells, which are achieved through
the prevention of premature trypsinogen activation, mitigation of
oxidative damage and inhibition of apoptotic or necrotic pathways
mediated by TNF-α and cathepsin B (Fig. 6). It was hypothesized that
regulating intracellular Ca2+ concentration and
preventing Ca2+ overload would be a promising strategy
for alleviating acute pancreatic damage. Furthermore, the cell
rescue capability of BLN suggests its potential application in
treating other severe diseases characterized by extensive cellular
damage such as stroke and hepatic encephalopathy. Notably, compared
with UTI, a marketed trypsin and serine protease inhibitor, BLN
exhibited superior therapeutic outcomes that strongly support its
future clinical translational potential. In theory, the synergistic
effects of BLN with protease inhibitors and antioxidants can cute
AP. To further enhance the potential efficacy of this drug, future
research will focus on optimizing carrier design to achieve optimal
therapeutic effects.
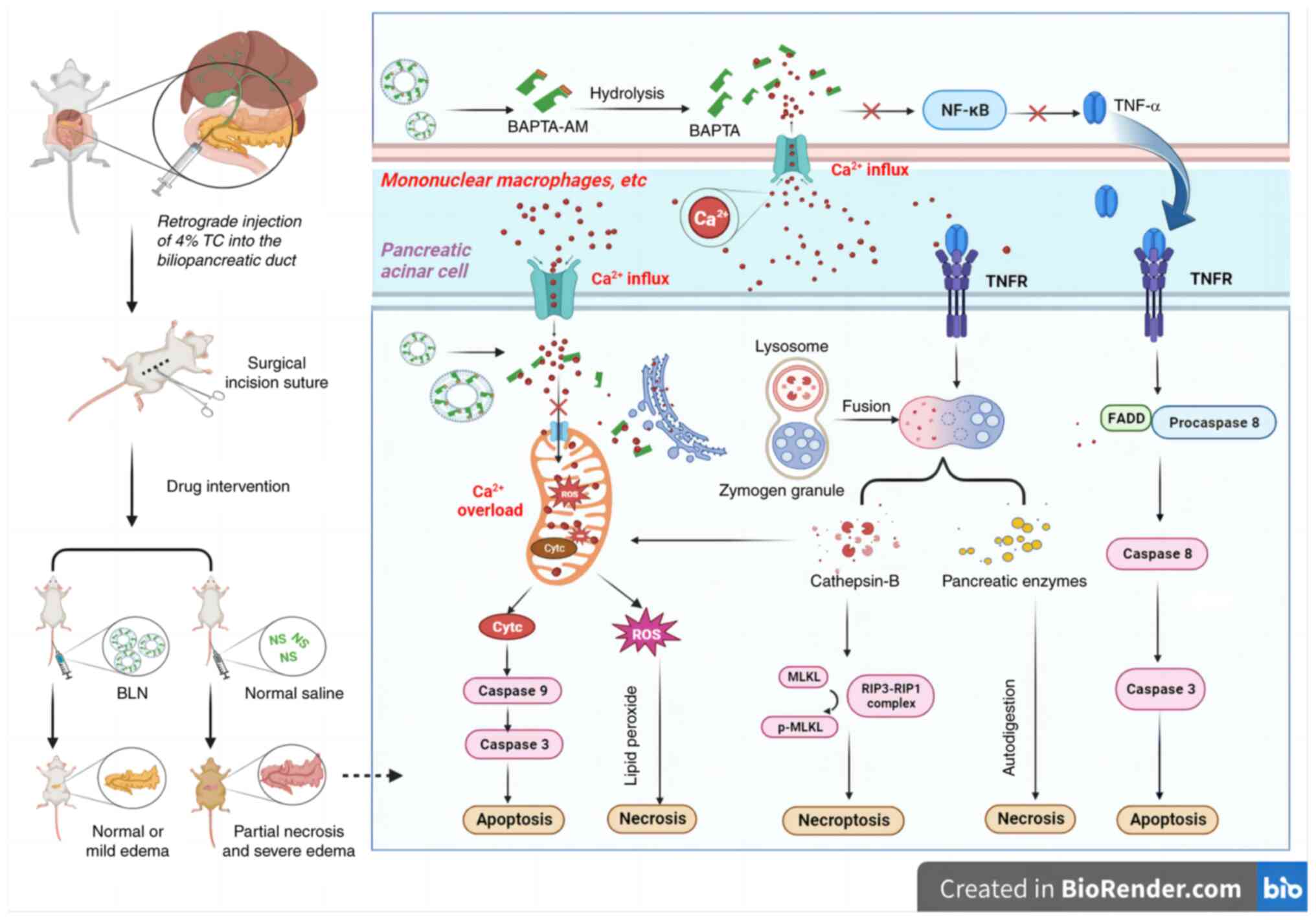 | Figure 6Schematic illustration of proposed
mechanism by which BLN protects pancreatic cells in the rat model
of AP through preventing the intracellular calcium overload. AP,
acute pancreatitis; BLN, BAPTA-AM loaded liposome nanoparticles;
BAPTA-AM, 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid
tetrakis (acetoxymethyl ester); p, phosphorylated; TC, sodium
taurocholate; ROS, reactive oxygen species; MLKL, mixed lineage
kinase domain like protein; RIP, receptor interaction protein;
FADD, fas-associating protein with death domain. |
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ZF, YS and WH conceived and planned the study. ZF,
DW, CZ, MX, YC, YZ and YH conducted all experiments and performed
data analysis and interpretation. ZF, WH and CZ confirm the
authenticity of all the raw data. ZF, YS and WH wrote the
manuscript, which was reviewed and edited by all co-authors. All
authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The animals used in the present study were provided
by the Zhejiang Center of Laboratory Animals [number of quality
certification, SCXK(zhe)-2021-0002; number of experimental
facilities certification, SYXK(zhe)-2022-0005]. Animal experiments
were approved by the Animal Experiments Ethical Inspection of
Zhejiang Center of Laboratory Animals (approval no. 2022R0006). All
animal studies complied with the Health Guide for Care and Use of
Laboratory Animals, Zhejiang Center of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by Zhejiang Province Medical and
Health Science and Technology Project (grant no. 2020370024) and
the Joint Funds of the Zhejiang Provincial Natural Science
Foundation of China under Grant (grant no. LYY21H310007).
References
|
1
|
Chan YC and Leung PS: Acute pancreatitis:
Animal models and recent advances in basic research. Pancreas.
34:1–14. 2007.
|
|
2
|
Jia A, Yang Z, Shi J, Liu J, Zhang K and
Cui Y: MiR-325-3p alleviates acute pancreatitis via targeting
RIPK3. Digest Dis Sci. 67:4471–4483. 2022.
|
|
3
|
Zhao Q, Zhang H, Huang J, Yu H, Li J, Che
Q, Sun Y, Jin Y and Wu J: Melatonin attenuates the inflammatory
response via inhibiting the C/EBP homologous protein-mediated
pathway in taurocholate-induced acute pancreatitis. Int J Mol Med.
42:3513–3521. 2018.
|
|
4
|
Baron TH, DiMaio CJ, Wang AY and Morgan
KA: American gastroenterological association clinical practice
update: Management of pancreatic necrosis. Gastroenterology.
158:67–75. 2020.
|
|
5
|
Boxhoorn L, Voermans RP, Bouwense SA,
Bruno MJ, Verdonk RC, Boermeester MA, van Santvoort HC and
Besselink MG: Acute pancreatitis. Lancet. 396:7262020.
|
|
6
|
Heckler M, Hackert T, Hu K, Halloran CM,
Büchler MW and Neoptolemos JP: Severe acute pancreatitis: Surgical
indications and treatment. Langenbecks Arch Surg. 406:521–535.
2021.
|
|
7
|
Leppaniemi A, Tolonen M, Tarasconi A,
Segovia-Lohse H, Gamberini E, Kirkpatrick AW, Ball CG, Parry N,
Sartelli M, Wolbrink DRJ, et al: 2019 WSES guidelines for the
management of severe acute pancreatitis. World J Emerg Surg.
14:272019.
|
|
8
|
Crockett SD, Wani S, Gardner TB,
Falck-Ytter Y, Barkun AN, Crockett S, Falck-Ytter Y, Feuerstein J,
Flamm S, Gellad Z, et al: American gastroenterological association
institute guideline on initial management of acute pancreatitis.
Gastroenterology. 154:1096–1101. 2018.
|
|
9
|
Portelli M and Jones CD: Severe acute
pancreatitis: Pathogenesis, diagnosis and surgical management.
Hepatob Pancreat Dis. 16:155–159. 2017.
|
|
10
|
Bang U, Semb S, Nojgaard C and Bendtsen F:
Pharmacological approach to acute pancreatitis. World J
Gastroenterol. 14:2968–2976. 2008.
|
|
11
|
Moggia E, Koti R, Belgaumkar AP, Fazio F,
Pereira SP, Davidson BR and Gurusamy KS: Pharmacological
interventions for acute pancreatitis. Cochrane Database Syst Rev.
4:D113842017.
|
|
12
|
Ward JB, Jenkins SA, Sutton R and Petersen
OH: Is an elevated concentration of acinar cytosolic free ionised
calcium the trigger for acute pancreatitis? Lancet. 346:1016–1019.
1995.
|
|
13
|
Mithöfer K, Fernández-Del Castillo C,
Frick TW, Lewandrowski KB, Rattner DW and Warshaw AL: Acute
hypercalcemia causes acute pancreatitis and ectopic trypsinogen
activation in the rat. Gastroenterology. 109:239–246. 1995.
|
|
14
|
Sakorafas GH and Tsiotou AG: Etiology and
pathogenesis of acute pancreatitis: Current concepts. J Clin
Gastroenterol. 30:343–356. 2000.
|
|
15
|
Yang AL and McNabb-Baltar J:
Hypertriglyceridemia and acute pancreatitis. Pancreatology.
20:795–800. 2020.
|
|
16
|
Weber H, Huhns S, Luthen F, Jonas L and
Schuff-Werner P: Calpain activation contributes to oxidative
stress-induced pancreatic acinar cell injury. Biochem Pharmacol.
70:1241–1252. 2005.
|
|
17
|
Orrenius S, Ankarcrona M and Nicotera P:
Mechanisms of calcium-related cell death. Adv Neurol.
71:1371996.
|
|
18
|
Orrenius S, Gogvadze V and Zhivotovsky B:
Calcium and mitochondria in the regulation of cell death. Biochem
Bioph Res Commun. 460:72–81. 2015.
|
|
19
|
Gerasimenko J, Peng S and Gerasimenko O:
Role of acidic stores in secretory epithelia. Cell Calcium.
55:346–354. 2014.
|
|
20
|
Xiao J, Lin H, Liu B and Jin J:
CaMKII/proteasome/cytosolic calcium/cathepsin B axis was present in
tryspin activation induced by nicardipine. Biosci Rep.
39:BSR201905162019.
|
|
21
|
Xie Z, Zhao M, Yan C, Kong W, Lan F,
Narengaowa, Zhao S, Yang Q, Bai Z, Qing H and Ni J: Cathepsin B in
programmed cell death machinery: Mechanisms of execution and
regulatory pathways. Cell Death Dis. 14:2552023.
|
|
22
|
Reinheckel T, Deussing J, Roth W and
Peters C: Towards specific functions of lysosomal cysteine
peptidases: Phenotypes of mice deficient for cathepsin B or
cathepsin L. Biol Chem. 382:7352001.
|
|
23
|
Sendler M, Dummer A, Weiss FU, Krüger B,
Wartmann T, Scharffetter-Kochanek K, van Rooijen N, Malla SR,
Aghdassi A, Halangk W, et al: Tumour necrosis factor α secretion
induces protease activation and acinar cell necrosis in acute
experimental pancreatitis in mice. Gut. 62:430–439. 2013.
|
|
24
|
Dawra R, Sah RP, Dudeja V, Rishi L,
Talukdar R, Garg P and Saluja AK: Intra-acinar Trypsinogen
activation mediates early stages of pancreatic injury but not
inflammation in mice with acute pancreatitis. Gastroenterology.
141:2210–2217. 2011.
|
|
25
|
Shen Y, Malik SA, Amir M, Kumar P,
Cingolani F, Wen J, Liu Y, Zhao E, Farris AB, Raeman R, et al:
Decreased hepatocyte autophagy leads to synergistic IL-1β and TNF
mouse liver injury and inflammation. Hepatology. 72:595–608.
2020.
|
|
26
|
Wu H, Zhang M, Li W, Zhu S and Zhang D:
Stachydrine attenuates IL-1β-induced inflammatory response in
osteoarthritis chondrocytes through the NF-κB signaling pathway.
Chem Biol Interact. 326:1091362020.
|
|
27
|
Criddle DN: Reactive oxygen species,
Ca(2+) stores and acute pancreatitis; a step closer to therapy?
Cell Calcium. 60:180–189. 2016.
|
|
28
|
Wang Y, Pu M, Yan J, Zhang J, Wei H, Yu L,
Yan X and He Z: 1,2-Bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic
Acid Acetoxymethyl ester loaded reactive oxygen species responsive
hyaluronic acid-bilirubin nanoparticles for acute kidney injury
therapy via alleviating calcium overload mediated endoplasmic
reticulum stress. ACS Nano. 17:472–491. 2023.
|
|
29
|
Quarato G, Llambi F, Guy CS, Min J, Actis
M, Sun H, Narina S, Pruett-Miller SM, Peng J, Rankovic Z and Green
DR: Ca2+-mediated mitochondrial inner membrane
permeabilization induces cell death independently of Bax and Bak.
Cell Death Differ. 29:1318–1334. 2022.
|
|
30
|
Tang Q, Jin M, Xiang J, Dong M, Sun H, Lau
C and Li G: The membrane permeable calcium chelator BAPTA-AM
directly blocks human ether a-go-go-related gene potassium channels
stably expressed in HEK 293 cells. Biochem Pharmacol. 74:1596–1607.
2007.
|
|
31
|
Fu Z, Fan Q, Zhou Y, Zhao Y and He Z:
Elimination of intracellular calcium overload by BAPTA-AM-loaded
liposomes: A promising therapeutic agent for acute liver failure.
ACS Appl Mater Interfaces. 11:39574–39585. 2019.
|
|
32
|
He Z, Tang H, You X, Huang K, Dhinakar A,
Kang Y, Yu Q and Wu J: BAPTA-AM nanoparticle for the curing of
acute kidney injury induced by ischemia/reperfusion. J Biomed
Nanotechnol. 14:868–883. 2018.
|
|
33
|
Hong X, Wang H, Yang J, Lin B, Min Q,
Liang Y, Huang P, Zhong Z, Guo S, Huang B and Xu YF: Systemic
injury caused by taurocholate-induced severe acute pancreatitis in
rats. Exp Ther Med. 24:4682022.
|
|
34
|
Laukkarinen JM, Van Acker GJ, Weiss ER,
Steer ML and Perides G: A mouse model of acute biliary pancreatitis
induced by retrograde pancreatic duct infusion of Na-taurocholate.
Gut. 56:1590–1598. 2007.
|
|
35
|
Aho HJ, Suonpää K, Ahola RA and Nevalainen
TJ: Experimental pancreatitis in the rat. Ductal factors in sodium
taurocholate-induced acute pancreatitis. Exp Pathol. 25:731984.
|
|
36
|
Lange JF, van Gool J and Tytgat GNJ:
Experimental pancreatitis in the rat: Role of bile reflux in sodium
taurocholate-induced acute haemorrhagic pancreatitis. Eur Surg Res.
18:369–374. 1986.
|
|
37
|
Tani S, Otsuki M, Itoh H, Nakamura T,
Fujii M, Okabayashi Y, Fujisawa T and Baba S: The protective effect
of the trypsin inhibitor urinastatin on cerulein-induced acute
pancreatitis in rats. Pancreas. 3:471–476. 1988.
|
|
38
|
Wang GP, Wen JM, Wen PM, Wilbur RRM, Zhou
SMP and Xiao XP: The effect of Somatostatin, Ulinastatin and Salvia
miltiorrhiza on severe acute pancreatitis treatment. Am J Med Sci.
346:371–376. 2013.
|
|
39
|
Sag S, Imamoglu M, Sarihan H, Yulug E,
Alver A, Geze Saatci S and Cay A: Effects of carbon dioxide
pneumoperitoneum on exocrine and endocrine functions, and oxidative
state of rat pancreas. Biotech Histochem. 96:257–262. 2021.
|
|
40
|
Wenhong D, Jia Y, Weixing W, Xiaoyan C,
Chen C, Sheng X, Hao J and Huang RS: Zerumbone Attenuates the
severity of acute necrotizing pancreatitis and pancreatitis-induced
hepatic injury. Mediat Inflamm. 2012:156507–156512. 2012.
|
|
41
|
Otani T and Matsukura A: Premature
trypsinogen activation during cerulein pancreatitis in rats occurs
inside pancreatic acinar cells. Pancreas. 20:421–422. 2000.
|
|
42
|
Saluja A, Dudeja V, Dawra R and Sah RP:
Early Intra-acinar events in pathogenesis of pancreatitis.
Gastroenterology. 156:1979–1993. 2019.
|
|
43
|
Mort JS and Buttle DJ: Cathepsin B. Int J
Biochem Cell Biol. 29:715–720. 1997.
|
|
44
|
Winer J, Jung CK, Shackel I and Williams
PM: Development and validation of real-time quantitative reverse
transcriptasepolymerase chain reaction for monitoring gene
expression in cardiac myocytes in vitro. Anal Biochem. 270:41–49.
1999.
|
|
45
|
Gerasimenko JV, Gerasimenko OV and
Petersen OH: The role of Ca2+ in the pathophysiology of
pancreatitis. J Physiol. 592:269–280. 2014.
|
|
46
|
Maléth J and Hegyi P: Ca2+ toxicity and
mitochondrial damage in acute pancreatitis: Translational overview.
Philos Trans R Soc Lond B Biol Sci. 371:201504252016.
|
|
47
|
Ismail OZ and Bhayana V: Lipase or amylase
for the diagnosis of acute pancreatitis? Clin Biochem.
50:1275–1280. 2017.
|
|
48
|
Szatmary P, Grammatikopoulos T, Cai W,
Huang W, Mukherjee R, Halloran C, Beyer G and Sutton R: Acute
pancreatitis: Diagnosis and treatment. Drugs. 82:1251–1276.
2022.
|
|
49
|
Das D, Mukherjee S, Das AS, Mukherjee M
and Mitra C: Aqueous extract of black tea (Camellia sinensis)
prevents ethanol+cholecystokinin-induced pancreatitis in a rat
model. Life Sci. 78:2194–2203. 2006.
|
|
50
|
Guicciardi ME, Deussing J, Miyoshi H,
Bronk SF, Svingen PA, Peters C, Kaufmann SH and Gores GJ: Cathepsin
B contributes to TNF-alpha-mediated hepatocyte apoptosis by
promoting mitochondrial release of cytochrome c. J Clin Invest.
106:1127–1137. 2000.
|
|
51
|
He R, Wang Z, Dong S, Chen Z and Zhou W:
Understanding Necroptosis in pancreatic diseases. Biomolecules.
12:8282022.
|
|
52
|
Sendler M, Maertin S, John D, Persike M,
Weiss FU, Krüger B, Wartmann T, Wagh P, Halangk W, Schaschke N, et
al: Cathepsin B activity initiates apoptosis via digestive protease
activation in pancreatic acinar cells and experimental
pancreatitis. J Biol Chem. 291:14717–14731. 2016.
|
|
53
|
Zhang X, Lin Q and Zhou Y: Progress of
study on the relationship between mediators of inflammation and
apoptosis in acute pancreatitis. Digest Dis Sci. 52:1199–1205.
2007.
|
|
54
|
Lankisch PG, Apte M and Banks PA: Acute
pancreatitis. Lancet. 386:85–96. 2015.
|
|
55
|
Song Y, Zhang Z, Yu Z, Xia G, Wang Y, Wang
L, Peng C, Jiang B and Liu S: Wip1 Aggravates the Cerulein-induced
cell autophagy and inflammatory injury by targeting STING/TBK1/IRF3
in acute pancreatitis. Inflammation. 44:1175–1183. 2021.
|
|
56
|
Criddle DN, Gerasimenko JV, Baumgartner
HK, Jaffar M, Voronina S, Sutton R, Petersen OH and Gerasimenko OV:
Calcium signalling and pancreatic cell death: Apoptosis or
necrosis? Cell Death Differ. 14:1285–1294. 2007.
|
|
57
|
Zhou X, Chen H, Wei X, He Y, Xu C and Weng
Z: Establishment of a mouse severe acute pancreatitis model using
retrograde injection of sodium taurocholate into the
Biliopancreatic duct. J Vis Exp. 182:e631292022.
|
|
58
|
Petersen OH: Ca2+-induced pancreatic cell
death: Roles of the endoplasmic reticulum, zymogen granules,
lysosomes and endosomes. J Gastroenterol Hepatol. 23(Suppl 1):
S31–S36. 2008.
|
|
59
|
Li J, Zhou R, Zhang J and Li Z: Calcium
signaling of pancreatic acinar cells in the pathogenesis of
pancreatitis. World J Gastroenterol. 20:16146–16152. 2014.
|
|
60
|
Wang Q, Bai L, Luo S, Wang T, Yang F, Xia
J, Wang H, Ma K, Liu M, Wu S, et al: TMEM16A
Ca2+-activated Cl− channel inhibition
ameliorates acute pancreatitis via the IP3R/Ca2+/NFκB/IL-6
signaling pathway. J Adv Res. 23:25–35. 2020.
|
|
61
|
Yan J, Wang Y, Zhang J, Liu X, Yu L and He
Z: Rapidly blocking the calcium Overload/ROS production feedback
loop to alleviate acute kidney injury via
microenvironment-responsive BAPTA-AM/BAC Co-delivery Nanosystem.
Small. 19:e22069362023.
|
|
62
|
Fanczal J, Pallagi P, Görög M, Diszházi G,
Almássy J, Madácsy T, Varga Á, Csernay Biró P, Katona X, Tóth E, et
al: TRPM2-mediated extracellular Ca2+ entry promotes
acinar cell necrosis in biliary acute pancreatitis. J Physiol.
598:1253–1270. 2020.
|
|
63
|
Ricardo Carvalho VP, Figueira da Silva J,
Buzelin MA, Antônio da Silva Júnior C, Carvalho Dos Santos D,
Montijo Diniz D, Binda NS, Borges MH, Senna Guimarães AL, Rita
Pereira EM and Gomez MV: Calcium channels blockers toxins attenuate
abdominal hyperalgesia and inflammatory response associated with
the cerulein-induced acute pancreatitis in rats. Eur J Pharmacol.
891:1736722021.
|
|
64
|
Zhang P, Yin X, Wang X, Wang J, Na G and
Ирина Павловна К: Paeonol protects against acute pancreatitis by
Nrf2 and NF-κB pathways in mice. J Pharm Pharmacol. 74:1618–1628.
2022.
|
|
65
|
Mayerle J, Sendler M, Hegyi E, Beyer G,
Lerch MM and Sahin-Tóth M: Genetics, cell biology, and
pathophysiology of pancreatitis. Gastroenterology. 156:1951–1968.
2019.
|
|
66
|
Werneburg NW, Guicciardi ME, Bronk SF and
Gores GJ: Tumor necrosis factor-alpha-associated lysosomal
permeabilization is cathepsin B dependent. Am J Physiol
Gastrointest Liver Physiol. 283:G947–G956. 2002.
|
|
67
|
Forsmark CE, Swaroop Vege S, Wilcox CM and
Campion EW: Acute pancreatitis. N Engl J Med. 375:1972–1981.
2016.
|
|
68
|
Hines OJ and Pandol SJ: Management of
severe acute pancreatitis. BMJ. 367:l62272019.
|
|
69
|
Li L, Tucker RW, Hennings H and Yuspa SH:
Chelation of intracellular Ca2+ inhibits murine keratinocyte
differentiation in vitro. J Cell Physiol. 163:105–114. 1995.
|
|
70
|
Tymianski M, Spigelman I, Zhang L, Carlen
PL, Tator CH, Charlton MP and Wallace MC: Mechanism of action and
persistence of Neuroprotection by Cell-permeant Ca2+ chelators. J
Cereb Blood Flow Metab. 14:911–923. 1994.
|
|
71
|
Toronyi É, Hamar J, Perner F and Szende B:
Prevention of apoptosis reperfusion renal injury by calcium channel
blockers. Exp Toxicol Pathol. 51:209–212. 1999.
|
|
72
|
Jang M, Shin M, Cho Y, Baik H, Kim S,
Hwang E and Kim C: 1,2-bis
(2-aminophenoxy)ethane-N,N,N'N'-tetraacetic acid (BAPTA-AM)
inhibits caffeine-induced apoptosis in human neuroblastoma cells.
Neurosci Lett. 358:189–192. 2004.
|
|
73
|
Hsu S, Jan C and Liang W: The
investigation of the pyrethroid insecticide lambda-cyhalothrin
(LCT)-affected Ca2+ homeostasis and -activated
Ca2+-associated mitochondrial apoptotic pathway in
normal human astrocytes: The evaluation of protective effects of
BAPTA-AM (a selective Ca2+ chelator). Neurotoxicology.
69:97–107. 2018.
|
|
74
|
Frosali S, Leonini A, Ettorre A, Di Maio
G, Nuti S, Tavarini S, Di Simplicio P and Di Stefano A: Role of
intracellular calcium and S-glutathionylation in cell death induced
by a mixture of isothiazolinones in HL60 cells. Biochim Biophys
Acta. 1793:572–583. 2009.
|
|
75
|
Nathens AB, Curtis JR, Beale RJ, Cook DJ,
Moreno RP, Romand J, Skerrett SJ, Stapleton RD, Ware LB and
Waldmann CS: Management of the critically ill patient with severe
acute pancreatitis. Crit Care Med. 32:2524–2536. 2004.
|
|
76
|
Maher MM, Lucey BC, Gervais DA and Mueller
PR: Acute pancreatitis: The role of imaging and interventional
radiology. Cardiovasc Inter Rad. 27:208–225. 2004.
|
|
77
|
Nordestgaard AG, Wilson SE and Williams
RA: Correlation of serum amylase levels with pancreatic pathology
and pancreatitis etiology. Pancreas. 3:159–161. 1988.
|
|
78
|
Moridani MY and Bromberg IL: Lipase and
pancreatic amylase versus total amylase as biomarkers of
pancreatitis: An analytical investigation. Clin Biochem. 36:31–33.
2003.
|
|
79
|
Song R, Yu D and Park J: Changes in gene
expression of tumor necrosis factor alpha and interleukin 6 in a
canine model of caerulein-induced pancreatitis. Can J Vet Res.
80:236–241. 2016.
|
|
80
|
Jiang CY, Wang W, Tang JX and Yuan ZR: The
adipocytokine resistin stimulates the production of proinflammatory
cytokines TNF-α and IL-6 in pancreatic acinar cells via NF-κB
activation. J Endocrinol Invest. 36:986–992. 2013.
|
|
81
|
Fang S, Li P, Zhu C, Han X, Bao P and Guo
W: Research progress of ulinastatin in the treatment of liver
diseases. Int J Clin Exp Patho. 13:2720–2726. 2020.
|