Introduction
Breast cancer, which starts as a local disease and
can metastasize to distant organs, is the second leading cause of
cancer-related mortality in women (1,2). The
conversion of early stage tumors into invasive malignancies has
been associated with the activation of the epithelial-mesenchymal
transition (EMT), defined as changes in cell phenotype from an
epithelial to a mesenchymal state, which is both a fundamental
event and a hallmark in tumorigenesis (3–9). The
acquisition of mesenchymal properties through EMT can promote the
detachment of cancer cells from the primary tumor and facilitate
their subsequent migration, thus allowing the progression of
metastasis to proceed (8,10). Recent studies have shown that
process is also associated with the acquisition of self-renewing,
tumor-initiating properties (11–13).
Despite the now recognized role of EMT in the metastatic cascade,
how stimuli-induced EMT occurs at the primary tumor site remains
largely unknown.
Epithelial cell tumors develop in a symbiotic manner
with the surrounding stroma. Tumor cells actively recruit cells,
including MSCs, into the tumor microenvironment, and these cells
may subsequently play an important role in facilitating cancer
progression (14). MSCs have been
shown to affect the morphology and proliferation of cells within
their vicinity through cell-to-cell interactions as well as through
the secretion of chemotactic cytokines and paracrine factors
(15–18). Accumulating evidence suggests that
the interactions between MSCs and breast cancer cells may impact
the phenotype of the cancer cells and promote their metastatic
potential (19–22). A pivotal study by Karnoub et
al reported that MSCs enhance breast cancer cell motility,
invasion and metastatic potential in vivo through CCL5
signaling, which confirms that these paracrine interactions play an
important role in the MSC-mediated metastatic spread (20). Further understanding of the
interactions between MSCs and breast cancer cells is required to
determine the role of MSCs in breast cancer progression or
therapeutics.
Among several growth factors that can act as
inducers of EMT, TGF-β has been found to play an important role at
particular stages of development and in disease processes such as
fibrosis and cancer metastasis (23–25).
TGF-β induces EMT activators: a group of transcription factors,
including ZEB1 and ZEB2, which repress epithelial gene expression
(26,27). Furthermore, ZEB1 and ZEB2 are
crucial targets of miR-200 family members (28–30),
and all miR-200 members are transcriptional targets of ZEB1 and
ZEB2 (31). Thus, ZEB factors and
miR-200 family members not only have opposite functions but also
reciprocally control the expression of each other. This
double-negative feedback loop between miR-200 family members and
ZEB1 allows for the plasticity that exists between the cell’s
epithelial and mesenchymal states (31,32).
Therefore, tilting to one side of the feedback loop instead of the
other, which allows for the stabilization of either an epithelial
or mesenchymal phenotype, may depend on several factors such as the
extracellular signals.
In this study, we co-cultured MCF7 human breast
cancer cells and hAD-MSCs in a transwell system to observe the
effect of hAD-MSCs on MCF7 cells. We aimed to elucidate the role of
hAD-MSCs paracrine signaling in the establishment and maintenance
of EMT in breast cancer cells and to detect the underlying
mechanisms.
Materials and methods
Culture of human adipose-derived
MSCs
Human adipose tissue was obtained from patients
undergoing tumescent liposuction following procedures approved by
the Ethics Committee at the Chinese Academy of Medical Sciences and
Peking Union Medical College. Isolation and cell culturing
protocols reported by Cao et al were used in this study
(33). hAD-MSCs at passage three
were employed.
Immunophenotype analysis
Prior to using them in experiments, MSCs were
evaluated for the expression of CD29, CD44, CD105, CD106, CD31,
CD34 and HLA-DR. The cells were detached and washed with
phosphate-buffered saline (PBS), then incubated with primary
antibodies for 30 min at 4°C. To detect intracellular antigens, the
cells were fixed in 2% paraformaldehyde at 4°C for 15 min and then
permeabilized with 0.1% saponin at room temperature for 1 h.
Working concentrations for primary antibodies (BD Biosciences, USA)
were 10–20 ng/ml. Same-species and isotype irrelevant antibodies
were used as negative control. After washing with PBS, the cells
were incubated with fluorescein isothiocyanate and
phycoerythrin-conjugated secondary antibodies at 4°C for 30 min.
The cells were then resuspended in PBS and analysed by a flow
cytometer (FACSCalibur; Becton-Dickinson, San Jose, CA). The
results were analysed by CellQuest Pro software (BD
Biosciences).
Culture of human tumor cell line
The human breast cancer cell line MCF7 was obtained
from the American Type Culture Collection (Manassas, VA, USA) and
grown in H-DMEM (Dulbecco’s modified Eagle’s medium; Gibco)
supplemented with 10% fetal bovine serum (FBS), 100 U/ml of
penicillin and 100 μg/ml of streptomycin.
Transwell co-culture experiment
In this assay, a transwell system with a
0.3-μm pore size permeable membrane (Corning Costar) was
used to separate MCF7 cells physically from hAD-MSCs. MCF7 cells
were seeded into the upper insert of a transwell system at a
density of 1×105 cells/well in 6-well plates in MSC
culture medium. hAD-MSCs were cultured in the lower chamber of this
co-culture system. The transwell co-culture system was used in all
experiments.
Cell cycle analysis
Briefly, 5.0×105 cells were harvested and
fixed in 75% cold ethanol, washed twice with cold PBS, and then
incubated in PBS containing 50 μg/ml propidium iodide (PI)
and 20 μg/ml RNaseA for 30 min at 37°C. PI and forward light
scattering were detected using a flow cytometer (FACSCalibur;
Becton-Dickinson). Experiments were performed in triplicate. A
percentage of cells in each phase of the cell cycle was
analyzed.
Cell proliferation assay
Cells were seeded in 96-well plates at
2.0×103 cells/well.
3-(4,-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay was used to detect viable proliferating cells at various time
points. The absorbance values for positive staining cells were
measured using a microplate reader (BioTek, USA) at a 570 nm
wavelength to determine the OD.
Soft-agar colony formation assay
The colony formation assay was performed to
determine the in vitro tumorigenesis of both MCF7 cells
before and after co-culturing with hAD-MSCs. Wells of a 6-well
plate (BD Falcon, USA) were pre-coated with 0.6% agar containing
H-DMEM and 10% FBS. Cells (1.0×103 cells/well) in 1.5 ml
of 0.3% agar containing H-DMEM and 10% FBS were then added to the
base. Cells were fed with fresh top agar every week. Colonies were
counted visually after two weeks by staining using a crystal violet
staining kit (Beyotime, China).
In vitro migration and invasion
assay
For invasion assays, MCF7 cells were cultured in
24-well Matrigel-coated invasion chambers (8-μm pore, BD
Biosciences). The lower chambers were filled with 0.75 ml H-DMEM
containing 10% FBS as a chemoattractant. A cell suspension of
5.0×104 cells in a volume of 0.5 ml H-DMEM was added to
the upper chamber. After the cells were incubated for 24 h at 37°C
in a humidified incubator with 5% CO2, the non-invading
cells that remained on the upper surface of the membrane were
removed by scraping. The invasive cells attached to the lower
surface of the membrane insert were fixed with 4% paraformaldehyde
at room temperature for 15 min and stained with crystal violet
staining solution for 30 min. The number of cells in each chamber
was then counted under a microscope.
For the transwell migration assays,
1.0×105 cells were plated into the upper chamber of the
membrane (8-μm pore, BD Biosciences) that had been
pre-coated with fibronectin (100 μg/ml) and 2.5% bovine
serum albumin (BSA). Cells were incubated for the indicated time
periods under standard cell culture conditions. Tumor cells
remaining on the top-side of the membrane were removed and cells
that had migrated to the bottom-side were fixed and stained as
described above. Five pre-selected fields per insert were
photographed. After the cells had been stained and photographed,
cells that had migrated to the bottom-side were washed using 33%
acetic acid, and then the absorbance values for positive staining
cells were measured using a microplate reader (BioTek) at a 570 nm
wavelength to determine the OD.
RNA-reverse transcription and real-time
PCR
Total RNA was extracted using the Trizol protocol
(Invitrogen, USA) and the cDNA was synthesized from the mRNA using
the PrimeScript II 1st Strand cDNA Synthesis system (Takara,
Japan). Relative expression of the genes of interest was assessed
by real-time PCR on an ABI StepOnePlusTM Fast Real-Time
PCR System (Applied Biosystems) using SYBR®-Green I-PCR
reaction mixture (Takara) and specific primers (Table I). The average of three independent
analyses for each gene and sample was calculated and normalized to
the endogenous reference control gene GAPDH.
 | Table IThe primers used for the Q-PCR
experiment. |
Table I
The primers used for the Q-PCR
experiment.
| Gene | Forward primer | Reverse primer |
|---|
| GAPDH |
GGTCACCAGGGCTGCTTTTA |
GGATCTCGCTCCTGGAAGATG |
| E-cadherin |
CCCACCACGTACAAGGGTC |
ATGCCATCGTTGTTCACTGGA |
| β-catenin |
GCTACTCAAGCTGATTTGATGGA |
GGTAGTGGCACCAGAATGGATT |
| ZO-1 |
CAACATACAGTGACGCTTCACA |
GACGTTTCCCCACTCTGAAAA |
| Fibronectin | CCCCATTC
CAGGACACTTCTG |
GCCCACGGTAACAACCTCTT |
| N-cadherin |
GAGGAGTCAGTGAAGGAGTCA |
GGCAAGTTGATTGGAGGGATG |
| Vimentin |
AGAACTTTGCCGTTGAAGCTG |
CCAGAGGGAGTGAATCCAGATTA |
| TWIST |
CGGACAAGCTGAGCAAGATT |
TGGAGGACCTGGTAGAGGAA |
| SNAIL |
AATCGGAAGCCTAACTACAGCG |
GTCCCAGATGAGCATTGGCA |
| ZEB1 |
AGTGATCCAGCCAAATGGAA |
TTTTTGGGCGGTGTAGAATC |
| ZEB2 |
AACAAGCCAATCCCAGGAG |
GTTGGCAATACCGTCATCCT |
Reverse transcription of miRNAs was performed using
the miScript Reverse Transcription Kit (Qiagen, China). Expression
of mature miRNAs was determined using miRNA-specific quantitative
real-time PCR (Qiagen). Real-time PCR data for mRNA and microRNA
were expressed relative to GAPDH or U6, respectively. The
expression levels were normalized to U6, an internal control, and
measured by the comparative Ct (ΔΔCt) method. The
miRNA-specific quantitative real-time PCR consisted of 40 cycles
(95°C for 5 sec and 60°C for 34 sec) after an initial denaturation
step (95°C for 10 sec).
Western blot analysis
The protein levels of TWIST, SNAIL, ZEB1, ZEB2, and
phosphorylated-SMAD2 in MCF7 cells cultured alone or co-cultured
with hAD-MSCs were analyzed by western blotting. Cells were
harvested in RIPA lysis buffer (Beyotime, China). After quantifying
by BCA assay, whole cell protein extracts were separated on an
SDS-10% polyacrylamide gel, and the proteins were transferred to a
nitrocellulose membrane (Bio-Rad). The membrane was blocked in TBS
containing 5% dried milk powder (w/v) and 0.1% Tween-20, and
hybridized with antibodies against TWIST (1:500, Abcam), SNAIL
(1:500, Santa Cruz), ZEB1 (1:500, Santa Cruz), ZEB2 (1:500, Santa
Cruz), phosphorylated SMAD2 (1:500, Santa Cruz) and β-actin (1:500,
Abcam), respectively. The levels of the above-mentioned proteins
were normalized to that of β-actin as a loading control.
Enzyme-linked immunosorbent assay
Concentrations of VEGF, HGF, NGF, FGF, SDF-1α,
TGFβ1, TGFβ2, and TGFβ3 were measured by ELISA (Shanghai Senxiong
Technology Industry, Shanghai, China), according to the
manufacturer’s instructions. Experiments were repeated at least
three times. Briefly, cells were treated under the appropriate
conditions and cell culture supernatants were collected,
centrifuged and filtered through a 0.45 μm Steriflip Filter
Unit (Millipore, USA). The absorbance (450 nm) for each sample was
analyzed by an ELISA reader and interpolated with a standard
curve.
Statistical analysis
Each experiment was repeated at least three times.
Statistical significance between treatment and control groups was
analyzed using the Student’s t-test. The data is represented as the
means ± SD. P<0.05 was regarded as a statistically significant
difference.
Results
Morphology and molecular phenotype of
hAD-MSCs
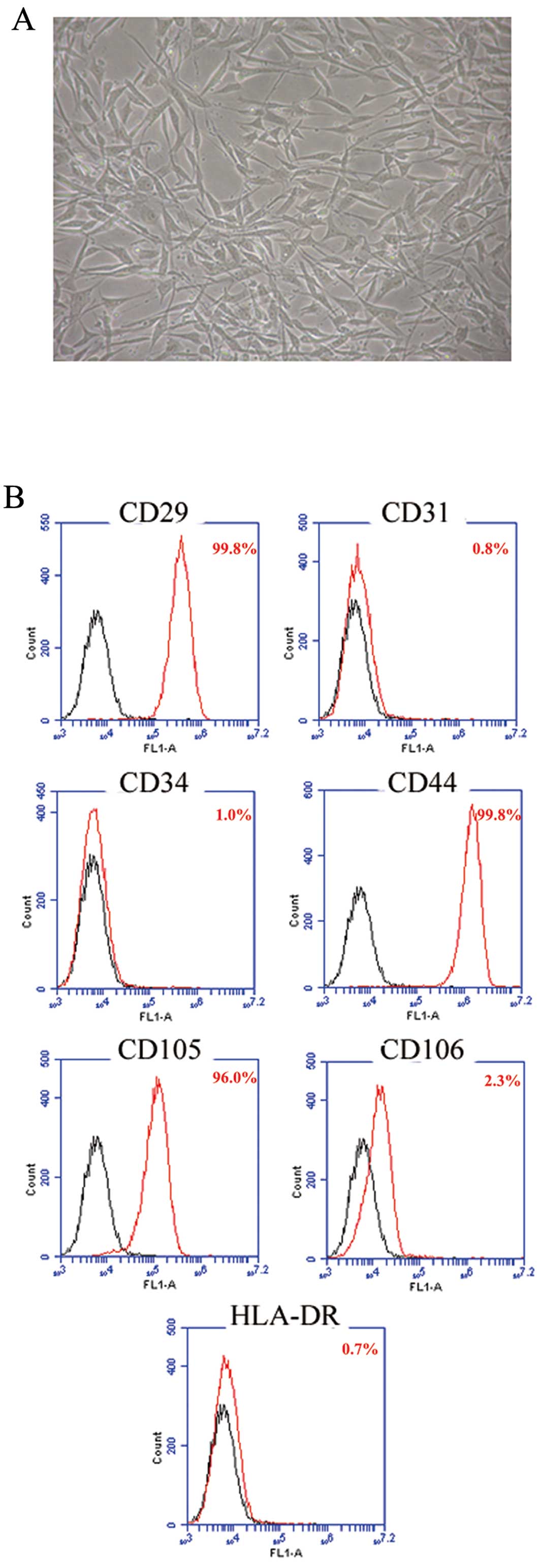
The cell morphology and the growth curve of hAD-MSCs
were evaluated after three passages. Cell morphology was
characterized by spindle or triangular-shaped adherent cells with
good refraction. The cell body contained a round nucleus with
uniform refractive index giving the nucleus a clear appearance and
one or more prominent nucleoli (Fig.
1A). Immunophenotype analysis showed that hAD-MSCs were
CD29+, CD44+, CD105+,
CD34−, CD31−, CD106− and
HLA-DR− (Fig. 1B).
These features are consistent with our previous findings (33).
Co-cultured MCF7 cells display EMT
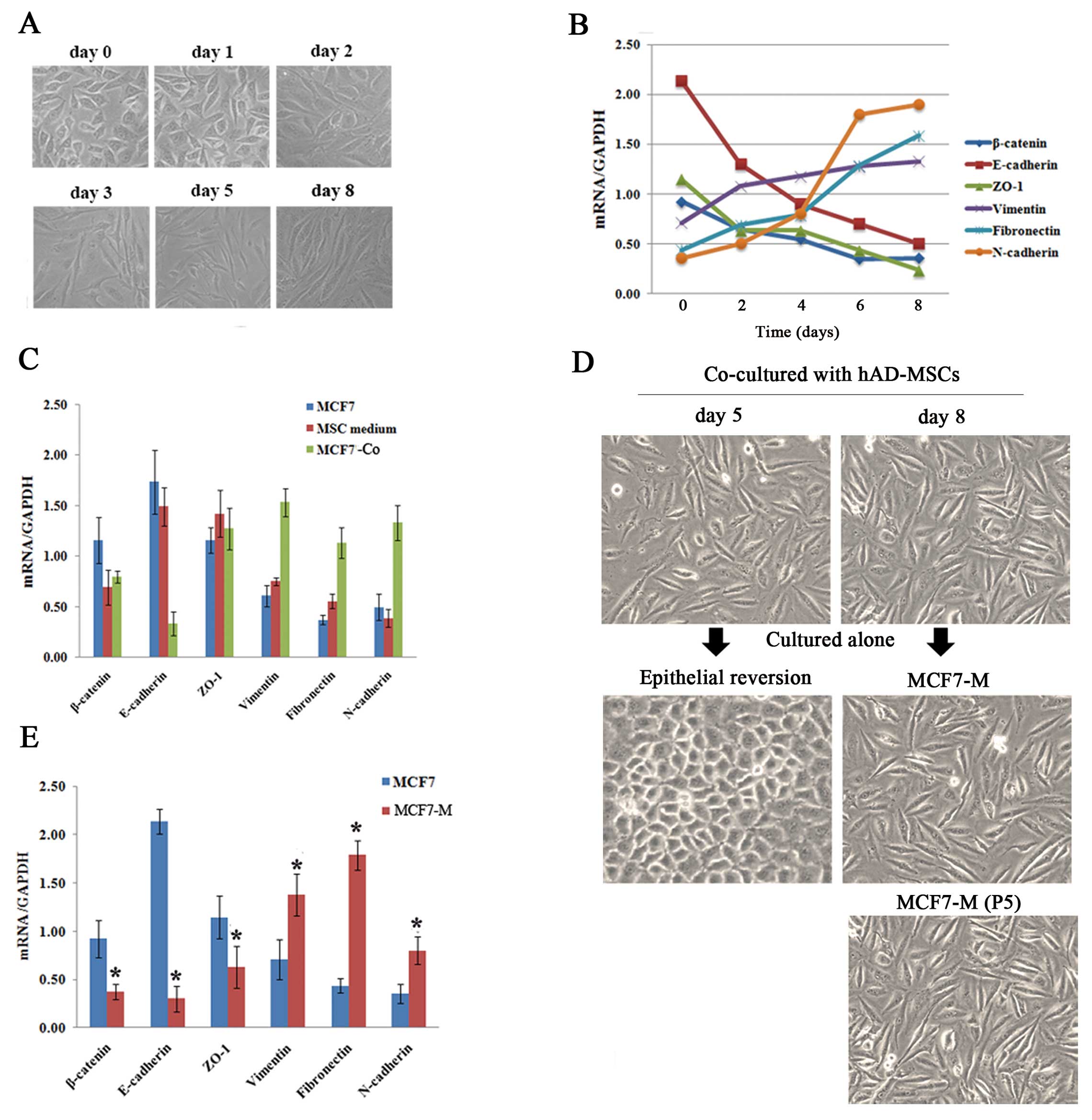
changes in a time-dependent manner
Fibroblast-like appearances that are consistent with
EMT were observed in MCF7 cells after co-culturing with hAD-MSCs
for 3 days (MCF7-Co) (Fig. 2A).
Quantification of the expression of EMT-related genes on days 0, 2,
4, 6 and 8 also showed a progressive decrease in epithelial makers
(E-cadherin, zonula occludens 1 (ZO-1), β-catenin) along with a
simultaneous increase in mesenchymal markers (N-cadherin, vimentin,
fibronectin) (Fig. 2B). To rule
out the effect of MSCs culture medium itself on MCF7 cells, MCF7
cells were cultured in MSC culture medium for 8 days, but unlike
co-culturing with hAD-MSCs, no EMT-related changes were observed
(Fig. 2C).
Then we cultured these mesenchymal MCF7-Co cells
obtained at different time points alone in standard MCF7 culture
medium, and we observed that the cells obtained at day 8 could
maintain a stable mesenchymal phenotype (MCF7-M) and propagate
after being passaged five times (Fig.
2D). Quantification by real-time PCR analysis demonstrated the
downregulation of epithelial markers and the upregulation of
mesenchymal markers in these MCF7-M cells (Fig. 2E).
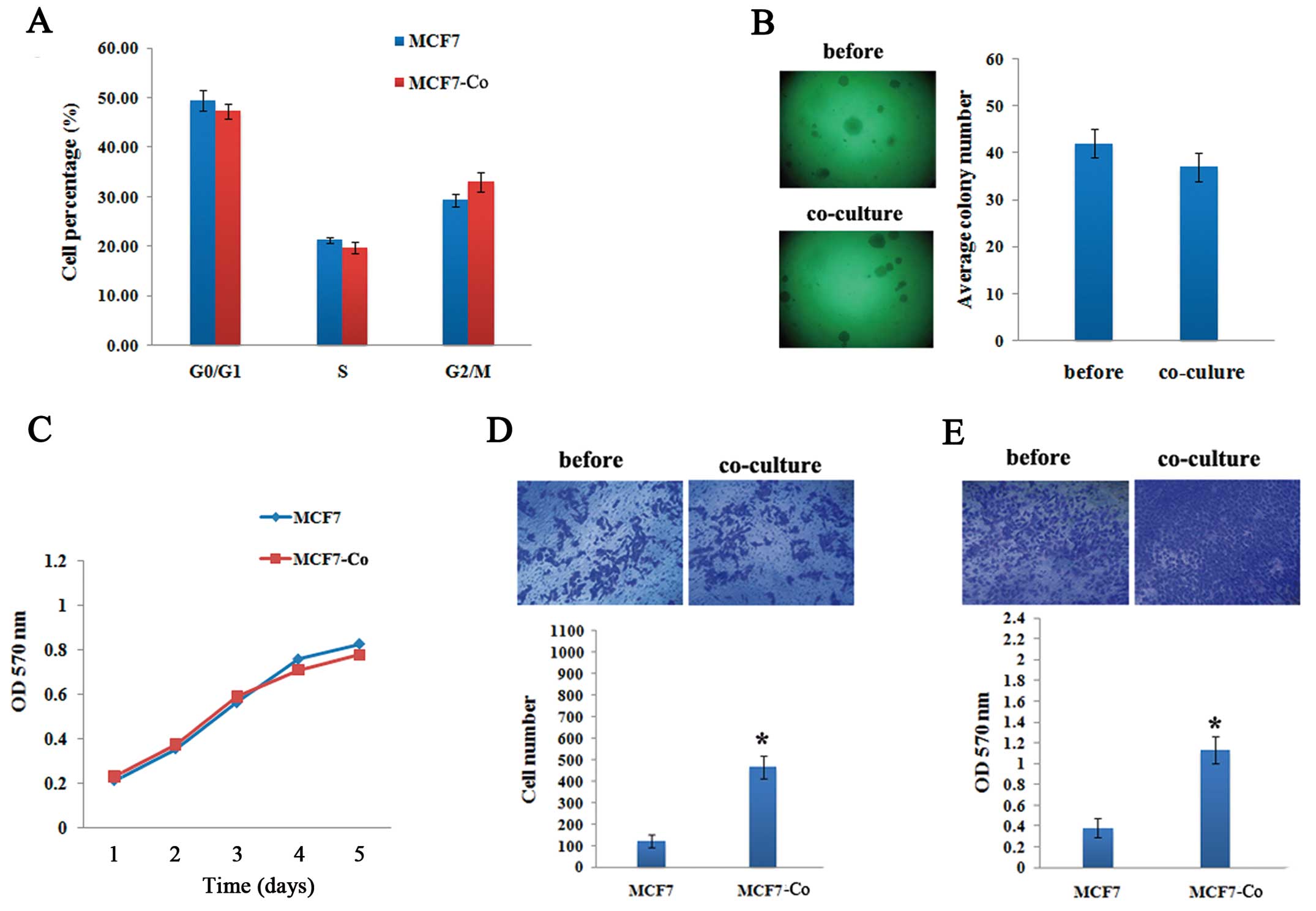
Effects of hAD-MSCs co-culturing on tumor
characteristics of breast cancer cells
To investigate the functional consequences of
hAD-MSCs co-culturing on breast cancer cells, we performed flow
cytometry, MTT, soft agar colony formation and transwell assays to
evaluate the malignant characteristics of both breast cancer cell
lines. No significant changes in cell cycle progression, cell
proliferative capacity and anchorage-independent growth were
observed in MCF7 cells after 72 h of co-culturing with hAD-MSCs
(Fig. 3A–C). In vitro
invasion and migration assays showed increased invasive and
migratory capacities at 3.8-fold and 2.97-fold respectively of
MCF7-Co compared with MCF7 cells (Fig.
3D and E).
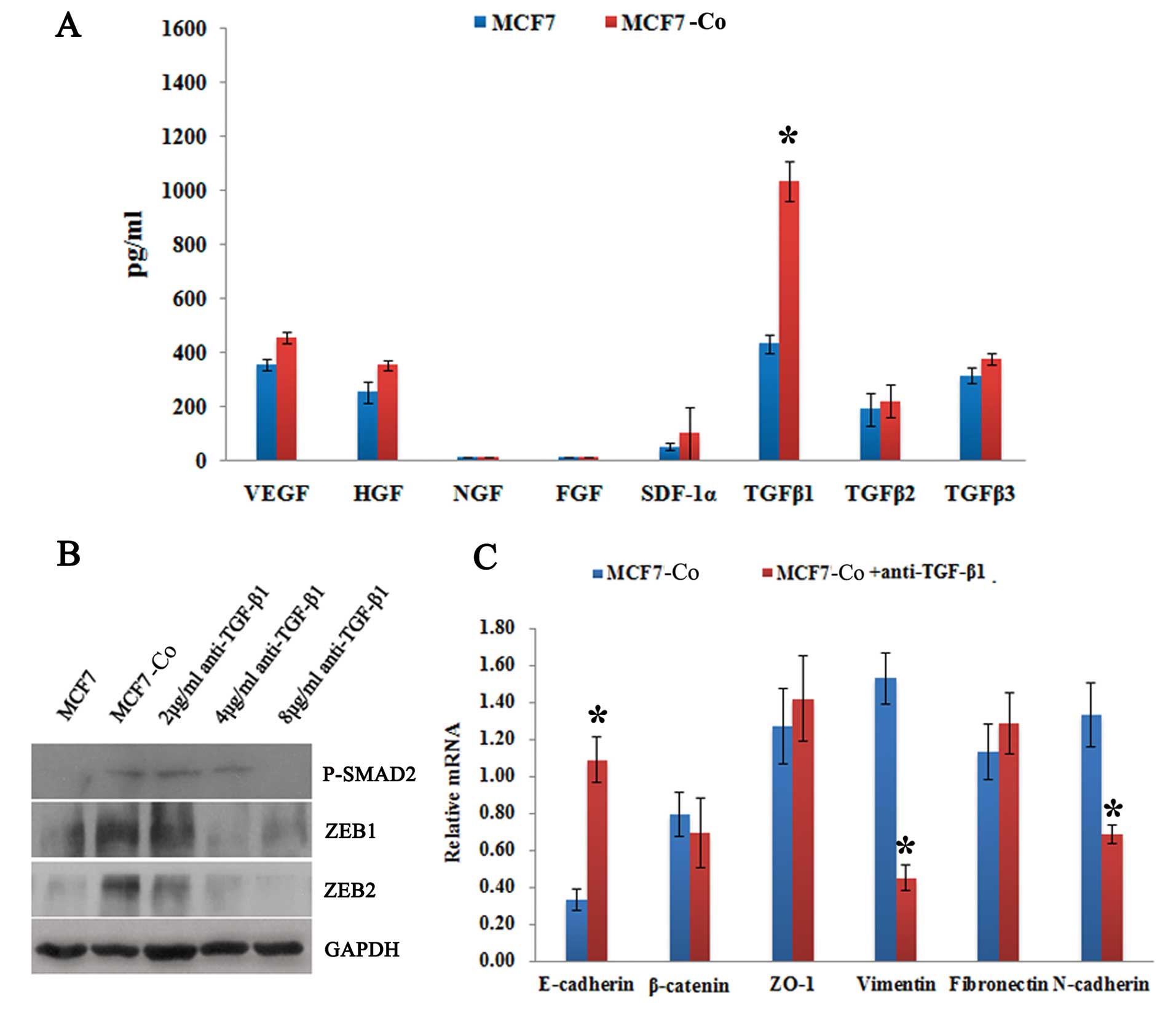
TGF-β1-stimulated EMT in MCF7 co-cultured
with hAD-MSCs
We hypothesized that the induction of EMT in MCF7
cells co-cultured with hAD-MSCs was due to cytokines secreted by
MSCs; thus, we tested the cell culture supernatant using the ELISA
assay. The results showed that the level of TGF-β1 was
significantly higher in the supernatant from co-culture system
compared with that from MCF7 cultured alone; differences in the
levels of TGF-β2 and TGF-β3 were very slight (Fig. 4A). Furthermore, the titrated
addition of various concentrations of anti-TGF-β1 antibody from day
0 to day 6 of the co-culture showed that the expression of
phosphorylated SMAD2 (P-SMAD2), which is under the regulation of
TGF-β1, was reduced in the MCF7 cells in a dose-dependent manner
(Fig. 4B). Concurrently, a marked
decrease in the level of vimentin transcript and a switch from
N-cadherin to E-cadherin was also observed (Fig. 4C). These results indicate that, in
our study, the hAD-MSC co-culture-induced EMT in MCF7 cells was
TGF-β1-dependent.
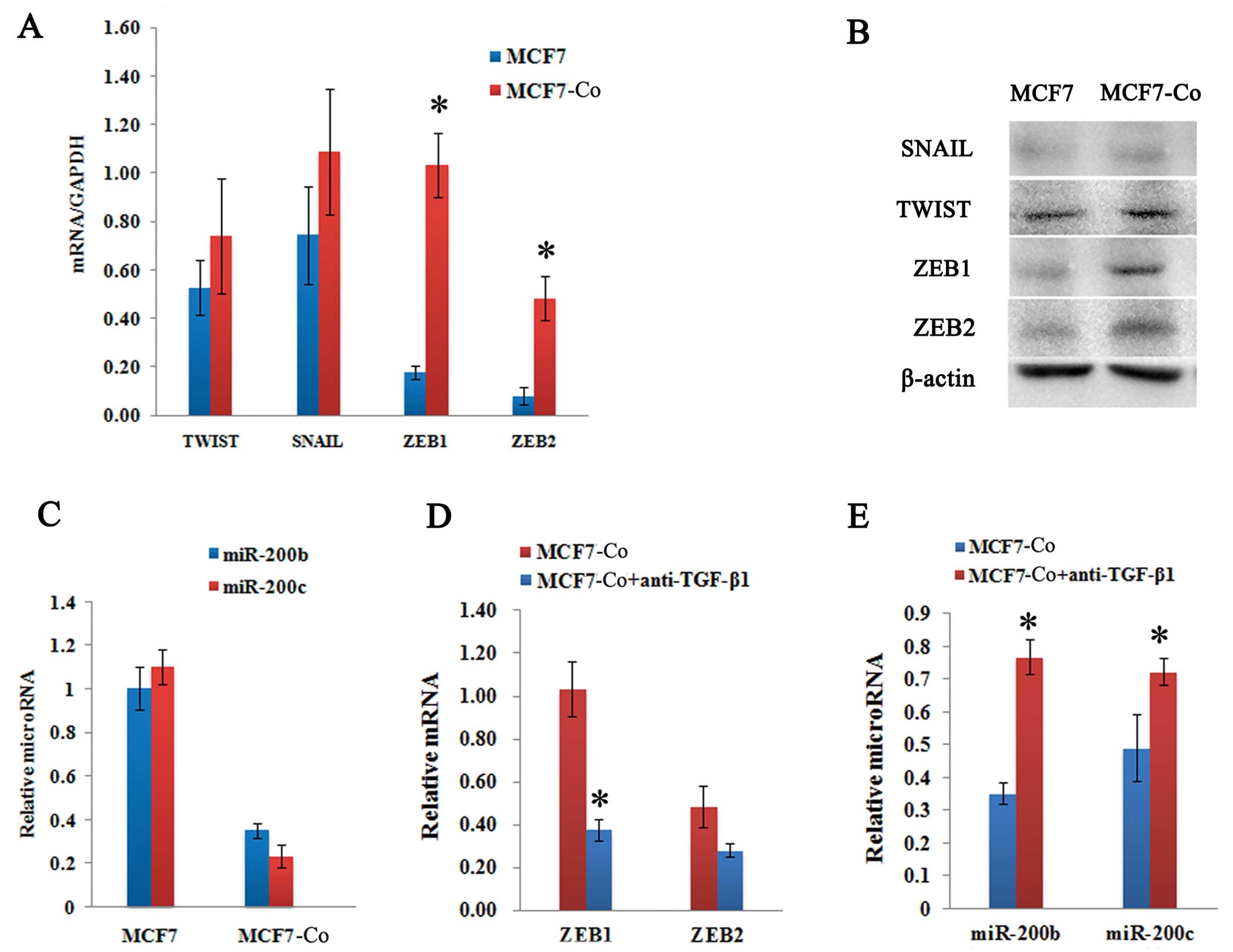
TGF-β targets the ZEB/miR-200 regulatory
loop
To test whether the ZEB/miR-200 regulatory loop was
involved in the induction of EMT, we determined the change in
ZEB1/2 and miR-200 expression levels in MCF7 cells before and after
co-culturing with hAD-MSCs. After 72 h of co-culturing, the
expression of ZEB1 and ZEB2 in MCF7-Co cells was significantly
upregulated as observed by both real-time PCR quantification
(Fig. 5A) and western blot
analysis (Fig. 5B). However, the
expression of SNAIL and TWIST did not exhibit significant
alterations (Fig. 5A and B). We
also observed that miR-200b and miR-200c were downregulated 3-fold
and 5-fold in MCF7-Co cells, respectively (Fig. 5C).
Furthermore, the addition of different
concentrations of anti-TGF-β1 antibody for an over 24 h period led
to a dose-dependent reduction of ZEB1/2 expression in the MCF7-Co
cells, which correlated with a decreased expression of
phosphorylated SMAD2 (P-SMAD2) (Fig.
4B). Moreover, miR-200b and miR-200c transcripts were
upregulated (Fig. 3E), and ZEB1
and ZEB2 mRNA levels were downregulated (Fig. 5D). These results are consistent
with the idea that the ZEB/miR-200 regulatory loop is regulated by
paracrine TGF-β signaling.
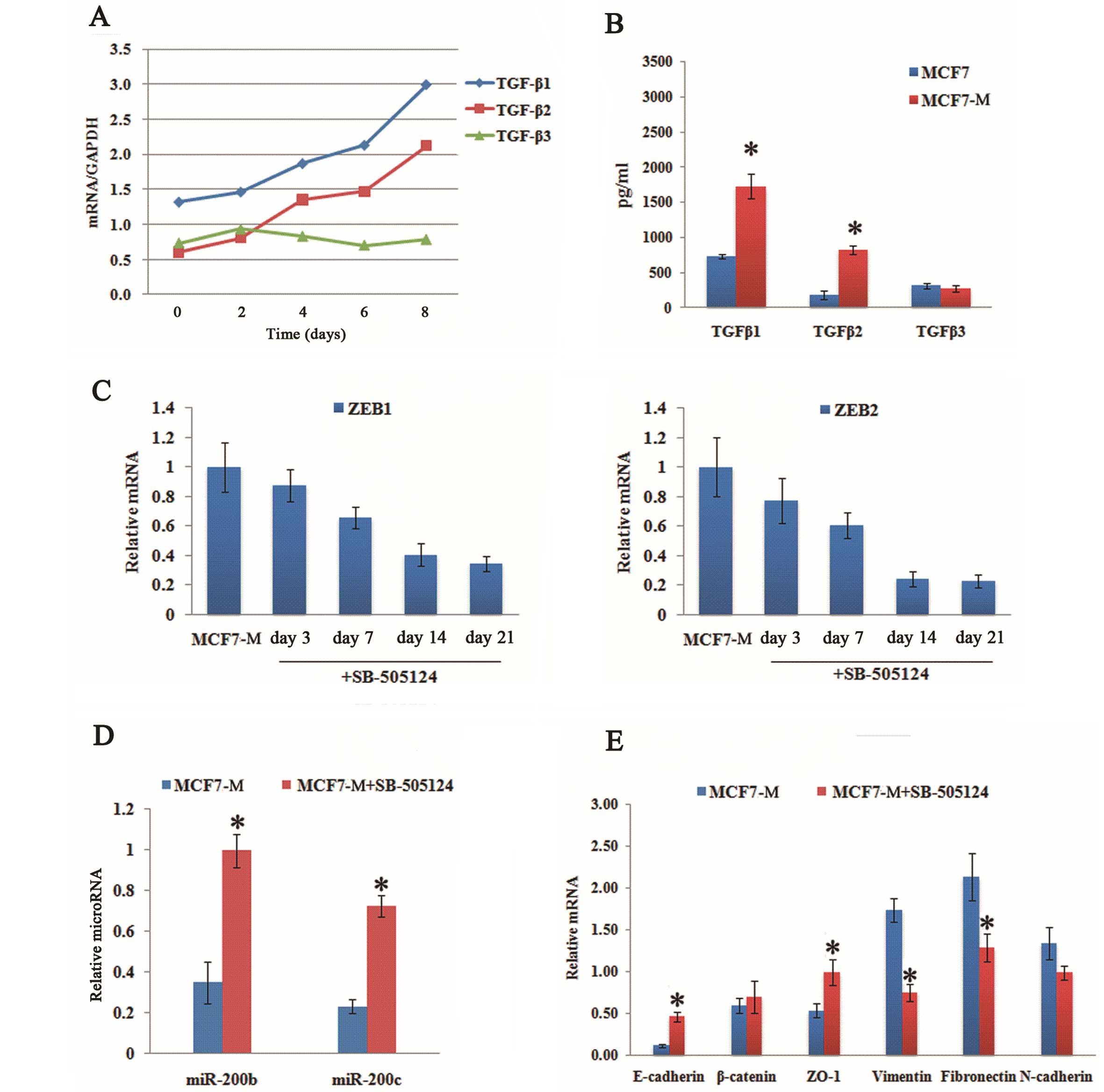
Autocrine TGF-β signaling is induced and
required for the maintenance of a mesenchymal state in MCF7
cells
It has been well documented in studies with other
EMT cell culture models that TGF-β can cooperate with other
signaling pathways to establish autocrine TGF-β signaling. We
hypothesized that prolonged co-culturing with hAD-MSCs might
initiate an autocrine TGF-β signaling in the MCF7 cells that would
ultimately enforce its mesenchymal state. To test this, we first
measured the expression of endogenous TGF-β1, TGF-β2, and TGF-β3
mRNA levels in MCF7 cells at varying time points after co-culturing
and we found that they were indeed progressively increased by
co-culturing (Fig. 6A). We also
observed that TGF-β1 and TGF-β2 proteins were being actively
secreted by stable mesenchymal state MCF7-M cells (Fig. 6B).
To determine whether the response of cells to this
endogenously synthesized TGF-β is important for mesenchymal
stability, we treated stable MCF7-M cells with an inhibitor of
TGF-β Receptor I, SB-505124. The addition of this inhibitor led to
a time-dependent decrease in ZEB mRNA levels (Fig. 6C), which is consistent with the
need for autocrine TGF-β production by MCF7-M cells for endogenous
ZEB transcription. Concomitant with the loss of ZEB expression, the
expression of miR-200 was increased (Fig. 6D), and accompanied by hallmark
epithelial features such as increased expression of E-cadherin and
ZO-1 (Fig. 6E). These data
collectively confirm that this epithelial reversion in MCF7-M cells
was caused by the inhibition of the TGF-β pathway.
Discussion
Previous studies investigating breast cancer cells
that were directly co-cultured with MSCs have shown significant
morphological alterations and the downregulation of E-cadherin
protein expression in the breast cancer cells (15,17).
In this study, we found that hAD-MSCs can not only play a role in
triggering EMT in MCF7 cells through paracrine TGF-β1 signaling,
but after prolonged co-culturing, they can also lead to the
maintenance of MCF7 cells in a stable mesenchymal state. These
results suggest that MSCs may promote breast cancer cell migration
by stimulating and facilitating the EMT.
Due to the fact that miR-200c and ZEB1 regulate each
other in a mutual, negative feedback loop (31), there is a critical threshold in the
balance between miR-200 and ZEB levels that determines whether
cells stably reside in a self-reinforcing epithelial or mesenchymal
state (34,35). Our findings have confirmed the
previously held opinion that TGF-β plays a pivotal role in this
double feedback loop between miR-200c and ZEB1 to ultimately
determine either an epithelial or mesenchymal phenotype (34). Paracrine TGF-β1 secreted by
hAD-MSCs results in an increased level of ZEB1 transcription in
MCF7 cells that can reach a point where ZEB1 transcription can
overcome the repression caused by miR-200. As ZEB1 proteins begin
to accumulate, they can then repress the miR-200 family members,
resulting in the progression of the EMT. The downregulation of
paracrine TGF-β1 signaling can reduce ZEB1 and ZEB2 expression,
upregulate miR-200b and miR-200c, and inhibit the progression of
the EMT. These results suggest that MSCs in the primary tumor
microenvironment may play a pivotal role in triggering EMT through
paracrine TGF-β signaling, which is followed by the targeting of
the ZEB/miR-200 regulatory loop in cancer cells. Our study provides
a mechanistic explanation for how MSCs might facilitate cancer
progression and metastasis in the tumor microenvironment.
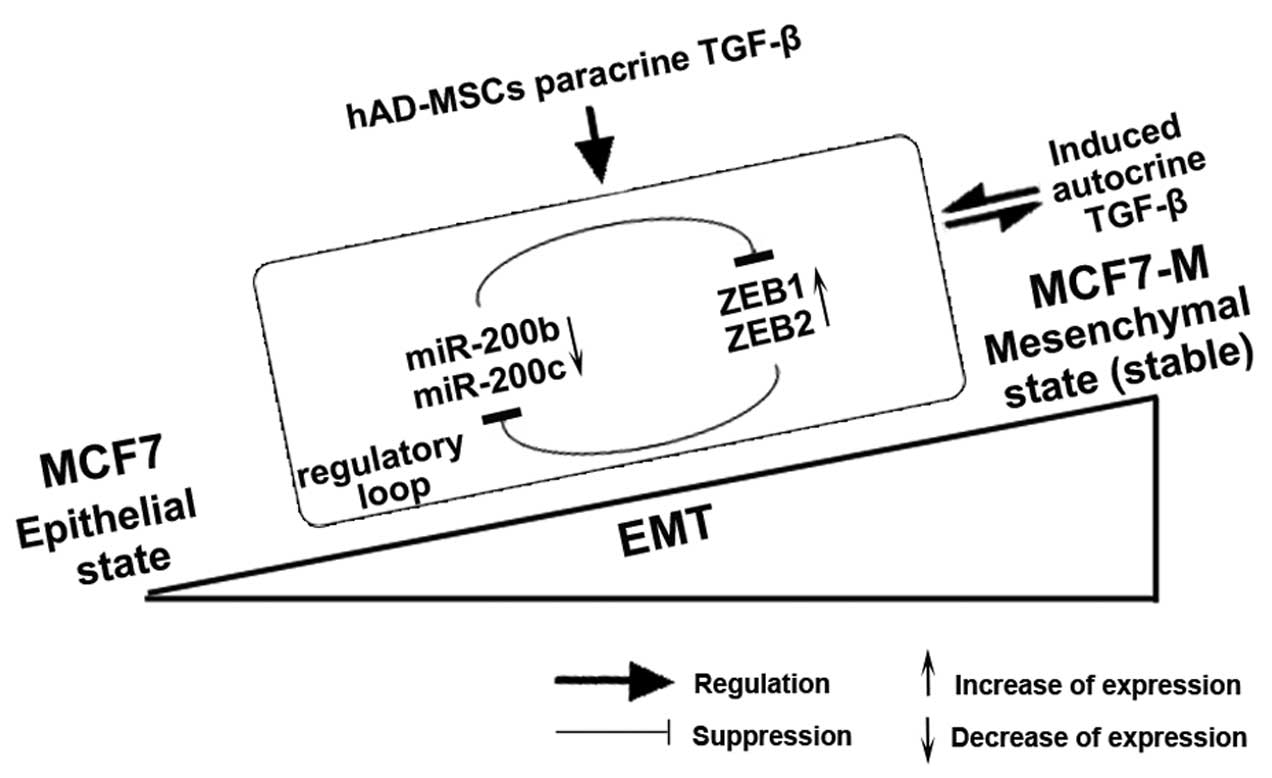
Our report shows that co-cultured MCF7 cells display
EMT changes in a time-dependent manner and that the transition
towards the mesenchymal state is only stabilized after 8 days of
co-culturing. We also found that autocrine TGF-β signaling is
initiated and required for the maintenance of a stable mesenchymal
state in MCF7 cells. Taken together, these results indicate that
threshold changes in ZEB1/2 and miR-200 levels may potentially
influence the autocrine TGF-β signaling, which is important in
determining the final outcome of the phenotypic state of the cell.
It has been shown that TGF-β2 is directly targeted by miR-141/200a
in breast and colon cancer cell lines (28), which suggests that the loss of
miR-200 family members during EMT might enhance autocrine TGF-β
signaling since the repression of TGF-β2 is also alleviated. The
subsequently initiated autocrine TGF-β signaling could drive and
sustain ZEB expression, which is necessary for stably maintaining
the cells in a self-reinforcing mesenchymal state. Thus, we deduce
that the paracrine TGF-β signaling by MSCs during the long-term
co-culture activates the autocrine TGF-β signaling in MCF7 cells
that is responsible for maintaining the MCF7 cells in a stable
mesenchymal state (Fig. 7).
Due to the clinical importance of EMT-induced
processes in understanding cancer progression, the inhibition of
EMT is an attractive therapeutic approach that could have
significant effects on disease outcome. Although the initial event
triggering these changes is still poorly understood, our study
suggests that targeting abnormal tumor-promoting paracrine
signaling between the epithelial tumor and MSCs may be a promising
approach for cancer prevention and treatment.
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition;
|
|
MSCs
|
mesenchymal stem cells;
|
|
hAD-MSCs
|
human adipose-derived MSCs;
|
|
MCF7-Co
|
MCF7 cells co-cultured with
hAD-MSCs;
|
|
MCF7-M
|
MCF7 cells with stable mesenchymal
state;
|
|
ZO-1
|
zonula occludens 1
|
Acknowledgements
The authors thank Professor Zengxuan
Song for the helpful discussion and critical reading of the
manuscript, Yi Lin for the critical reading of the manuscript,
Lianming Liao for the helpful discussion, and Kanghua Li and
Jiansuo Zhou for their technical assistance. This project was
supported by grants from the ‘863 Projects’ of the Ministry of
Science and Technology of PR China (no. 2011AA020100), the National
Natural Science Foundation of China (no. 30830052 and 30911130363),
the National Key Scientific Program of China (no. 2011CB964900),
and the Program for Changjiang Scholars and Innovative Research
Team in University-PCSIRT (no. IRT0909).
References
|
1.
|
Gupta GP and Massague J: Cancer
metastasis: building a framework. Cell. 127:679–695. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Weigelt B, Peterse JL and van’t Veer LJ:
Breast cancer metastasis: markers and models. Nat Rev Cancer.
5:591–602. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Kang Y and Massague J:
Epithelial-mesenchymal transitions: twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Trimboli AJ, Fukino K, de Bruin A, et al:
Direct evidence for epithelial-mesenchymal transitions in breast
cancer. Cancer Res. 68:937–945. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Baum B, Settleman J and Quinlan MP:
Transitions between epithelial and mesenchymal states in
development and disease. Semin Cell Dev Biol. 19:294–308. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Hugo H, Ackland ML, Blick T, et al:
Epithelial-mesenchymal and mesenchymal - epithelial transitions in
carcinoma progression. J Cell Physiol. 213:374–383. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: at the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Brabletz T, Jung A, Spaderna S, Hlubek F
and Kirchner T: Opinion: migrating cancer stem cells - an
integrated concept of malignant tumour progression. Nat Rev Cancer.
5:744–749. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Mani SA, Guo W, Liao MJ, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Gupta PB, Chaffer CL and Weinberg RA:
Cancer stem cells: mirage or reality? Nat Med. 15:1010–1012. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Polyak K and Weinberg RA: Transitions
between epithelial and mesenchymal states: acquisition of malignant
and stem cell traits. Nat Rev Cancer. 9:265–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Hu M and Polyak K: Molecular
characterisation of the tumour microenvironment in breast cancer.
Eur J Cancer. 44:2760–2765. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Fierro FA, Sierralta WD, Epunan MJ and
Minguell JJ: Marrow-derived mesenchymal stem cells: role in
epithelial tumor cell determination. Clin Exp Metastasis.
21:313–319. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Hombauer H and Minguell JJ: Selective
interactions between epithelial tumour cells and bone marrow
mesenchymal stem cells. Br J Cancer. 82:1290–1296. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Sasser AK, Mundy BL, Smith KM, et al:
Human bone marrow stromal cells enhance breast cancer cell growth
rates in a cell line-dependent manner when evaluated in 3D tumor
environments. Cancer Lett. 254:255–264. 2007. View Article : Google Scholar
|
|
18.
|
Chen J, Zhang ZG, Li Y, et al: Intravenous
administration of human bone marrow stromal cells induces
angiogenesis in the ischemic boundary zone after stroke in rats.
Circ Res. 92:692–699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Kumar S, Chanda D and Ponnazhagan S:
Therapeutic potential of genetically modified mesenchymal stem
cells. Gene Ther. 15:711–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Karnoub AE, Dash AB, Vo AP, et al:
Mesenchymal stem cells within tumour stroma promote breast cancer
metastasis. Nature. 449:557–563. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Dwyer RM, Potter-Beirne SM, Harrington KA,
et al: Monocyte chemotactic protein-1 secreted by primary breast
tumors stimulates migration of mesenchymal stem cells. Clin Cancer
Res. 13:5020–5027. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Molloy AP, Martin FT, Dwyer RM, et al:
Mesenchymal stem cell secretion of chemokines during
differentiation into osteoblasts, and their potential role in
mediating interactions with breast cancer cells. Int J Cancer.
124:326–332. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Zeisberg M and Kalluri R: The role of
epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med.
82:175–181. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Pardali K and Moustakas A: Actions of
TGF-beta as tumor suppressor and pro-metastatic factor in human
cancer. Biochim Biophys Acta. 1775:21–62. 2007.PubMed/NCBI
|
|
25.
|
Derynck R and Akhurst RJ: Differentiation
plasticity regulated by TGF-beta family proteins in development and
disease. Nat Cell Biol. 9:1000–1004. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: an alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Burk U, Schubert J, Wellner U, et al: A
reciprocal repression between ZEB1 and members of the miR-200
family promotes EMT and invasion in cancer cells. EMBO Rep.
9:582–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Gregory PA, Bert AG, Paterson EL, et al:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Bracken CP, Gregory PA, Kolesnikoff N, et
al: A double-negative feedback loop between ZEB1-SIP1 and the
microRNA-200 family regulates epithelial-mesenchymal transition.
Cancer Res. 68:7846–7854. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Gregory PA, Bracken CP, Bert AG and
Goodall GJ: MicroRNAs as regulators of epithelial-mesenchymal
transition. Cell Cycle. 7:3112–3118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Cao Y, Sun Z, Liao L, Meng Y, Han Q and
Zhao RC: Human adipose tissue-derived stem cells differentiate into
endothelial cells in vitro and improve postnatal neovascularization
in vivo. Biochem Biophys Res Commun. 332:370–379. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Gregory PA, Bracken CP, Smith E, et al: An
autocrine TGF-beta/ZEB/miR-200 signaling network regulates
establishment and maintenance of epithelial-mesenchymal transition.
Mol Biol Cell. 22:1686–1698. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Brabletz S and Brabletz T: The ZEB/miR-200
feedback loop - a motor of cellular plasticity in development and
cancer? EMBO Rep. 11:670–677. 2010. View Article : Google Scholar : PubMed/NCBI
|