Introduction
Breast cancer (BC) is one of the most frequently
diagnosed cancers globally and ranks as the leading cause of
cancer-related mortality in female patients, with an estimated 2.3
million new cases in 2020 (1).
Metastatic (M)BC is characterized by an incurable nature and 53.3%
overall survival (OS) rate at 5 years and has limited effective
treatment options (2).
Consequently, identification of crucial molecular targets
implicated in metastasis holds promise for the formulation of novel
therapeutic strategies tailored to patients with MBC. SET domain
bifurcated 1 (SETDB1), belonging to the histone H3 lysine 9
methyltransferase (HKMT) family, serves a role in gene silencing
via catalytic H3K9 methylation (3,4).
Numerous studies have identified SETDB1 as an oncogene linked to
metastasis and unfavorable prognosis in various solid tumors,
including non-small cell lung cancer (NSCLC) (5,6),
hepatocellular carcinoma (7),
melanoma (8) and BC (9). Notably, SETDB1 amplification is
detected in 9.1% of BC cases (10), with triple-negative BC (TNBC)
exhibiting the most substantial increase in expression of HKMTs
(11). In BC, SETDB1 promotes
MYC and cyclin D1 mRNA expression by augmenting internal ribosome
entry site (IRES)-mediated translation. Simultaneously, the
c-MYC-polycomb family transcriptional repressor axis reciprocally
regulates SETDB1 expression, amplifying cellular proliferation and
self-renewal capabilities (12).
Acting as a ΔNp63α interactor, SETDB1 contributes to p63 protein
stability, thereby promoting the self-renewal of BC stem cells
(13). Additionally, SETDB1
induces epithelial-mesenchymal transition (EMT) and enhances
migration and invasion by downregulating SMAD7 (9) and its effects can be reversed by
microRNA (miRNA/miR)-7 (14) and
-381-3p (15).
Given the association between protein localization
and its physiological role in biological processes, determining
localization of SETDB1 in cancer cells and the underlying
mechanisms in metastasis is key. Both endogenous and exogenous
SETDB1 have been identified in the cytoplasm (16). The nucleocytoplasmic trafficking
of SETDB1 depends on chromosome region maintenance 1 (CRM1)
protein, encompassing an N-terminal N255 region with a SUMO
interaction motif site binding to promyelocytic leukemia nuclear
body (17). Treatment with the
CRM1 inhibitor, leptomycin B (LMB), obstructs SETDB1 export to the
cytoplasm and combination with the proteosome inhibitor MG132
contributes to the accumulation of SETDB1 in the nuclear region
(16). Wnt3a activation of
canonical Wnt signaling induces cytoplasmic localization of SETDB1,
whereas use of IWP2, a small molecule inhibitor of Wnt signaling,
leads to the reduction of cytoplasmic SETDB1 expression (18). While SETDB1 is key for gene
transcription, it also serves a role in catalyzing non-histone
methylation in the context of cancer. SETDB1-mediated
trimethylation of AKT on K64 residue enhances AKT ubiquitination by
TNF receptor associated factor 6), promoting AKT membrane
translocation and kinase activation (19). Elevated expression of
SETDB1-mediated AKT K64 methylation is associated with poorer
prognosis in patients with NSCLC (20). However, the function and
mechanism of cytoplasmic SETDB1 in BC remain elusive.
The present study aimed to investigated the
expression pattern of SETDB1 in both breast cancer samples and cell
lines. We also examined the biological functions of cytoplasmic
SETDB1 in breast cancer cells and explored the potential mechanisms
of cytoplasmic SETDB1 involved in EMT and Warburg effect.
Materials and methods
Human tissue specimens and
immunohistochemistry (IHC
All primary BC samples (n=172) along with normal
breast epithelium tissue specimens at 50 mm adjacent to the
corresponding cancer surgically resected in the first diagnosis and
breast fibroadenoma tissue (n=50) were obtained from Affiliated
Hospital of Jining Medical University (Jining, China, between
August 2008 and August 2010. The average age of patients was
50.51±12.29 years. All subjects were female patients with Invasive
Ductal Carcinoma (IDC) by breast biopsy or pathology, without
chemotherapy or radiotherapy before operation. Male patients or
non-IDC including invasive lobular carcinoma, inflammatory breast
cancer and metaplastic breast cancer were excluded. The acquisition
was conducted with informed consent from the patients, adhering to
protocols approved by the Ethics Committee of Jining Medical
University. IHC detection was performed on 4-μm-thick 4%
formalin-fixed (24 h, room temperature) and paraffin-embedded
tissue slices. Before staining, slides were deparaffinized with
xylene and tissues were rehydrated with graded ethanol, then the
slides were dried at 60°C for 1 h. The slides were submerged in
citrate repairing buffer (10 mmol/l, pH 6.0) in microwave at 90°C
for 30 min and allowed to cool to room temperature. After being
soaked in methanol containing 3% hydrogen peroxide for 10 min, the
sections were washed with 1X Tris Buffered Saline (TBS) three
times, 5 min) and blocked with 10% goat serum (Beyotime Institute
of Biotechnology; cat. no. #C0265) for 30 min at 37°C. The slides
were incubated with polyclonal rabbit anti-SETDB1 antibody
(Proteintech Group, Inc.; cat. no. #11231-1-AP, dilution 1:100)
overnight at 4°C. IgG antibody was utilized as a negative control
in BC tissues. SETDB1 staining in normal breast epithelium and
breast fibroadenoma tissues was used as positive control. After
incubation, the slides were washed with 1x TBS and incubated with
ready-to-use secondary anti-rabbit biotinylated antibody (Vector
Laboratories; cat. no. #BP-9100-50) at RT for 1 h. After washed
again, the sections were treated with 3,3′-diaminobenzidine
(ZSGB-BIO, #ZLI-9019). In addition, tissues were stained with
hematoxylin at room temperature for 3 min, dehydrated, cleared and
mounted with Distyrene Plasticizer Xylene (VMR International, LLC;
cat. no. #100504-938). Micrographs were captured by a light
microscope. The assessment of SETDB1 staining involved the
independent evaluation by two experienced pathologists, assigning
scores of 0, 1, 2 and 3, corresponding to negative, weak, medium
and high expression, respectively. Intensity scores were determined
based on the percentage of positively stained tumor cells (<10,
1; 11-50, 2; ≥51%, 3). The final score was calculated as follows:
Staining score x intensity score. Patients were then categorized as
having either low or high SETDB1 expression, determined by the
cut-off score (nuclear SETDB1 expression: 2.53; cytoplasmic SETDB1
expression: 2.62).
Cell culture and drug treatment
All breast cancer cell lines including MCF7, T47D,
BT549, MDA-MB-231 were obtained from the Type Culture Collection of
Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM
(Gibco, #11965092) or RPMI-1640 medium (cat. no. #11875093) with
10% fetal bovine serum (FBS; all Gibco, #16140071). The cultures
were maintained in a humidified incubator at 37°C with 5%
CO2. LMB (Beyotime Institute of Biotechnology; cat. no.
#S1726) or recombinant human Wnt3a protein (Proteintech, #HZ-1296)
was introduced into the serum-free medium at 37°C with a
concentration of 200 nM or 50 ng/ml for 24 h, respectively, while
0.1% DMSO served as the control.
Lentiviral vector infections and small
interfering (si)RNA transfection
The lentiviral SETDB1 overexpression and short
hairpin RNA vectors were constructed and introduced into cell
lines. A stably overexpressed (MCF7/SETDB1) and a stably depleted
SETDB1 cell line (BT549/shSETDB1) were established as described in
a previous study (21). The
lactate dehydrogenase A (LDHA) overexpression plasmid was
constructed by cloning the coding region of human LDHA into the
pLVX-IRES-PURO-3xFlag vector generated by OBiO Technology
(Shanghai) Corp., Ltd. For siRNA transfections, three siRNAs
targeting the human LDHA gene, along with negative control
(siCtrl), were procured from JianRan as follows: siLDHA#1 (5′-GCU
GAU UUA UAA UCU UCU A-3′), siLDHA#2 (5′-GAA UAA GAU UAC AGU UGU
U-3′) and siLDHA#3 (5′-GAC UGAU AAA GAU AAG GAA-3′). A total of
1×105 cells/well was seeded into six-well plate; 24 h
later, the siRNAs were transfected into cells using Lipofectamine
RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc.; cat. no.
#13778) for 48 h at 37°C.
Immunofluorescence staining
Cultured cells were fixed in 4% paraformaldehyde for
10 min at room temperature, followed by washing in PBS and
permeabilization with 0.1% Triton X-100 for 30 min at RT.
Subsequently, cells were blocked with 10% goat serum (Beyotime
Institute of Biotechnology; cat. no. #C0265) for 1 h at RT and
exposed to SETDB1 antibody (cat. no. #HPA018142; Sigma-Aldrich;
Merck KGaA; 1:100) at 4°C overnight, followed by Alexa Fluor
488-conjugated secondary antibody (Abcam, #ab150073, dilution:
1:200) for 1 h at room temperature. For visualizing the
cytoskeleton, nuclear staining with Hoechst 33342 (Thermo Fisher
Scientific, #62249, dilution: 1:200) at RT in the dark for 5 min
was used, along with F-actin staining using phalloidin (Abcam,
#ab176753; 1:1,000) for 1 h at RT. Images were captured using
Axionvision software v4.8 (zeiss.com) and a
Carl Zeiss fluorescence microscope (magnification, ×200).
Subcellular fraction extraction and
western blot analysis
Subcellular fractions of nuclei or cytoplasm were
isolated by Minute™ Cytoplasmic and Nuclear Fractionation kit
(Invent Biotechnologies, Inc.; cat. no. #SC-003) according to the
manufacturers' protocol. The extracted proteins were examined via
western blot using β-actin (Cell Signaling Technology, Inc. cat.
no. #4970S) serving as marker for total and cytoplasmic protein,
Lamin A antibody (CSignaling Technology, Inc.; cat. no. #86846S)
serving as marker for nuclear protein. Western blot was performed
as described in a previous study (21). Primary antibody against p53 (cat.
no. #60283-2-Ig, 1:1,000), Kras (cat. no. #12063-1-AP, 1:1,000),
pan-Ras (cat. no. #60309-1-Ig, 1:1,000), AKT (cat. no. #60203-2-Ig,
1:1,000), phosphorylated (p-)AKT (Ser473; cat. no. #80455-1-RR,
1:1,000), p-AKT (Thr308; cat. no. #29163-1-AP, 1:1,000), c-MYC
(cat. no. #67447-1-Ig; 1:1,000), LDHA (cat. no. #21799-1-AP,
1:1,000) and GAPDH (cat. no. #60004-1-Ig, 1:2,000) were obtained
from ProteinTech Group, Inc.; p-c-MYC (Ser293; cat. no.
#PA5-105447, 1:2,000) was obtained from Invitrogen (Thermo Fisher
Scientific, Inc.) and p-MYC (T58 + S62; cat. no. #13342, 1:1,000)
was obtained from Signalway Antibody LLC. Antibodies against
E-cadherin, N-cadherin, vimentin, Snail and Slug were obtained from
EMT Antibody Sampler kit (cat. no. #9782; Cell Signaling
Technology, Inc.; 1:1,000).
Reverse transcription-quantitative
(RT-q)PCR analysis
Total RNA was extracted from cells with RNAiso plus
obtained (Takara Biotechnology Co., Ltd.; cat. no. #9108) and its
concentration was determined using a NanoDrop spectrophotometer.
RNA was reverse-transcribed to cDNA using PrimeScript RT Master Mix
(Takara Biotechnology Co., Ltd.; cat. no. #DRRO36A) according to
the manufacturer's instructions. cDNA was amplified utilizing SYBR
Premix Ex Taq (Takara Biotechnology, cat no. #RR420A) with specific
primer pairs for the c-MYC, LDHA and β-actin genes. The primer
pairs (Sangon Biotech Co., Ltd.) were as follows: MYC forward,
5′-CGA CGA GAC CTT CAT CAA AAA C-3′ and reverse, 5′-CTT CTC TGA GAC
GAG CTT GG-3′; LDHA forward, 5′-TCA GCC CGA TTC CGT TAC CTA ATG-3′
and reverse, 5′-CAC CAG CAA CAT TCA TTC CAC TCC-3′ and β-actin
forward, 5′-CCC AGA TCA TGT TTG AGA CC-3′ and reverse, 5′-AGG GCA
TAC CCC TCG TAG AT-3′. The thermocycle conditions consisted of one
cycle at 95°C for 10 min, followed by 40 cycles of amplification at
95°C for 15 sec, and then 60°C for 1 min. Following normalization
to β-actin, the expression levels were quantified using 2-∆∆Cq
method (22), with all
experiments conducted in triplicate.
Wound healing assay
BT549, T47D and MCF7 cells were seeded in six-well
plates reaching 70-80% confluence with 2 ml complete medium.
Following cell adhesion, a scratch was introduced using a pipette
tip and the dislodged cells were washed with PBS. BT549 cells were
cultured in serum-free medium with DMSO or LMB treatment for 24 or
48 h. T47D and MCF7 cells were cultured in RPMI-1640 or DMEM with
10% FBS for 24 h or 48 h. Images of the scratched area were
captured with a light microscope (magnification, ×100) at 24 and 48
h. Accurate wound measurements were taken to calculate the wound
closure=(wound width at 0 h-wound width at 24 or 48 h)/wound width
at 0 h.
Migration and invasion assay
The migration assay used Transwell chambers
(Corning, Inc.; cat. no. #3422); for the invasion assay, inserts
were precoated with Matrigel (BD Biosciences; #354234) and at 37°C
for 4 h. In the upper chambers, a total of 5×104 cells
were seeded in 200 μl serum-free DMEM medium; in the lower
chamber, 500 μl DMEM medium containing 10% FBS was added.
Cells were incubated at 37°C for 24 h, then fixed with methanol for
10 min at RT and stained with 0.1% crystal violet for 20 min at
room temperature. Following PBS washes and air-drying, cells were
photographed using a light microscope (magnification, ×200) and
counting was performed manually across five semi-random,
non-overlapping areas and average value was calculated.
Metabolite detection
Detection of glucose, pyruvate, lactate production
and ATP content in cells was performed using glucose dehydrogenase
(cat. no. M011), pyruvate (cat. no. A081), lactate (cat. no. A019)
and ATP assay kits (cat. no. A095; all Nanjing Jiancheng
Bioengineering Institute) according to the manufacturer's
instructions.
Transcriptome sequencing and
bioinformatics analysis
RNA-seq was performed by BGI, Inc. Briefly, total
RNA extraction was performed using TRIzol reagent (Thermo Fisher
Scientific, #15596026) and Optimal Dual-mode mRNA Library Prep Kit
(BGI, Inc.; #LR00R96) was used for constructing the mRNA library
according to the manufacturer's instructions. mRNA was isolated
using 50 μl magnetic beads (Beckman; cat. no. #A63880) with
Oligo (dT) attached and fragmented using a Frag/Prime Buffer (BGI,
Inc.; cat. no. #LR00R96-E). Subsequently, through random
hexamer-primed RT), first-strand cDNA was generated (25°C, 10 min;
42°C, 15 min; 70°C, 15 min; 4°C, hold), followed by second-strand
cDNA synthesis (16°C, 30 min; 72°C, 15 min; 4°C, hold). Following
repair incubation (20°C, 15 min), A-Tailing Mix and RNA Index
Adapters (BGI Plug-In Adapter Kit) were introduced. The cDNA
underwent PCR amplification (1 cycle: 98°C, 1 min; 16 cycles: 98°C,
10 sec, 60°C, 30 sec, 72°C, 30 sec, 72°C, 5 min; hold, 4°C),
purification using 50 μl Ampure XP Beads (Beckman, #A63880)
at RT for 5 min and dissolution in 52 μl Elution Buffer
(BGI, Inc.; #LR00R96-N) at RT for 5 min. The double-stranded PCR
products were denatured by heat and circularized using the splint
oligo sequence (5′-GAA CGA CAT GGC TAC GAT CCG ACT TAA GTC GGA GGC
CAA GCG GTC TTA GGA AGA CAA CAA CTC CTT GGC TCT CAC A-3′). The
single-strand circle DNA was formatted as the final library and
amplified with phi29 to create DNA nanoballs (DNBs). Using the
BGIseq 500 platform (BGI, China), DNBs were fed into the patterned
nanoarray, producing single-end 50 base reads. Bioinformatics
analysis was conducted by BGI, China. Differentially expressed
genes (DEGs) were identified using the limma package in R software
v3.3.2 (https://www.r-project.org/), with a
cutoff of |log2FC|>1 and FDR<0.05. Gene Ontology (GO;
geneontology.org/) and Kyoto Encyclopedia of
Genes and Genomes (KEGG; genome.jp/kegg/) pathway analysis was
performed using the R software v3.3.2 module profiler package
(https://cran.r-project.org/web/packages/profileR/index.html)
with a criterion of P<0.05.
Immunoprecipitation (IP) and liquid
chromatography-mass spectrometry (LC-MS)
Cytoplasmic protein (100 μg/IP reaction) was
extracted as aforementioned and incubated with SETDB1 antibody (2
ug per IP (Proteintech Group, Inc.; cat. no. 11231-1-AP) and 30
μl Protein G agarose beads (cat. no. #37478; Cell Signaling
Technology, Inc.) by rocking at 4°C overnight. The beads were
washed with 1 ml Nonidet P-40 (Thermo Fisher Scientific, Inc.; cat.
no. #85124) buffer three times and resuspended in 2X SDS loading
buffer. Subsequently, LC-MS was performed by APPLIED PROTEIN
TECHNOLOGY, Inc (Shanghai Zhongkexin Life Biotechnology Co., Ltd.).
Briefly, proteins were integrated into buffer (4% SDS, 100 mM DTT,
100 mM Tris) and digested with trypsin and the resulting peptides
were collected as a filtrate. DTT (Sigma-Aldrich; Merck KGaA; cat.
no. #3483-12-3) and indole-3-acetic acid (IAA) (Sigma-Aldrich,
#87-51-4) were added to alkylate proteins. Protein digestion was
performed using trypsin and halted by trifluoracetic acid. Gel
pieces were excised from SDS-PAGE and subjected to three
extractions with 60.0% Acetonitrile (Sigma-Aldrich, #76-05-8)/0.1%
Trifluoroacetic Acid (both Sigma-Aldrich, #76-05-1). Each fraction
was injected for nano LC with tandem mass spectrometry (LC-MS/MS)
analysis. The resulting peptides were analyzed using Thermo Fisher
Orbitrap Elite with Waters NanoAcuity Ultra-Performance LC) (Thermo
Fisher Scientific, Inc.). MS/MS data were searched against the
Uniprot Human protein database (uniprot.org/)
using Mascot 2.5.1 (https://www.matrixscience.com/) and data analysis was
performed using Scaffold 4.4.8 software (https://researchexperts.utmb.edu/en/equipments/scaffold-version-448-informatics-software).
Peptides and modified peptides were accepted if they passed 1% FDR
threshold.
Statistical analysis
Statistical analysis was conducted utilizing IBM
SPSS Statistics 20 (ibm.com/products/spss-statistics) or GraphPad Prism 10
software (https://www.graphpad.com/). Data from
triplicate tests are presented as the mean ± SD. χ2 or
Fisher's exact test was used to compare clinicopathological
features between SETDB1 high and low groups. Correlations were
estimated by Spearman's correlation. Two-tailed unpaired Student's
t test and one-way ANOVA with Tukey's post hoc test were used to
determine the statistical significance of differences between
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Cytoplasmic expression of SETDB1 is
positively associated with lymph node metastasis and more
aggressive subtypes in patients with BC
To assess the SETDB1 expression, IHC was performed
on paraffin-embedded BC samples. SETDB1 was expressed in both the
nucleus and cytoplasm of BC tissues, whereas only nuclear staining
was observed in breast fibroadenoma tissue. SETDB1 was not
expressed in normal breast epithelium cells (Fig. S1). Based on staining score,
patients were categorized into SETDB1 high or low expression
groups. No significant differences were observed between nuclear
staining of SETDB1 and clinicopathological factors, including age,
tumor size, TNM stage, lymph node metastasis, progression and
survival (Table I). However, a
positive correlation was identified between cytoplasmic SETDB1 and
lymph node metastasis (Table
II). Furthermore, high cytoplasmic SETDB1 was more prevalent in
patients with high HER2 expression (42.86%) and TNBC subtypes
(43.90%) compared with luminal subtype (28.18%; Table III). Taken together, these
findings indicate that cytoplasmic SETDB1 is associated with more
aggressive BC subtypes and metastasis.
 | Table ICorrelation between nuclear SETDB1
expression and clinicopathological factors. |
Table I
Correlation between nuclear SETDB1
expression and clinicopathological factors.
| Factor | All (n=172) | SETDB1 expression
| r | P-value |
|---|
| Low (n=86) | High (n=86) |
|---|
| Age, years | | | | -0.062 | 0.418 |
| ≤35 | 18 | 8 | 10 | | |
| 36-51 | 77 | 37 | 40 | | |
| >51 | 77 | 41 | 36 | | |
| Tumor size, cm | | | | -0.005 | 0.990 |
| <2 | 96 | 48 | 48 | | |
| 2-5 | 69 | 34 | 35 | | |
| >5 | 7 | 4 | 3 | | |
| TNM stage | | | | -0.003 | 0.966 |
| I | 62 | 32 | 30 | | |
| II | 77 | 36 | 41 | | |
| III-IV | 33 | 18 | 15 | | |
| Number of LN
metastases | | | | 0.036 | 0.635 |
| 0 | 100 | 50 | 50 | | |
| 1-3 | 42 | 24 | 18 | | |
| 4-9 | 20 | 10 | 10 | | |
| >9 | 10 | 2 | 8 | | |
| Tumor
progression | | | | -0.173 | 0.261 |
| Absent | 55 | 27 | 28 | | |
| Present | 8 | 6 | 2 | | |
| Death | | | | -0.02 | 0.877 |
| No | 52 | 27 | 25 | | |
| Yes | 11 | 6 | 5 | | |
| Table IICorrelation between cytoplasmic
SETDB1 expression and clinicopathological factors. |
Table II
Correlation between cytoplasmic
SETDB1 expression and clinicopathological factors.
| Factor | All (n=172) | SETDB1 C expression
| r | P-value |
|---|
| Low (n=114) | High (n=58) |
|---|
| Age, years | | | | -0.032 | 0.673 |
| ≤35 | 18 | 11 | 7 | | |
| 36-51 | 77 | 51 | 26 | | |
| >51 | 77 | 52 | 25 | | |
| Tumor size, cm | | | | -0.100 | 0.197 |
| <2 | 96 | 60 | 36 | | |
| 2-5 | 69 | 48 | 21 | | |
| >5 | 7 | 6 | 1 | | |
| TNM stage | | | | -0.133 | 0.082 |
| I | 62 | 38 | 24 | | |
| II | 77 | 49 | 28 | | |
| III-IV | 33 | 27 | 6 | | |
| Number of LN
metastases | | | | 0.167 | 0.029a |
| 0 | 100 | 72 | 28 | | |
| 1-3 | 42 | 27 | 15 | | |
| 4-9 | 20 | 11 | 9 | | |
| >9 | 10 | 4 | 6 | | |
| Tumor
progression | | | | -0.223 | 0.102 |
| Absent | 55 | 39 | 16 | | |
| Present | 8 | 8 | 0 | | |
| Death | | | | 0.020 | 1.000 |
| No | 52 | 39 | 13 | | |
| Yes | 11 | 8 | 3 | | |
| Table IIICorrelation between cytoplasmic
SETDB1 expression and breast cancer subtype. |
Table III
Correlation between cytoplasmic
SETDB1 expression and breast cancer subtype.
| Subtype | Total | SETDB1 expression
| r | P-value |
|---|
| Low (%) | High (%) |
|---|
| Luminal | 110 | 79 (71.82) | 31 (28.18) | 0.154 | 0.044a |
| HER2 | 21 | 12 (57.14) | 9 (42.86) | | |
| Triple
negative | 41 | 23 (56.10) | 18 (43.90) | | |
Localization of endogenous and exogenous
SETDB1 protein in BC cells
To confirm SETDB1 localization, immunofluorescence
was performed in MCF7 and BT549 cells. Endogenous SETDB1 was
predominantly expressed in the nucleus in MCF7 cells with punctuate
signals, while both nuclear and cytoplasmic signals in a diffuse
pattern were observed in BT549 cells (Fig. 1A). Subcellular distribution of
endogenous SETDB1 was evaluated by separately extracting nuclear
and cytoplasmic fractions and immunoblotting. Although the total
protein expression of SETDB1 was higher in MCF7 and T47D compared
with BT549 and MDA-MB-231 cells, its localization was predominantly
nuclear rather than cytoplasmic (Fig. 1B). Conversely, endogenous SETDB1
was primarily detected in the cytoplasm of BT549 and MDA-MB-231. To
determine subcellular localization of exogenous SETDB1, a stably
overexpressed SETDB1 cell line (MCF7/SETDB1) was established as
described in a previous study (21). Exogenous SETDB1 was primarily
found in the cytoplasm, with only a few signals detected in the
nucleus (Fig. 1C). Taken
together, these findings suggested a potential association between
distribution of SETDB1 protein and its function in BC, indicating a
potential mechanism underlying SETDB1 transport from the nucleus to
the cytoplasm.
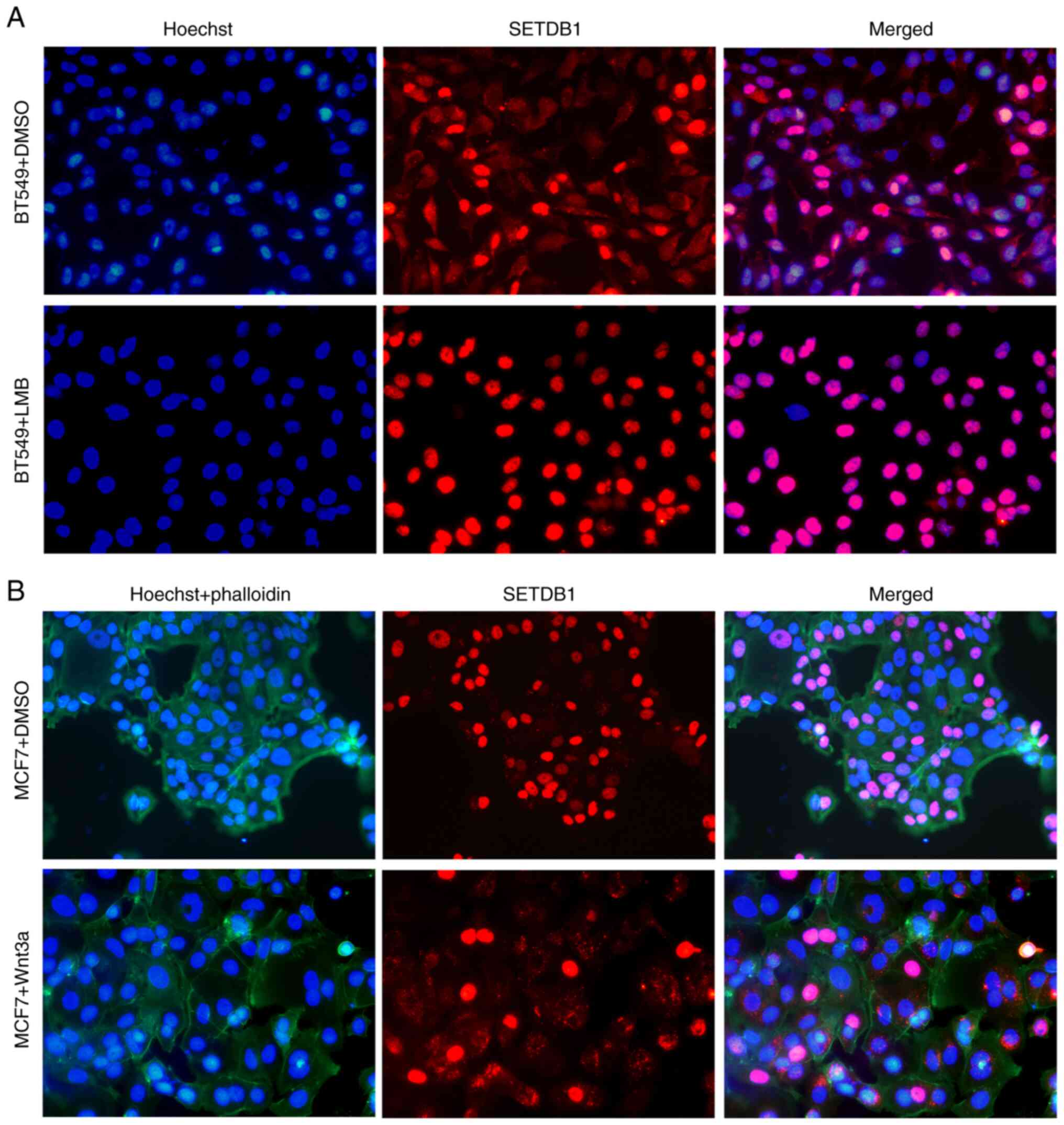
LMB blocks, while Wnt3a induces SETDB1
transport from nucleus to cytoplasm
SETDB1 encompassing nuclear localization signals
(NLSs) and nuclear export signals (NESs) which indicate that SETDB1
is able to shuttle between the cytoplasm and nucleus (16). CRM1 protein facilitates
transportation of SETDB1 from the nucleus to the cytoplasm
(16). To validate the impact of
CRM1 on SETDB1 transportation, LMB was used to bind CRM1 and
inhibit its activity in BT549 cells. Following 8 h treatment with
LMB, SETDB1 was predominantly detected in the nucleus (Fig. 2A). In comparison, with DMSO as a
control, SETDB1 was found in both the nucleus and cytoplasm,
confirming that CRM1-mediated SETDB1 shuttling between the nucleus
and cytoplasm was inhibited by LMB. Cytoplasmic SETDB1 expression
depends on canonical Wnt signaling during myoblast differentiation
(18). Recombinant Wnt3a protein
was used to activate Wnt signaling. Following 24 h Wnt3a treatment
in MCF7 cells, increased cytoplasmic localization of SETDB1 was
observed compared with the control group (Fig. 2B). These results suggested a
mechanism that regulates SETDB1 transportation between nucleus and
cytoplasm, involving the interplay of CRM1 and Wnt signaling
pathways.
CRM1 inhibitor decreases migration and
invasion and reverses SETDB1-induced EMT
To investigate the impact of SETDB1 shuttling from
the nucleus to the cytoplasm on migration and invasion abilities of
BC, wound healing and Transwell assays were performed following
treatment with LMB or DMSO. The wound healing rate in BT549 cells
was significantly lower following LMB treatment at both 24 and 48 h
compared with the control group (Fig. 3A). Similarly, Transwell assay
revealed that LMB significantly reduced migration and invasion
abilities in BT549 (Fig. 3B) as
well as MCF7/SETDB1 cells (Fig.
3C). Morphological change was observed in MCF7/SETDB1 cells
following LMB treatment for 36 h from long spindle forms to
polygonal shapes (Fig. 3D).
Furthermore, immunoblotting showed that mesenchymal marker vimentin
expression decreased, while epithelial marker E-cadherin expression
increased in MCF7/SETDB1 + LMB cells (Fig. 3E), indicating that LMB treatment
reversed SETDB1-induced EMT by inhibiting cytoplasmic SETDB1. In
summary, cytoplasmic SETDB1 served critical roles in migration,
metastasis and EMT.
Cytoplasmic SETDB1 enhances the Warburg
effect and LMB treatment reverses SETDB1-induced Warburg
effect
To determine the molecular mechanism underlying
involvement of cytoplasmic SETDB1 in BC cells, total RNA was
extracted from MCF7/NC + DMSO, MCF7/SETDB1 + DMSO and MCF7/SETDB1 +
LMB cells. RNA-sequencing analysis revealed 1,575 up- and 2,132
downregulated genes in the MCF7/SETDB1 + LMB group compared with
the MCF7/SETDB1 + DMSO group (Fig.
4A). KEGG analysis demonstrated that DEGs were enriched in
various metabolism pathways, such as 'glutathione metabolism',
'butanoate metabolism' and 'pentose and glucuronate
interconversions' (Fig. 4B).
Additionally, GO analysis revealed enrichment in immune-related
pathways, including 'adaptive immune response', 'immune response to
tumor cell' and 'positive regulation of immune system process'
(Fig. 4C). Reprogrammed
metabolism, a hallmark of cancer, involves aerobic glycolysis,
commonly known as the Warburg effect (23). To assess potential involvement of
SETDB1 in the Warburg effect, levels of metabolic indicators,
including glucose, pyruvate, lactate and ATP, were measured.
Knockdown of SETDB1 in BT549 cells led to a significant decrease in
the levels of these metabolites compared with the control group
(Fig. 4D). Furthermore, in the
absence of LMB, overexpressed SETDB1 increased levels of glucose,
pyruvate, lactate and ATP in MCF7 cells. However, the metabolite
levels decreased in the presence of LMB (Fig. 4E). These findings suggested that
exogenous SETDB1 may enhance the Warburg effect by localizing to
the cytoplasm and its impact was mitigated by inhibiting
cytoplasmic SETDB1 with LMB.
Cytoplasmic SETDB1 upregulates c-MYC/LDHA
signaling and activates its downstream signaling pathway
Numerous studies (24,25) have demonstrated the key role of
diverse pathways in the Warburg effect, including the
PI3K/AKT/mTOR, hypoxia-inducible factor-1 (HIF-1), Kras, p53 and
MYC pathways. Therefore, cytoplasmic protein isolation and
immunoblotting analysis were conducted in MCF7/SETDB1 cells in the
presence or absence of LMB. Decreased in p53 expression and an
increase in Kras, pan-Ras, AKT and p-AKT (Ser473) were noted in
SETDB1-overexpressing cells. However, no significant changes were
found between the MCF7/SETDB1 + DMSO and MCF7/SETDB1 + LMB groups
(Fig. 5A). Conversely,
cytoplasmic expression of c-MYC and its phosphorylated proteins
significantly increased in MCF7/SETDB1 + DMSO cells; this effect
was attenuated after LMB treatment (Fig. 5B). Consistently, RT-qPCR results
demonstrated elevated c-MYC mRNA expression in
SETDB1-overexpressing cells, which was decreased following LMB
treatment (Fig. 5C). These data
revealed that cytoplasmic SETDB1 upregulated c-MYC expression and
activated its downstream signaling pathway. LDHA, a key enzyme in
the Warburg effect, is known to be controlled by c-MYC (26). Consequently, the cytoplasmic
content of LDHA was examined through immunoblotting, revealing
increased LDHA expression in MCF7/SETDB1 cells and decreased levels
of LDHA in cells treated with LMB (Fig. 5D). In summary, these results
suggested that cytoplasmic SETDB1 upregulated the c-MYC/LDHA
pathway and inhibition of cytoplasmic SETDB1 abolished this
effect.
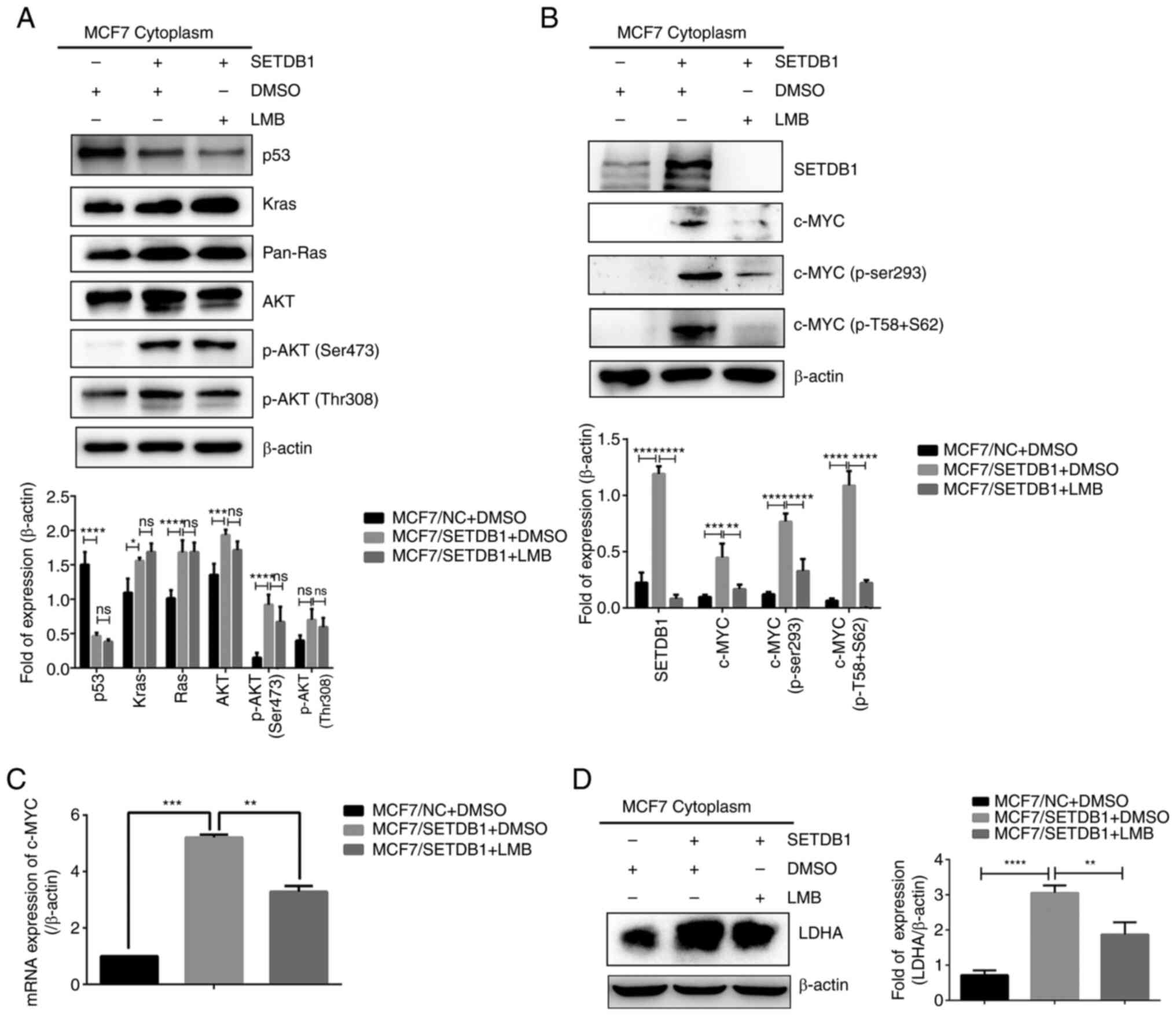 | Figure 5Cytoplasmic SETDB1 upregulates
c-MYC/LDHA signaling pathway. (A) Cytoplasmic protein levels of
p53, Kras, pan-Ras, AKT, p-AKT (Ser473) and p-AKT (Thr308) as well
as (B) SETDB1, c-MYC, p-c-MYC (Ser293) and p-c-MYC (T58 + S62). (C)
mRNA expression of c-MYC and (D) cytoplasmic protein expression of
LDHA in MCF7/SETDB1 cells following LMB treatment.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. SETDB1, SET
domain bifurcated 1; LMB, leptomycin B; LDHA, lactate dehydrogenase
A; pan-, pan-reactive; p-, phosphorylated. |
LDHA overexpression promotes migration
and invasion and induces EMT in BC cells
SETDB1, functioning as a histone lysine
methyltransferase, has been reported to mediate lysine methylation
of non-histone proteins in various types of cancer, such as
non-small cell lung cancer (19,20,27). To investigate methylation of
cytoplasmic proteins catalyzed by SETDB1, IP and LC-MS analysis
were performed to identify methylated residues binding with SETDB1
(Fig. S2A). A total of 5,826
peptides were detected and 513 methylated residues were identified.
GO analysis revealed enrichment in cellular process, metabolic
process, localization and immune system process for these proteins
(Fig. S2B). Notably, the
di-methylation of LDHA at lysine 155 (K155) was identified
(Fig. S2C). Furthermore, IP
confirmed the binding of LDHA and SETDB1 (Fig. S3). To determine the function of
LDHA in BC cells, a stably expressed T47D/LDHA cell line was
established and the infection efficiency was verified by
immunoblotting and RT-qPCR (Fig.
6A). Due to the Flag tag in the vector, two bands of LDHA were
evident in overexpressing cells, with the red band representing
exogenously expressed LDHA and the black band indicating
endogenously expressed LDHA. Wound healing and Transwell assay
revealed that LDHA overexpression significantly promoted migration
and invasion in T47D cells compared with the control (Fig. 6B and C). T47D/LDHA cells
exhibited distinct morphological changes, displaying a
spindle-shaped morphology (Fig.
6D). Furthermore, immunoblotting demonstrated that epithelial
marker E-cadherin was decreased, while mesenchymal makers
N-cadherin and vimentin, as well as EMT-associated transcription
factors Snail and Slug, were increased in LDHA-overexpressing T47D
cells (Fig. 6E). Collectively,
these data demonstrated that LDHA enhanced migration and invasion
by inducing EMT in BC cells.
 | Figure 6LDHA promotes migration and invasion
and induces epithelial-mesenchymal transition in T47D cells. (A)
Verification of LDHA expression in T47D cells by immunoblotting and
reverse transcription-quantitative PCR. Red arrow: exogenous LDHA;
black arrow: endogenous LDHA. (B) Wound healing (Magnification,
×100) and (C) Transwell assay (Magnification, ×200) detected
migration and invasion in T47D/LDHA cells. (D) Morphology of
LDHA-overexpressing T47D cells. Magnification, ×200. (E) Protein
levels of E-cadherin, N-cadherin, vimentin, Snail and Slug in
T47D/LDHA cells. **P<0.01, ***P<0.001,
****P<0.0001. LDHA, lactate dehydrogenase A; NC,
negative control. |
Knockdown of LDHA decreases
SETDB1-induced EMT and Warburg effect in BC cells
To assess the crucial role of LDHA in the function
of SETDB1 in BC cells, LDHA was knocked down in
SETDB1-overexpressing MCF7 cells. Following the confirmation of
LDHA expression via immunoblotting and RT-qPCR (Fig. 7A), siLDHA2# and siLDHA3# showed
significant decreases in both mRNA and protein level of LDHA and
thus were chosen for subsequent investigation. LDHA knockdown
significantly decreased migration and invasion abilities in
SETDB1-overexpressing cells (Fig.
7B). Compared with control cells, wound healing assay
demonstrated a significant decrease in cell migration following
LDHA depletion (Fig. 7C).
Additionally, levels of glucose, pyruvate, lactate and ATP were
significantly decreased in LDHA knockdown cells (Fig. 7D). Notably, LDHA knockdown led to
an increase in the epithelial marker E-cadherin, while expression
of mesenchymal markers N-cadherin and vimentin, as well as Snail
and Slug, were decreased (Fig.
7E). In summary, these findings demonstrated the essential role
of LDHA in SETDB1-induced EMT and Warburg effect in BC cells.
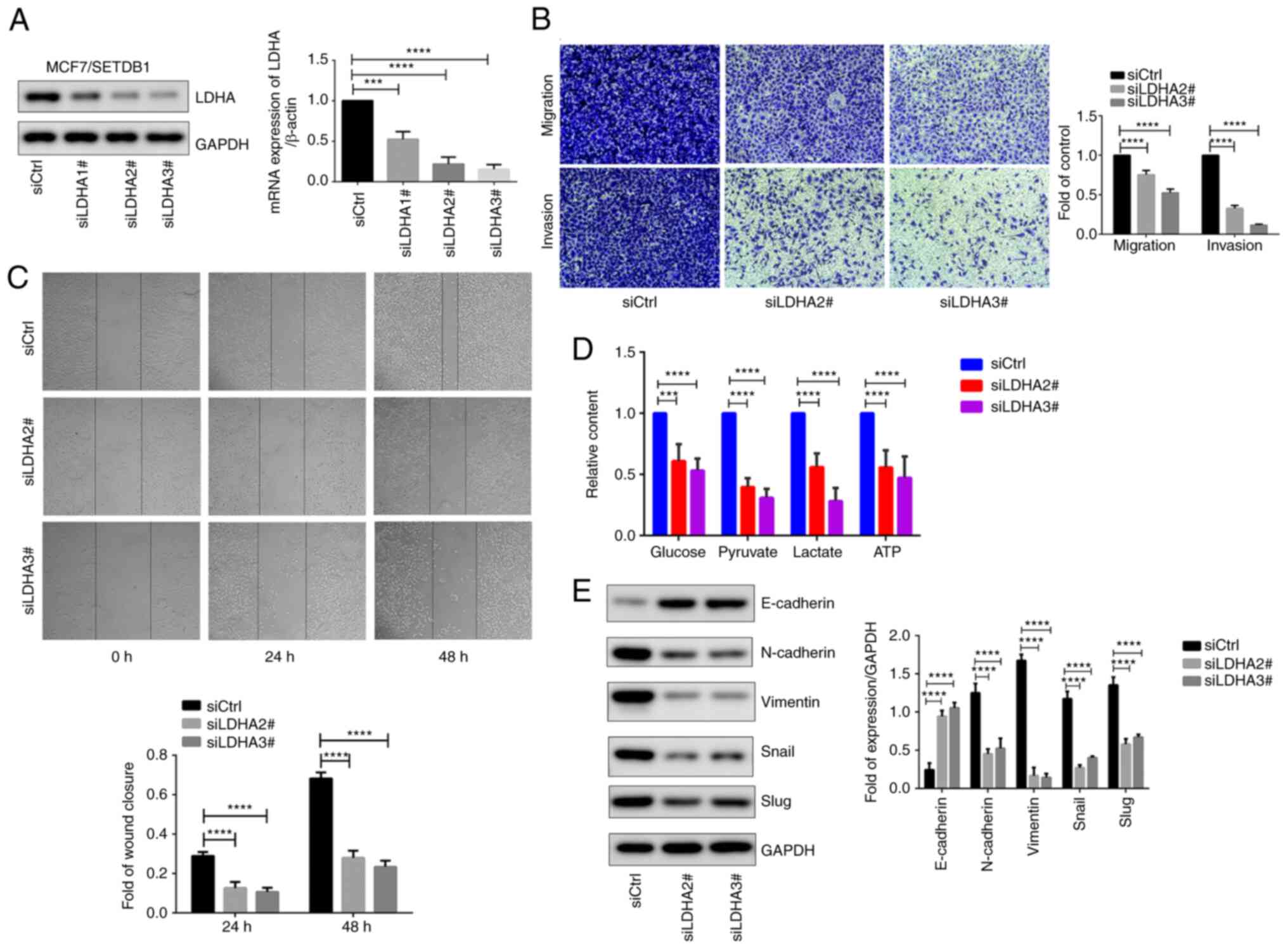 | Figure 7LDHA is key for SETDB1-induced
migration, invasion, EMT and Warburg effect. (A) Detection of LDHA
expression with immunoblotting and reverse
transcription-quantitative PCR after transfecting three LDHA siRNAs
in MCF7/SETDB1 cells. (B) Transwell assay. Magnification, ×200. (C)
Wound healing, Magnification, ×100. (D) Metabolite levels in
MCF7/SETDB1 cells following knockdown of LDHA. (E) Expression of
EMT-associated genes and transcription factors in LDHA-deficient
MCF7/SETDB1 cells. ***P<0.001,
****P<0.0001. LDHA, lactate dehydrogenase A; SETDB1,
SET domain bifurcated 1; EMT, epithelial-mesenchymal transition;
siCtrl, short hairpin RNA control. |
Discussion
HKMTs constitute a group of enzymes that regulate
transcription, chromatin architecture and cellular differentiation
by catalyzing site-specify methylation of lysine residues on
histone proteins (28).
Alterations, genetic translocation and modified gene expression
associated with these HKMTs are frequently observed in cancer
(29). Additionally, subcellular
localization of several histone-modifying enzymes is known to
regulate their functions. Enhancer of zeste Homolog 2 (EZH2) is
primarily localized to the nucleus. However, there is evidence to
suggest that EZH2 can also shuttle between the nucleus and
cytoplasm and its subcellular localization may influence its
activity and function (30).
Although SETDB1 is exported from the nucleus to the cytoplasm
(17), its role and regulation
of cytoplasmic localization in BC are not yet fully understood.
Here, IHC confirmed expression of SETDB1 protein in both the
nucleus and cytoplasm in BC samples. Only cytoplasmic SETDB1
expression was correlated with lymph node metastasis and aggressive
BC subtypes. Furthermore, SETDB1 was predominantly expressed in the
nucleus in MCF7 and T47D cells with limited migration and invasion
ability. SETDB1 expression was mostly cytoplasmic in BT549 and
MDA-MB-231 cells which are known of triple-negative breast cancer
cell lines and characterized by strong migration and invasion
ability (31).
The N-terminal region containing NES and NLS is
responsible for cytoplasmic SETDB1 localization (17). Moreover, SETDB1 may undergo
degradation by the proteosome and be exported to cytosol, mediated
by CRM1 protein. Thus, combined treatment with LMB and MG132
contributes to the accumulation of SETDB1 in the nucleus (16). Conversely, Wnt3a treatment
involving the Wnt/β-catenin pathway induces SETDB1 export to the
cytoplasm (17). Tsusaka et
al (32) reported that
activating Transcription Factor 7-Interacting Protein 1, a known
binding partner of SETDB1, antagonizes the nuclear localization of
SETDB1 by binding to its N-terminal region and subsequently
increases its ubiquitination. Consistent with these findings, the
present study confirmed that LMB treatment inhibited SETDB1 export
to the cytoplasm, and Wnt3a treatment contributed to the
accumulation of cytoplasmic SETDB1.
To determine the function of cytoplasmic SETDB1 in
BC migration and migration, in vitro experiments were
performed in the presence or absence of CRM1 inhibitor treatment. A
significant decrease in migration and invasion abilities was
observed in BC cells with LMB treatment. Mechanistically,
cytoplasmic SETDB1 was involved in various metabolism and
immune-related pathways, such as 'pentose and glucuronate
interconversions', 'immune response to tumor cell'. It has been
reported that SUMOylated SETDB1 suppresses expression of lipid
metabolism-associated target genes and downregulates lipid storage
in adipocytes (33). SETDB1 also
serves a crucial role in inhibiting endogenous retroviruses in
primordial germ cells (34) and
exogenous retroviruses in committed B lineage cells. Its loss also
results in B lymphocyte failure and molecular abnormality (35). Moreover, loss of SETDB1 in pro-B
cells leads to suppression of retrotransposon sequences such as
endogenous murine leukemia virus and blocks B cell development
(36). Studies have demonstrated
the importance of SETDB1 in T cell development, partly due to
suppression of Fc receptor ⅡB for IgG) (37,38). However, whether cytoplasmic
SETDB1 is involved in metabolic reprogramming in BC remain
unknown.
Altered tumor metabolism has gained widespread
acceptance as a hallmark of cancer (23) and enhances cell migration in most
types of cancer (39). The
Warburg effect, also known as aerobic glycolysis, occurs when
cancer cells utilize glucose at an abnormally high rate in the
presence of oxygen (40). More
aggressive mesenchymal BC cell lines such as BT549 and MDA-MB-231
exhibit a greater Warburg effect with high rate of glycolytic to
oxidative ATP flux compared with less aggressive epithelial lines
such as MCF7 and T47D (39). To
investigate whether SETDB1 regulates the Warburg effect, metabolite
levels were detected. The results of metabolite detection showed
that SETDB1-deficient cells exhibited a lower Warburg effect than
controls. However, SETDB1 overexpression increased the contents of
metabolite, which could be reversed by LMB treatment. To the best
of our knowledge, the present study is the first to demonstrate the
function of cytoplasmic SETDB1 in the Warburg effect. However,
little is known about the underlying pathway by which SETDB1
affects the Warburg effect.
Several key signaling pathways involved in the
Warburg effect, including PI3K/AKT/mTOR, Ras/Myc, reactive oxygen
species/HIF-1, p53, and Kras (24). AKT binds to the Tudor domain of
SETDB1, which binds demethylated lysine of H4-K20 and is involved
in DNA repair (41). SETDB1
coordinates with AKT to suppress FOXO1 by enhancing AKT activation,
while SETDB1 deficiency contributes to an increase in PTEN
expression and induces apoptosis in spermatogonial stem cells
(42). In paclitaxel-treated
lung cancer cells, P53 works with histone lysine
N-methyltransferase suppressor of Variegation 3-9 Homolog 1 to
enhance H3K9me3 occupancy and inhibit SETDB1 transcription by
directly binding to its promoter (43). Moreover, upregulation of SETDB1
suppresses apoptosis induced by 5-fluorouracil treatment in
colorectal cancer cells by binding to the TP53 promoter region and
inhibiting TP53 transcription (44). There is a positive regulation
between SETDB1 and c-MYC in BC cells: SETDB1 increases c-MYC
expression by IRES-mediated translation and c-MYC protein enhances
SETDB1 expression by binding to its promoter (12). Consistent with a previous study
(24), the present study
demonstrated that overexpression of cytoplasmic SETDB1 increased
Kras, Ras, AKT and c-MYC expression. Furthermore, in the presence
of LMB, decreased expression of c-MYC and its phosphorylated
protein, as well as c-MYC-targeted gene LDHA in MCF7/SETDB1 cells,
was observed. LDHA is known as an enzyme that catalyzes the final
step of the Warburg effect by converting pyruvate to lactate
(45). LDHA promotes migration
and invasion of cancer cells by inducing EMT (46,47). Consistently, the present study
confirmed that LDHA overexpression enhanced migration and invasion
in BC cells and regulated EMT-associated genes and transcription
factors. Therefore, the present study investigated how SETDB1
regulates (24) LDHA and whether
LDHA is key for SETDB1-induced EMT and Warburg effect.
KMTs methylate non-histone proteins and certain SET
domain-containing proteins are known to exclusively target
non-histone substrates (48).
SETDB1 is key for the methylation of the viral protein Tat at
lysine residues 50 and 51 to inhibit the transcription of human
immunodeficiency virus-1 long terminal repeat (49). In hepatocellular carcinoma,
SETDB1 catalyzes di-methylation of p53 at 370 lysine residues,
which decreases p53 protein stability by murine double minute
2-mediated ubiquitination (50).
SETDB1 has been reported to interact with the Pleckstrin Homology
domain of AKT and mediate methylation of AKT K140 to promote cell
growth and glycolysis and drug resistance in a carcinogen-induced
skin cancer model (19).
Furthermore, SETDB1-mediated AKT K64 methylation is linked to
carcinogenesis and a poor prognosis in patients with NSCLC
(20). Here, LDHA bound to
SETDB1 and dimethylation of LDHA at K155 was observed, suggesting
that LDHA may be a novel non-histone substrate of SETDB1. Moreover,
LDHA depletion reversed SETDB1-induced EMT and metabolic
reprogramming.
There are several limitations in the present study.
Firstly, the role of di-methylation of LDHA, catalyzed by SETDB1,
in the induction of EMT remains unclear. Secondly, while inhibition
of migration and invasion in gastric carcinoma cells by LMB
treatment has been reported (51), the precise molecular mechanism
underlying this phenomenon remains elusive. The present study
suggested that SETDB1 may be a target gene of CMR1 inhibitor;
however, further investigation is required to determine the impact
of LMB on BC cells. Thirdly, a distant metastasis nude mouse model
is required to confirm in vivo efficacy of CMR1 inhibitor on
SETDB1-induced metastasis.
In conclusion, the present study demonstrated that
cytoplasmic SETDB1 was associated with BC metastasis and the
function of cytoplasmic SETDB1 in migration, EMT and metabolic
reprogramming was blocked by a CMR1 inhibitor. Mechanically,
cytoplasmic SETDB1 positively regulated the c-MYC/LDHA pathway and
enhanced snail family transcriptional repressors. SETDB1 may be a
potential target for individuals with MBC. Additionally,
medications that aim at different regions, such as the NES/NLS
regions responsible for SETDB1 nucleocytoplasmic shuttling, may
offer therapeutic options (52).
The present study provided insight into the function and mechanism
of cytoplasmic SETDB1 in MBC and suggested that a CRM1 inhibitor
may be a suitable option for patients with high cytoplasmic SETDB1
expression.
Supplementary Data
Availability of data and materials
The data generated in the present study may be found
in the Gene Expression Omnibus under accession number GSE253717 or
at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE253717.
Authors' contributions
SH and SY conceived and designed the study. TW
collected samples and patient information. WY, YW, TW, YX, XJ and
HQ performed experiments and analyzed the data. SH, SY and WY wrote
and revised the manuscript. SH and WY confirm the authenticity of
all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All primary BC samples were obtained from Affiliated
Hospital of Jining Medical University under protocols approved by
the Ethics Committee of Jining Medical University (approval no.
2021B090; Jining, China. Written informed consent was obtained from
all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by National Natural Science
Foundation of China (grant no. 82103573), Shandong Provincial
Natural Science Foundation, China (grant no. ZR2019BH047), Research
Fund for Lin He's Academician Workstation of New Medicine and
Clinical Translation in Jining Medical University (grant no.
JYHL2018FZD07) and National Natural Cultivation Project of Jining
Medical University (grant no. JYP2019KJ27).
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hotton J, Lusque A, Leufflen L, Campone M,
Levy C, Honart JF, Mailliez A, Debled M, Gutowski M, Leheurteur M,
et al: Early locoregional breast surgery and survival in de novo
metastatic breast cancer in the multicenter national ESME cohort.
Ann Surg. 277:e153–e161. 2023. View Article : Google Scholar
|
|
3
|
Wang H, An W, Cao R, Xia L,
Erdjument-Bromage H, Chatton B, Tempst P, Roeder RG and Zhang Y:
mAM facilitates conversion by ESET of dimethyl to trimethyl lysine
9 of histone H3 to cause transcriptional repression. Mol Cell.
12:475–487. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schultz DC, Ayyanathan K, Negorev D, Maul
GG and Rauscher FJ III: SETDB1: A novel KAP-1-associated histone
H3, lysine 9-specific methyltransferase that contributes to
HP1-mediated silencing of euchromatic genes by KRAB zinc-finger
proteins. Genes Dev. 16:919–932. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rodriguez-Paredes M, Martinez de Paz A,
Simó-Riudalbas L, Sayols S, Moutinho C, Moran S, Villanueva A,
Vázquez-Cedeira M, Lazo PA, Carneiro F, et al: Gene amplification
of the histone methyltransferase SETDB1 contributes to human lung
tumorigenesis. Oncogene. 33:2807–2813. 2014. View Article : Google Scholar :
|
|
6
|
Cruz-Tapias P, Zakharova V,
Perez-Fernandez OM, Mantilla W, Ramírez-Clavijo S and Ait-Si-Ali S:
Expression of the major and pro-oncogenic H3K9 lysine
methyltransferase SETDB1 in non-small cell lung cancer. Cancers
(Basel). 11:11342019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wong CM, Wei L, Law CT, Ho DW, Tsang FH,
Au SL, Sze KM, Lee JM, Wong CC and Ng IO: Up-regulation of histone
methyltransferase SETDB1 by multiple mechanisms in hepatocellular
carcinoma promotes cancer metastasis. Hepatology. 63:474–487. 2016.
View Article : Google Scholar
|
|
8
|
Fazio M, van Rooijen E, Mito JK, Modhurima
R, Weiskopf E, Yang S and Zon LI: Recurrent co-alteration of HDGF
and SETDB1 on chromosome 1q drives cutaneous melanoma progression
and poor prognosis. Pigment Cell Melanoma Res. 34:641–647. 2021.
View Article : Google Scholar :
|
|
9
|
Ryu TY, Kim K, Kim SK, Oh JH, Min JK, Jung
CR, Son MY, Kim DS and Cho HS: SETDB1 regulates SMAD7 expression
for breast cancer metastasis. BMB Rep. 52:139–144. 2019. View Article : Google Scholar :
|
|
10
|
Strepkos D, Markouli M, Klonou A,
Papavassiliou AG and Piperi C: Histone methyltransferase SETDB1: A
common denominator of tumorigenesis with therapeutic potential.
Cancer Res. 81:525–534. 2021. View Article : Google Scholar
|
|
11
|
Liu L, Kimball S, Liu H, Holowatyj A and
Yang ZQ: Genetic alterations of histone lysine methyltransferases
and their significance in breast cancer. Oncotarget. 6:2466–2482.
2015. View Article : Google Scholar
|
|
12
|
Xiao JF, Sun QY, Ding LW, Chien W, Liu XY,
Mayakonda A, Jiang YY, Loh XY, Ran XB, Doan NB, et al: The
c-MYC-BMI1 axis is essential for SETDB1-mediated breast
tumourigenesis. J Pathol. 246:89–102. 2018. View Article : Google Scholar
|
|
13
|
Regina C, Compagnone M, Peschiaroli A,
Lena A, Annicchiarico-Petruzzelli M, Piro MC, Melino G and Candi E:
Setdb1, a novel interactor of ΔNp63, is involved in breast
tumorigenesis. Oncotarget. 7:28836–28848. 2016. View Article : Google Scholar
|
|
14
|
Zhang H, Cai K, Wang J, Wang X, Cheng K,
Shi F, Jiang L, Zhang Y and Dou J: MiR-7, inhibited indirectly by
lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of
breast cancer stem cells by downregulating the STAT3 pathway. Stem
Cells. 32:2858–2868. 2014. View Article : Google Scholar
|
|
15
|
Wu M, Fan B, Guo Q, Li Y, Chen R, Lv N,
Diao Y and Luo Y: Knockdown of SETDB1 inhibits breast cancer
progression by miR-381-3p-related regulation. Biol Res. 51:392018.
View Article : Google Scholar
|
|
16
|
Tachibana K, Gotoh E, Kawamata N, Ishimoto
K, Uchihara Y, Iwanari H, Sugiyama A, Kawamura T, Mochizuki Y,
Tanaka T, et al: Analysis of the subcellular localization of the
human histone methyltransferase SETDB1. Biochem Biophys Res Commun.
465:725–731. 2015. View Article : Google Scholar
|
|
17
|
Cho S, Park JS and Kang YK: Regulated
nuclear entry of over-expressed Setdb1. Genes Cells. 18:694–703.
2013. View Article : Google Scholar
|
|
18
|
Beyer S, Pontis J, Schirwis E, Battisti V,
Rudolf A, Le Grand F and Ait-Si-Ali S: Canonical Wnt signalling
regulates nuclear export of Setdb1 during skeletal muscle terminal
differentiation. Cell Discov. 2:160372016. View Article : Google Scholar
|
|
19
|
Guo J, Dai X, Laurent B, Zheng N, Gan W,
Zhang J, Guo A, Yuan M, Liu P, Asara JM, et al: AKT methylation by
SETDB1 promotes AKT kinase activity and oncogenic functions. Nat
Cell Biol. 21:226–237. 2019. View Article : Google Scholar
|
|
20
|
Wang G, Long J, Gao Y, Zhang W, Han F, Xu
C, Sun L, Yang SC, Lan J, Hou Z, et al: SETDB1-mediated methylation
of Akt promotes its K63-linked ubiquitination and activation
leading to tumorigenesis. Nat Cell Biol. 21:214–225. 2019.
View Article : Google Scholar
|
|
21
|
Yang W, Su Y, Hou C, Chen L, Zhou D, Ren
K, Zhou Z, Zhang R and Liu X: SETDB1 induces epithelial-mesenchymal
transition in breast carcinoma by directly binding with Snail
promoter. Oncol Rep. 41:1284–1292. 2019.
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar
|
|
24
|
DeBerardinis RJ and Chandel NS:
Fundamentals of cancer metabolism. Sci Adv. 2:e16002002016.
View Article : Google Scholar
|
|
25
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47. 2016.
View Article : Google Scholar
|
|
26
|
Shim H, Dolde C, Lewis BC, Wu CS, Dang G,
Jungmann RA, Dalla-Favera R and Dang CV: c-Myc transactivation of
LDH-A: Implications for tumor metabolism and growth. Proc Natl Acad
Sci USA. 94:6658–6663. 1997. View Article : Google Scholar
|
|
27
|
Shi MY, Wang Y, Shi Y, Tian R, Chen X,
Zhang H, Wang K, Chen Z and Chen R: SETDB1-mediated CD147-K71
di-methylation promotes cell apoptosis in non-small cell lung
cancer. Genes Dis. 11:978–992. 2023. View Article : Google Scholar
|
|
28
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar
|
|
29
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar
|
|
30
|
Lee ST, Li Z, Wu Z, Aau M, Guan P,
Karuturi RK, Liou YC and Yu Q: Context-specific regulation of NF-κB
target gene expression by EZH2 in breast cancers. Mol Cell.
43:798–810. 2011. View Article : Google Scholar
|
|
31
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View Article : Google Scholar :
|
|
32
|
Tsusaka T, Shimura C and Shinkai Y: ATF7IP
regulates SETDB1 nuclear localization and increases its
ubiquitination. EMBO Rep. 20:e482972019. View Article : Google Scholar :
|
|
33
|
Zheng Q, Cao Y, Chen Y, Wang J, Fan Q,
Huang X, Wang Y, Wang T, Wang X, Ma J and Cheng J: Senp2 regulates
adipose lipid storage by de-SUMOylation of Setdb1. J Mol Cell Biol.
10:258–266. 2018. View Article : Google Scholar
|
|
34
|
Liu S, Brind'Amour J, Karimi MM, Shirane
K, Bogutz A, Lefebvre L, Sasaki H, Shinkai Y and Lorincz MC: Setdb1
is required for germline development and silencing of
H3K9me3-marked endogenous retroviruses in primordial germ cells.
Genes Dev. 28:2041–2055. 2014. View Article : Google Scholar
|
|
35
|
Collins PL, Kyle KE, Egawa T, Shinkai Y
and Oltz EM: The histone methyltransferase SETDB1 represses
endogenous and exogenous retroviruses in B lymphocytes. Proc Natl
Acad Sci USA. 112:8367–8372. 2015. View Article : Google Scholar
|
|
36
|
Pasquarella A, Ebert A, Pereira de Almeida
G, Hinterberger M, Kazerani M, Nuber A, Ellwart J, Klein L,
Busslinger M and Schotta G: Retrotransposon derepression leads to
activation of the unfolded protein response and apoptosis in pro-B
cells. Development. 143:1788–1799. 2016.
|
|
37
|
Martin FJ, Xu Y, Lohmann F, Ciccone DN,
Nicholson TB, Loureiro JJ, Chen T and Huang Q: KMT1E-mediated
chromatin modifications at the FcγRIIb promoter regulate thymocyte
development. Genes Immun. 16:162–169. 2015. View Article : Google Scholar
|
|
38
|
Takikita S, Muro R, Takai T, Otsubo T,
Kawamura YI, Dohi T, Oda H, Kitajima M, Oshima K, Hattori M, et al:
A histone methyltransferase ESET is critical for T cell
development. J Immunol. 197:2269–2279. 2016. View Article : Google Scholar
|
|
39
|
Yizhak K, Le Dévédec SE, Rogkoti VM,
Baenke F, de Boer VC, Frezza C, Schulze A, van de Water B and
Ruppin E: A computational study of the Warburg effect identifies
metabolic targets inhibiting cancer migration. Mol Syst Biol.
10:7442014. View Article : Google Scholar
|
|
40
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar
|
|
41
|
Gao H, Yu Z, Bi D, Jiang L, Cui Y, Sun J
and Ma R: Akt/PKB interacts with the histone H3 methyltransferase
SETDB1 and coordinates to silence gene expression. Mol Cell
Biochem. 305:35–44. 2007. View Article : Google Scholar
|
|
42
|
Liu T, Chen X, Li T, Li X, Lyu Y, Fan X,
Zhang P and Zeng W: Histone methyltransferase SETDB1 maintains
survival of mouse spermatogonial stem/progenitor cells via
PTEN/AKT/FOXO1 pathway. Biochim Biophys Acta Gene Regul Mech.
1860:1094–1102. 2017. View Article : Google Scholar
|
|
43
|
Noh HJ, Kim KA and Kim KC: p53
down-regulates SETDB1 gene expression during paclitaxel
induced-cell death. Biochem Biophys Res Commun. 446:43–48. 2014.
View Article : Google Scholar
|
|
44
|
Chen K, Zhang F, Ding J, Liang Y, Zhan Z,
Zhan Y, Chen LH and Ding Y: Histone methyltransferase SETDB1
promotes the progression of colorectal cancer by inhibiting the
expression of TP53. J Cancer. 8:3318–3330. 2017. View Article : Google Scholar
|
|
45
|
Osaka N and Sasaki AT: Beyond Warburg:
LDHA activates RAC for tumour growth. Nat Metab. 4:1623–1625. 2022.
View Article : Google Scholar
|
|
46
|
Hou X, Shi X, Zhang W, Li D, Hu L, Yang J,
Zhao J, Wei S, Wei X, Ruan X, et al: LDHA induces EMT gene
transcription and regulates autophagy to promote the metastasis and
tumorigenesis of papillary thyroid carcinoma. Cell Death Dis.
12:3472021. View Article : Google Scholar
|
|
47
|
Cai H, Li J, Zhang Y, Liao Y, Zhu Y, Wang
C and Hou J: LDHA promotes oral squamous cell carcinoma progression
through facilitating glycolysis and epithelial-mesenchymal
transition. Front Oncol. 9:14462019. View Article : Google Scholar
|
|
48
|
Herz HM, Garruss A and Shilatifard A: SET
for life: Biochemical activities and biological functions of SET
domain-containing proteins. Trends Biochem Sci. 38:621–639. 2013.
View Article : Google Scholar
|
|
49
|
Van Duyne R, Easley R, Wu W, Berro R,
Pedati C, Klase Z, Kehn-Hall K, Flynn EK, Symer DE and Kashanchi F:
Lysine methylation of HIV-1 Tat regulates transcriptional activity
of the viral LTR. Retrovirology. 5:402008. View Article : Google Scholar
|
|
50
|
Fei Q, Shang K, Zhang J, Chuai S, Kong D,
Zhou T, Fu S, Liang Y, Li C, Chen Z, et al: Histone
methyltransferase SETDB1 regulates liver cancer cell growth through
methylation of p53. Nat Commun. 6:86512015. View Article : Google Scholar
|
|
51
|
Zhu H, Yang Y, Wang L, Xu X, Wang T and
Qian H: Leptomycin B inhibits the proliferation, migration, and
invasion of cultured gastric carcinoma cells. Biosci Biotechnol
Biochem. 84:290–296. 2020. View Article : Google Scholar
|
|
52
|
Batham J, Lim PS and Rao S: SETDB-1: A
potential epigenetic regulator in breast cancer metastasis. Cancers
(Basel). 11:11432019. View Article : Google Scholar
|