Introduction
Gliomas are the most lethal brain tumors, they
account for ∼50% of all central nervous system tumors (1). According to the histopathological and
clinical criteria established by the World Health Organization
(WHO), tumors are graded on a scale of I to IV, in which
glioblastoma multiforme (GBM) is the most common and aggressive
form of brain tumor in adult (2),
its median survival ranges from 9 to 12 months. The etiology of
glioma is largely unknown, despite the advanced surgical, radiation
and medical therapies (3). To
clarify the genetic mechanisms in glioma pathogenesis has become an
important challenge for development of new therapies.
Increasing evidence show that post-transcriptional
regulation of gene expression is mediated by microRNAs (miRNAs)
playing an important role in a variety of cancers including gliomas
(4–7). miRNAs are a class of regulatory small
RNAs with 19–24 nucleotides (nt) that enable to downregulate
expression of their target genes by partially complementing with
targeting mRNAs. The miRNA target site has been considered to be
the 3′ untranslated region (3′ UTR) of mRNA, however, recent
studies have shown that mRNAs may also bind the coding regions or
the 5′ untranslated regions (5′ UTRs) (8,9). Due
to the miRNAs being implicated in the pathogenesis of various
cancers (6), they are considered
to be novel therapeutic targets. Many independent studies provide
evidence that miRNAs can act as oncogenes to reduce expression of
their target tumor suppressor genes in tumors, or tumor suppressors
to produce higher levels of target oncogene expression, leading to
neoplasia (6,10).
Two miRNA clusters, miR-23a∼27a∼24-2 and
miR-23b∼27b∼24-1 are found to be deregulated in a variety of tumors
(11,12). The former mainly produces mature
miR-23a-3p, miR-27a-3p and miR-24-3p; the latter mainly produces
miR-23b-3p, miR-27b-3p and miR-24-3p. miR-24-3p is a master
regulator (11). It is reported
that miR-24-3p was upregulated consistently during terminal
differentiation of hematopoietic cell line into a variety of
lineage (13), also during muscle
and neuronal cell differentiation (14,15).
Several studies have reported that miR-24-3p might function
differently in cell proliferation in different cells (12). For example, miR-24-3p was able to
inhibit cell proliferation in HeLa cells (16), in contrast, it promotes cell
proliferation of transforming growth factor β-treated
hepatocellular carcinoma cells (HuH7), as well as A549 lung
carcinoma cells (17). miR-27-3p
has been reported to regulate adipogenesis, myeloblasts
differentiation (18), skeletal
muscle development (19) and
osteoblast differentiation (20).
In breast cancer cells, miR-27a-3p could enhance the expression of
specificity protein (Sp) transcription factors which is
overexpressed in tumours where they contribute to the proliferative
and angiogenic phenotype by regulating number of angiogenic
proteins (21). miR-27a-3p along
with miR-27b-3p were upregulated in activated hepatic stellate
cells and promoted cell proliferation (22). The two clusters contain three
families of miRNAs, although each family of the miRNAs is
paralogous, they have complex expression patterns. The miRNAs of
miR-23a∼27a∼24-2 cluster are derived from one primary transcript
(23), in contrast, the miRNAs
derived from miR-23b∼27b∼24-1 cluster have different primary
transcripts for each pre-miRNA (24). Therefore the miRNA expression
patterns vary from different biological conditions (11). miR-24-3p was discovered to be
upregulated in the glioblastoma cell lines and miR-27a-3p was often
upregulated in glioblastoma tissue (25,26).
However, the function of these miRNAs in gliomas is still
unclear.
MXI1, a member of Mad (Mxi1) family, encodes a
protein with 228 amino acids. It can form a heterodimer with the
highly stably expressed protein Max to function as the antagonist
of c-Myc, a transcription factor belonging to the basic
helix-loop-helix-ZIP family, which is essential to cellular growth
and development and it is involved in the processes including
control of cell proliferation and induction of apoptosis (27,28).
The MXI1 gene localizes at chromosome 10q24-q25, deletion of this
sequence has been demonstrated in 60–80% of human glioblastoma
tumors (29–31) and in 20–30% of human prostate
tumors (32,33). Numerous studies have shown both
MXI1 and MAD can inhibit the function of c-Myc, so that they have
been considered to be tumor suppressor genes (28). The loss of MXI1 function might lead
to tumor progression (34,35).
Although miR-24-3p and miR-27a-3p derive from two
duplicated gene clusters of miR-23a∼27a∼24-2 and miR-23b∼27b∼24-1,
the roles of the two clusters and the two miRNAs in tumors have not
been completely characterized. In this study, both miR-24-3p and
miR-27a-3p have been shown to promote glioma cell proliferation. We
have identified three miRNAs from the two miRNA clusters which can
synergistically regulate MXI1. Furthermore, we have demonstrated
that miR-24-3p and miR-27a-3p promoted cell proliferation by
directly regulating MXI1, and miR-27a-3p was found significantly
upregulate in glioma tissues.
Materials and methods
Cell culture
Human glioma cell lines (U87 and U251) were
purchased from American Type Culture Collection (Manassas, VA,
USA). U87 and U251 cells were cultured in DMEM (Invitrogen Life
Technologies, USA) containing 10% fetal bovine serum (FBS,
Invitrogen Life Technologies), 100 U/ml penicillin and 100
μg/ml streptomycin (Invitrogen Life Technologies) at 37°C
with 5% CO2.
Human tissue samples
Archival frozen human glioma tissue samples
(including a total of 17 tumors and 17 peritumoral tissues) were
obtained from Shantou Hospital of Sun Yat-sen University in
accordance with the Committee on Human Research approved
procedures. All of the samples were obtained with informed consent
of the patients and were histologically confirmed. Total-RNA from
tissues was isolated by TRIzol (Invitrogen Life Technologies)
according to the manufacturer’s instructions.
Vector construction and transfection
To express miRNA, genomic fragments containing human
miRNA precursors with about 80 bp of flanking sequences in both
sides were amplified using the primers listed in Table I and cloned into the modified
pLL3.7 vector under the control of the human U6 promoter.
 | Table IThe primers used in our study. |
Table I
The primers used in our study.
| Gene name | Primer sequence (5′
to 3′) |
|---|
| Sensor24 | S1:
TCGAATAACTGTTCCTGCTCCCTGAGCCACGATCTGTTCCTGCTCCCTGAGCCA
AS1:
ACGCGTTGGCTCAGGGAGCAGGAACAGATCGTGGCTCAGGGAGCAGGAACAGTTAT
S2: ACGCGTCTGTTCCTGCTCCCTGAGCCATCACCTGTTCCTGCTCCCTGAGCCAC
AS2: GATCGTGGCTCAGGGAGCAGGAACAGGTGATGGCTCAGGGAGCAGGAACAG |
| Sensor23a | S1:
TCGAATAAGGAAATCCCTCTAATGTGATCGATGGAAATCCCTCTAATGTGAT
AS1:
ACGCGTATCACATTAGAGGGATTTCCATCGATCACATTAGAGGGATTTCCTTAT
S2: ACGCGTGGAAATCCCTCTAATGTGATTCACGGAAATCCCTCTAATGTGATC
AS2: GATCGTGGCTCAGGGAGCAGGAACAGGTGATGGCTCAGGGAGCAGGAACAG |
| Sensor27a | S1:
TCGAATAAGCGGAACTTACGACTGTGAACGATGCGGAACTTACGACTGTGAA
AS1:
ACGCGTTTCACAGTCGTAAGTTCCGCATCGTTCACAGTCGTAAGTTCCGCTTAT
S2: ACGCGTGCGGAACTTACGACTGTGAATCACGCGGAACTTACGACTGTGAAC
AS2: GATCGTTCACAGTCGTAAGTTCCGCGTGATTCACAGTCGTAAGTTCCGC |
| GAPDH | S:
CCCATGTTCGTCATGGGTGT
AS: TGGTCATGAGTCCTTCCACGATA |
| MXI1 | S:
ATTCCACTAGGACCAGACTGCACC
AS: GCTGGTGGTACTTATATTGTCCAC |
| MYC | S:
TCAAGAGGCGAACACACAAC
AS: GGCCTTTTCATTGTTTTCCA |
| MXI1-cDNA | S:
ATGGAGCGGGTGAAGATGATCAAC
AS: TGCACTGTTATGTCATGCTGGGT |
| MXI1-cDNA | S:
GACGCTGGATTTTTTTCGGGTAGTGG
AS: CTTACGCACAAGAGTTCCGTAGCTG |
| MXI1-3′UTR
Full | S:
CTCGAGTAGAACCCAGCATGACATAACAGTG
AS: GGATCCTTCTTCGTTCACAGTTTTTATTTCTTC |
| MXI1-UTR1 | S:
CCGCTCGAGGACATAACAGTGCAGGGCAAAATA
AS: CGGGATCCAAACAGCCAGGGGTAAGGTCTC |
| MXI1-UTR2 | S:
CCGCTCGAGATTGATAGATCTTTATGTTTAGATAGGGCTGGGCAAG
AS: CGGGATCCTCTTCGTTCACAGTTTTTATTTCTTC |
| MXI1-UTR1-MUT | S:
AGGCCAAGGTGACTCGGACAGCAGCATTTTTATTTC
AS: AATGCTGCTGTCCGAGTCACCTTGGCCTGCTAATCT |
| MXI1-UTR2-MUT | S:
CCGCTCGAGATTGATAGATCTTTATGTTTAGATAGGGCTGGGCAAG
AS: CGGGATCCTCTTCGTAGTGTCATTTTATTTCTTCCAATTAACTT |
To construct luciferase reporter vectors, full
length 3′ UTR sequence of MXI1, first half and second half of MXI1
3′ UTR or a mutated sequence of the 3′ UTR sequence were amplified
using the primers listed in Table
I and cloned downstream of Renilla luciferase in the
psiCHECK-2 vector (Promega, USA). miRNA sensors were constructed
according to the reported method (36) by inserting tandem four copies of
the complementary sequences of mature miRNAs at the 3′ UTR region
of the Renilla luciferase in psiCHECK-2 vector.
MXI1-cDNA was obtained from NCBI CDS database
(NM_005962.4) and constructed into XhoI and BamHI sites after the
CMV promoter of the cDNA expression vector pcDNA-neo, which is
reconstructed from pcDNA3.1+. The primers are represented in
Table I. miR-24-3p and miR-27a-3p
mimics, and the negative control mimics were synthesized and
purified by Gene Science & Health, China.
RNA extraction and real-time quantitative
RT-PCR
Total-RNA was extracted from frozen brain specimens
using TRIzol reagent (Invitrogen Life Technologies) and were
reverse-transcribed using ReverTraAce-α transcriptase (Toyobo,
Japan). The sequences of the forward and reverse primers for
MXI1/c-MYC listed in Table I. The
RNA was quantified and checked for purity by spectrophotometry at
260 and 280 nm and subsequently amplified by PCR using the Taq DNA
polymerase (Takara, Japan). The expression of human miR-27a-3p and
miR-24-3p was quantitated in human tissues using SYBR®
Premix Ex Taq™ (Tli RNaseH Plus) (Takara). Using U6 RNA as an
internal standard and sets of primers were purchased from Gene
Science & Health, China. The comparative Ct (ΔΔCt) method was
used to determine the expression fold change.
Western blotting
U87 or 293T cells were lysed in RAPI lysis buffer
(Bioteke, China) and concentration was determined by bicinchoninic
acid protein assay kit (Beyotime, China). Heat-denatured protein
samples (20 μg per lane) were loaded onto a 10%
SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to an
Immobilon-P membrane (Millipore, USA). The membrane was incubated
with a primary goat polyclonal antibody against human Mxi1 (1:500
dilution) or mouse monoclonal antibody against human β-actin
(1:2000 dilution) and then incubated for 1 h with a rabbit
anti-goat (1:10,000 dilution) or goat anti-mouse (1:10,000
dilution) secondary antibody. The bound antibody was detected with
the use of enhanced chemiluminescence detection reagents (Pierce,
USA) according to the manufacturer’s instructions.
The primary antibody and second antibody of Mxi1
were bought from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA) and Abcam Inc. While the primary antibody of β-actin and
second antibodies were purchased from Sigma-Aldrich, Inc. and
Jackson ImmunoResearch Inc.
Dual luciferase reporter assays
293T cells (2.5×104) in 100 μl
growth medium were plated in 96-well plates. The next day, the
cells were transfected with 100 ng psiCHECK2-MXI1-3′ UTR or
psiHECK2-MXI1-3′ UTR-MUT and 300 ng pre-miRNAs or 40 nM miRNA
mimics using Lipofectamine 2000 (Invitrogen Life Technologies) or
FuGene (Roche, Switzerland). The cells were harvested 48 h after
transfection and assayed using the Dual-Luciferase Reporter Assay
kit (Promega) according to the manufacturer’s instructions.
Transfection was repeated in triplicate.
MTT (dimethyl thiazolyl diphenyl
tetrazolium) assay
U87 cells were seeded at 3×103 cells per
well in 96-well plates. Negative control or miR-24-3p mimics were
transfected with Lipofectamine 2000 (Invitrogen Life Technologies)
or FuGene HD (Roche) according to the manufacturer’s instructions.
At different time points (24, 48, 72 and 98 h) post-transfection,
MTT reagent (5 mg/ml) was added directly to the medium and
incubated at 37°C, 5% CO2 incubator for 4 h. Then
supernatants were removed and 150 μl DMSO was added to
dissolve the formazan crystals, and thoroughly mixed for 10 min.
Optical densities at 490 nm were measured using culture medium as a
blank.
Lentivirus packaging
VSV-G pseudotyped lentiviruses were produced by
co-transfection of 293T cells. One day before transfection, 293T
cells were seeded in 100-mm dishes. 2.3 μg pMK-VSVG, 5
μg pMDL-G/P-RRE, 3.8 μg pRSV-REV and 7.6 μg
miRNA expression vector (pre-miR-24-2 or pre-miR-27a) were mixed
and transfected by Lipofectamine 2000 according to the
manufacturer’s instructions. The cells grew to about 80% after 72 h
post-transfection, the production medium containing lentivirus was
harvested, centrifuged to remove cell debris and viral supernatant
was used for infection.
Cell growth curve
U87 stable cell lines were established which
expressed miR-24-3p and miR-27a-3p, with empty vector as a control.
In day 0, 500 cells were seeded in 96-well plates. MTT assay were
performed on day 1, 3, 5 and 7. Absorbance was normalized by day 1
and cell growth curve of U87 stable cells were measured in 7
days.
Statistical analysis
Data are presented as the mean ± standard deviation
(SD) of at least three separate experiments. The data were analyzed
using the SPSS 12.0 Windows version software. Statistical analyses
were done by analysis of variance or Student’s t-test. p-value
<0.05 was considered statistically significant
(*p<0.05; **p<0.01;
***p<0.001).
Results
miR-24-3p and miR-27a-3p promote cell
proliferation in glioma cells
The miR-24-3p and miR-27a-3p have been found to be
abundant in glioma cells (11,26).
In order to evaluate the role of these two miRNAs in cell
proliferation, we first constructed miR-24-3p or miR-27a-3p
expression vectors by inserting miRNA precursor containing some
flanking sequences at both sides into the pLL3.7 vector. To measure
whether our miRNA constructs could efficiently produce mature
miRNAs, miRNA sensors were constructed according to the reported
method (36) by inserting tandem
four copies of the complementary sequences of mature miRNAs at the
3′ UTR region of the Renilla luciferase (Rluc) in psiCHECK-2
vector. Co-transfection of each miRNA expression vector (empty
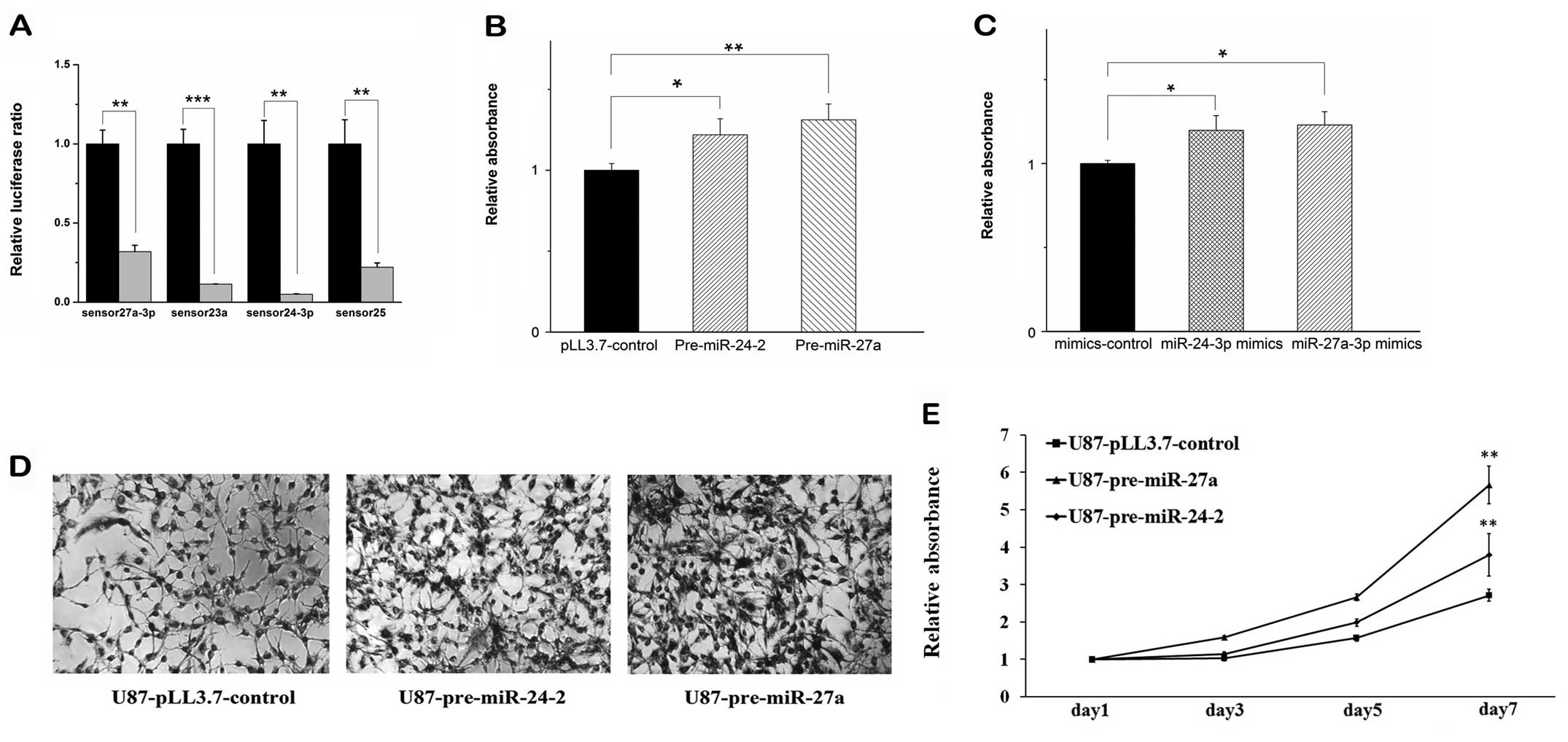
vector as control) and cognate sensor followed with dual luciferase
assay showed that the relative luciferase ratio declines
significantly while comparing with that of the control (Fig. 1A), indicating that our constructs
produced mature miRNAs efficiently. Then we performed transient
transfection of these vectors containing pre-miR-24-2 and
pre-miR-27a into U87 glioma cells, respectively. MTT assay at 72 h
post-transfection showed that overexpression of both pre-miR-24-2
and pre-miR-27a in U87 glioma cells were able to promote the cell
proliferation, respectively (Fig.
1B).
To confirm these results, the mimics of the
miR-24-3p and miR-27a-3p were performed for the same MTT experiment
using the validated constructs we have measured before. It was
shown that the mimics have similar effects to the overexpression of
the pre-miRNAs (Fig. 1C). Then, we
established stable U87 glioma cell lines that overexpress
pre-miR-24-2 (U87-pre-miR-24-2) or pre-miR-27a (U87-pre-miR-27a).
MTT assay revealed that overexpression of pre-miR-24-2 or
premiR-27a significantly increased cell proliferation in seven days
(Fig. 1D and E).
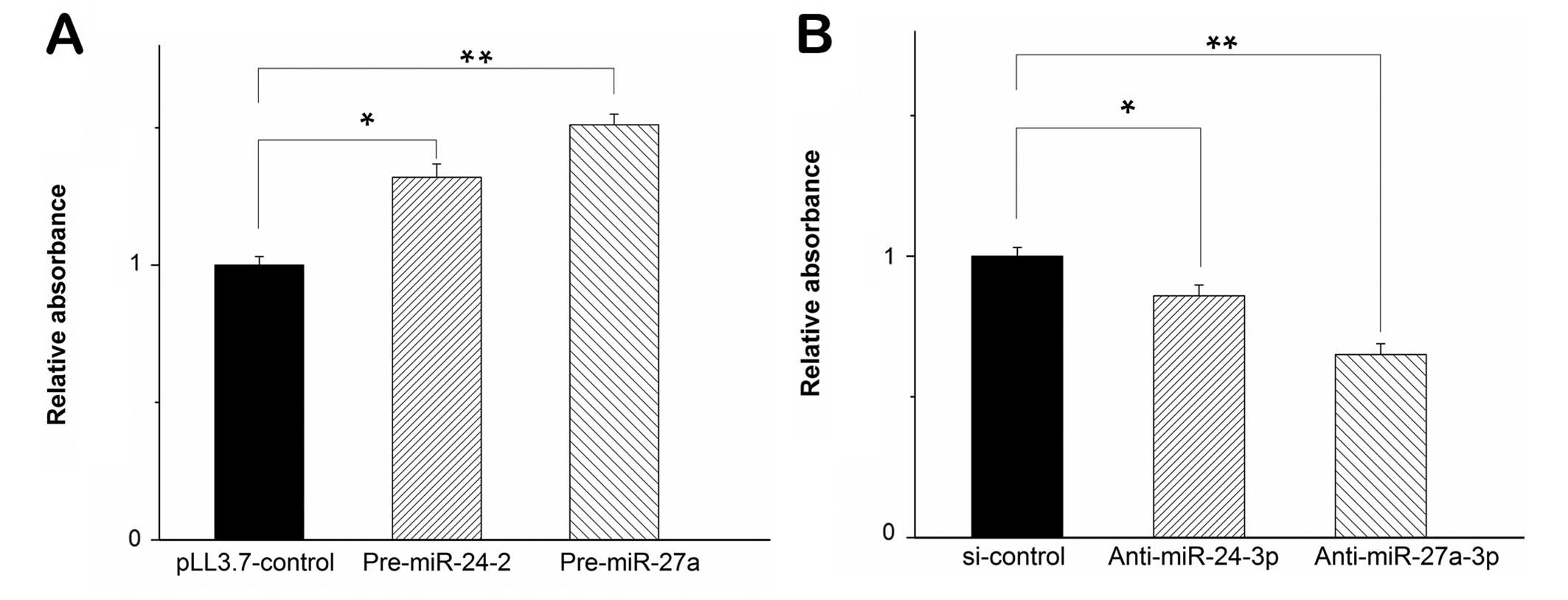
Similarly, we performed the experiments in U251
cells. When the two miRNAs were transfected in U251 cells, MTT
assay showed that the cell proliferation increased (Fig. 2A). The result indicated that
miR-24-3p and miR-27a-3p induced cell proliferation in diverse
glioma cell types. To substantiate the role of miR-24-3p and
miR-27a-3p in cell proliferation of glioma cells, we inhibited the
maturation of miR-24-3p and miR-27a-3p by shRNAs complementary
against miR-24-3p and miR-27a-3p (marked as anti-miR-24-3p and
anti-miR-27a-3p). We used these shRNAs to test cell proliferation
in U251 cells. At 72 h post-transfection, the level of miR-24-3p
and miR-27a-3p decreased significantly when measured by
quantitative RT-PCR (qPCR, data not shown) and MTT assay revealed
that inhibition of miR-24-3p by anti-miR-24-3p and miR-27a-3p by
anti-miR-27a-3p in U251 cells decreased the cell proliferation,
respectively (Fig. 2B). Taken
together, these results indicated that miR-24-3p and miR-27a-3p
promoted cell proliferation in glioma cells.
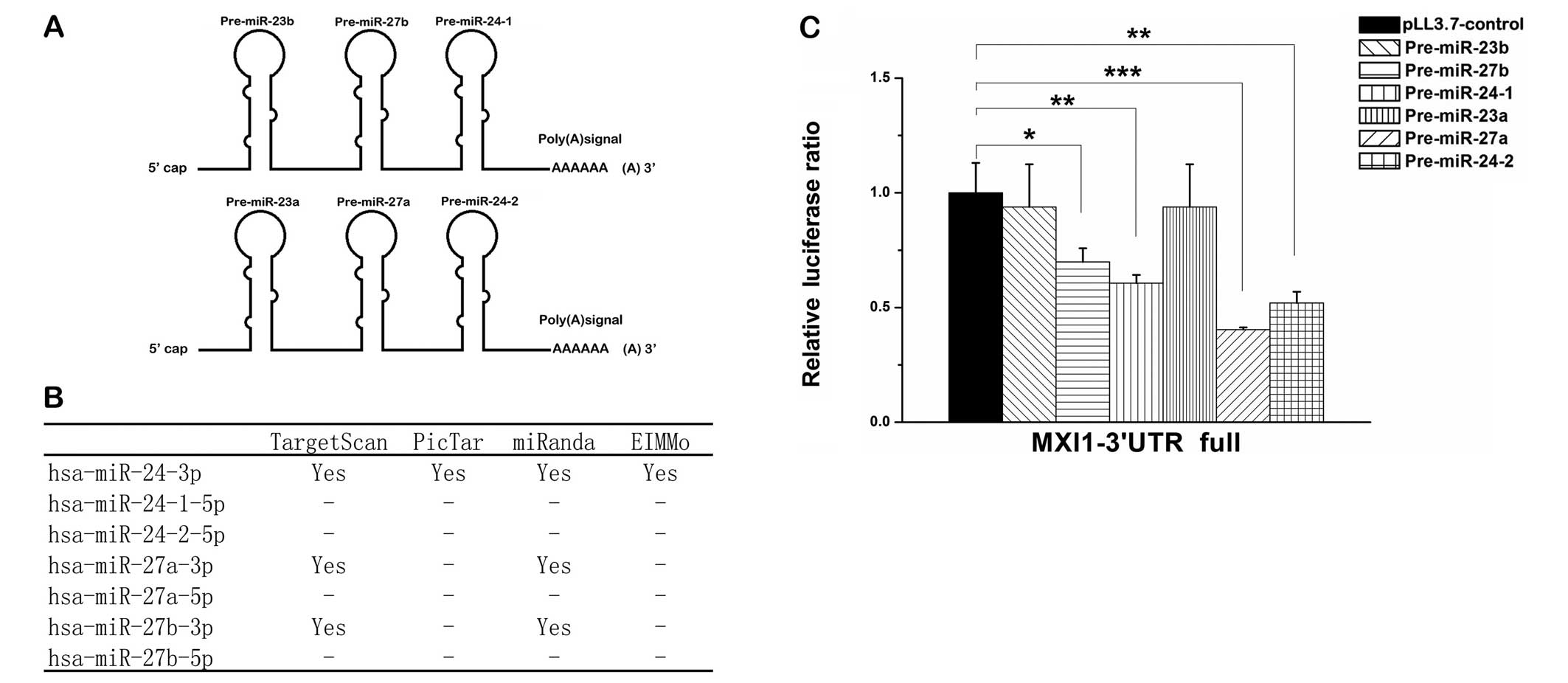
Three miRNAs from two duplicated gene
clusters all target the MXI1 gene
The two miRNAs, miR-24-3p and miR-27a-3p are
produced from two miRNA clusters, miR-23a∼miR-27a∼miR-24-2 at
chromosome 19p13 which is intronic and miR-23b∼miR-27b∼miR-24-1 at
chromosome 9q22 which is intergenic (Fig. 3A). miR-27a and miR-27b are two
paralogous sequences of miR-27 that differ at only one position
(22). To investigate how
miR-24-3p and miR-27a-3p promote cell proliferation of glioma
cells, we predicted miR-24-3p and miR-27a-3p target genes using
prediction software including TargetScan, miRanda, PicTar and
EIMMo. Among the predicted target genes, miR-24-3p and miR-27a-3p,
as well as miR-27b-3p, had been shown to interact with MXI1 gene
(Fig. 3B), which is a tumor
suppressor gene deleted in 60–80% of human glioblastoma tumors
(29–31). To test the predicted interactions,
we first constructed Renilla luciferase reporter vector
containing the full 3′ UTR of MXI1 gene. Co-transfections with the
miRNA expression vector plus the 3′ UTR report vector into 293T
cells were preformed to detect the interactions. Each miRNA
precursor from the clusters miR-23a∼miR-27a∼miR-24-2 and
miR-23b∼miR-27b∼miR-24-1 were tested and we found that
pre-miR-24-1, pre-miR-24-2, pre-miR-27a and pre-miR-27b
significantly suppressed the luciferase activity (Fig. 3C). Since the miRNA precursors
pre-miR-24-2 and pre-miR-27a from the same cluster suppressed the
luciferase activity more strongly than pre-miR-24-1 and pre-miR-27b
which located in the other cluster, and the predicted target sites
of miRNA from pre-miR-24-1 and pre-miR-24-2, or miRNAs from
pre-miR-27a and pre-miR-27b are identical, we chose the
pre-miR-24-2 and pre-miR-27a from the same cluster for further
investigation.
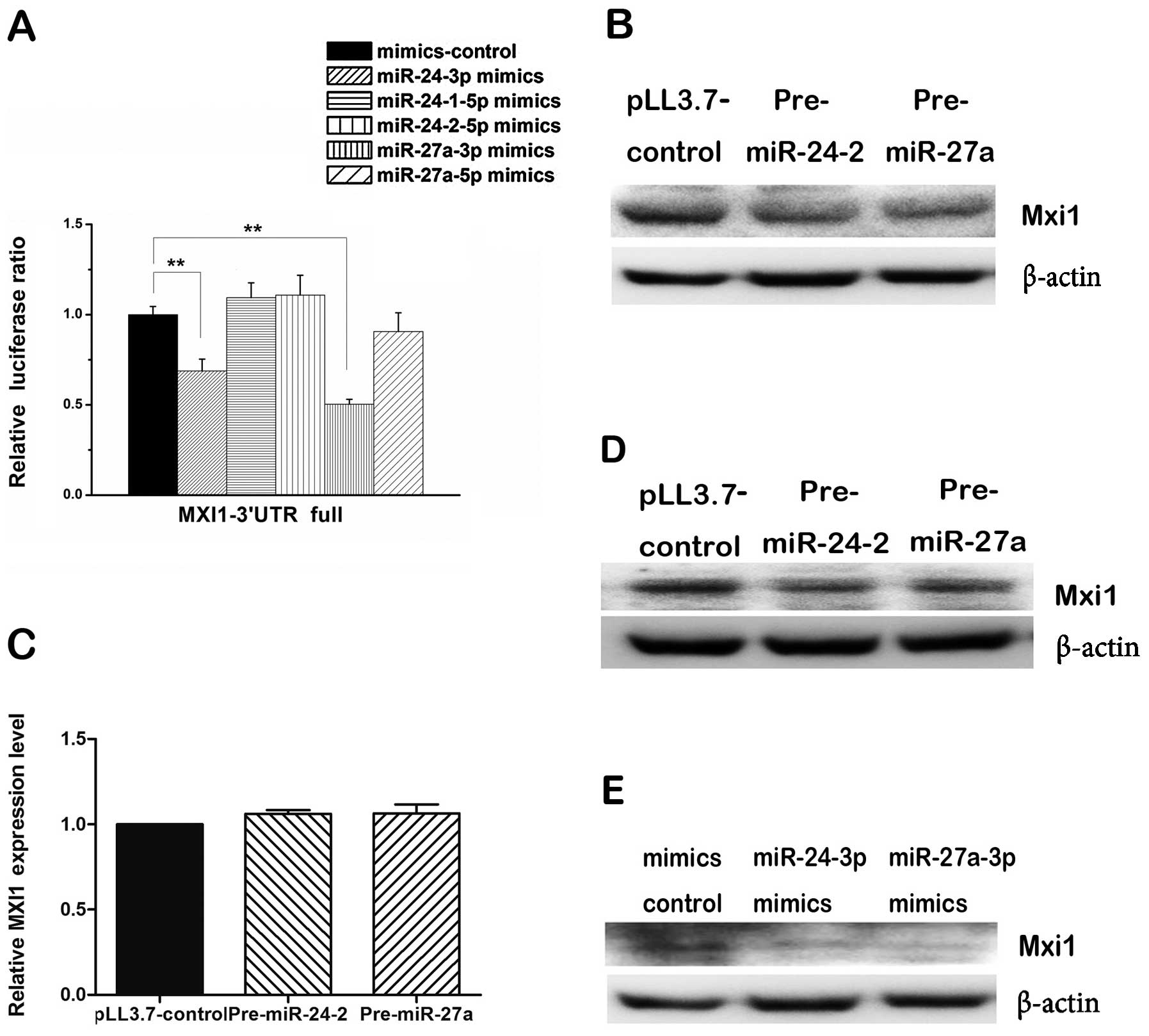
To determine which mature miRNA from the two arms of
each pre-miRNA might regulate the MXI1 3′ UTR, we co-transfected
synthetic mimics of each mature miRNA plus reporter vector
containing full length of MXI1 3′ UTR into 293T cells and measure
the luciferase ratio. We found that miR-24-3p and miR-27a-3p mimics
were able to reduce significantly the luciferase activity compared
with that of the control, however, miR-24-1-5p, miR-24-2-5p and
miR-27a-5p mimics could not (Fig.
4A), indicating that miR-24-3p and miR-27a-3p can regulate MXI1
via its 3′ UTR. While measuring the level of endogenous Mxi1
proteins, western blot assay showed that both pre-miR24-2 and
pre-miR-27a consistently and substantially downregulated the
expression of MXI1 in 293T cells (Fig.
4B) and U87 cells (Fig. 4D),
the mimics of the miR-24-3p and miR-27a-3p showed the same effect
in 293T cells (Fig. 4E). However,
qPCR analyses showed pre-miR-24-2 and pre-miR-27a did not change
the mRNA level of MXI1 gene (Fig.
4C). These results suggest that miR-24-3p and miR-27a-3p could
downregulate the expression of MXI1 gene at post-transcriptional
level.
To validate whether MXI1 is directly targeted by
miR-24-3p and miR-27a-3p, we mutated the predicted target sites in
MXI1 3′ UTR of the two miRNAs. The predicted results showed that
there was one putative target site for miR-24-3p at MXI1 3′ UTR
(870–892) and two putative target sites for miR-27a-3p: one was on
MXI1 3′ UTR (238–258), the other was on MXI1 3′ UTR (2352–2374).
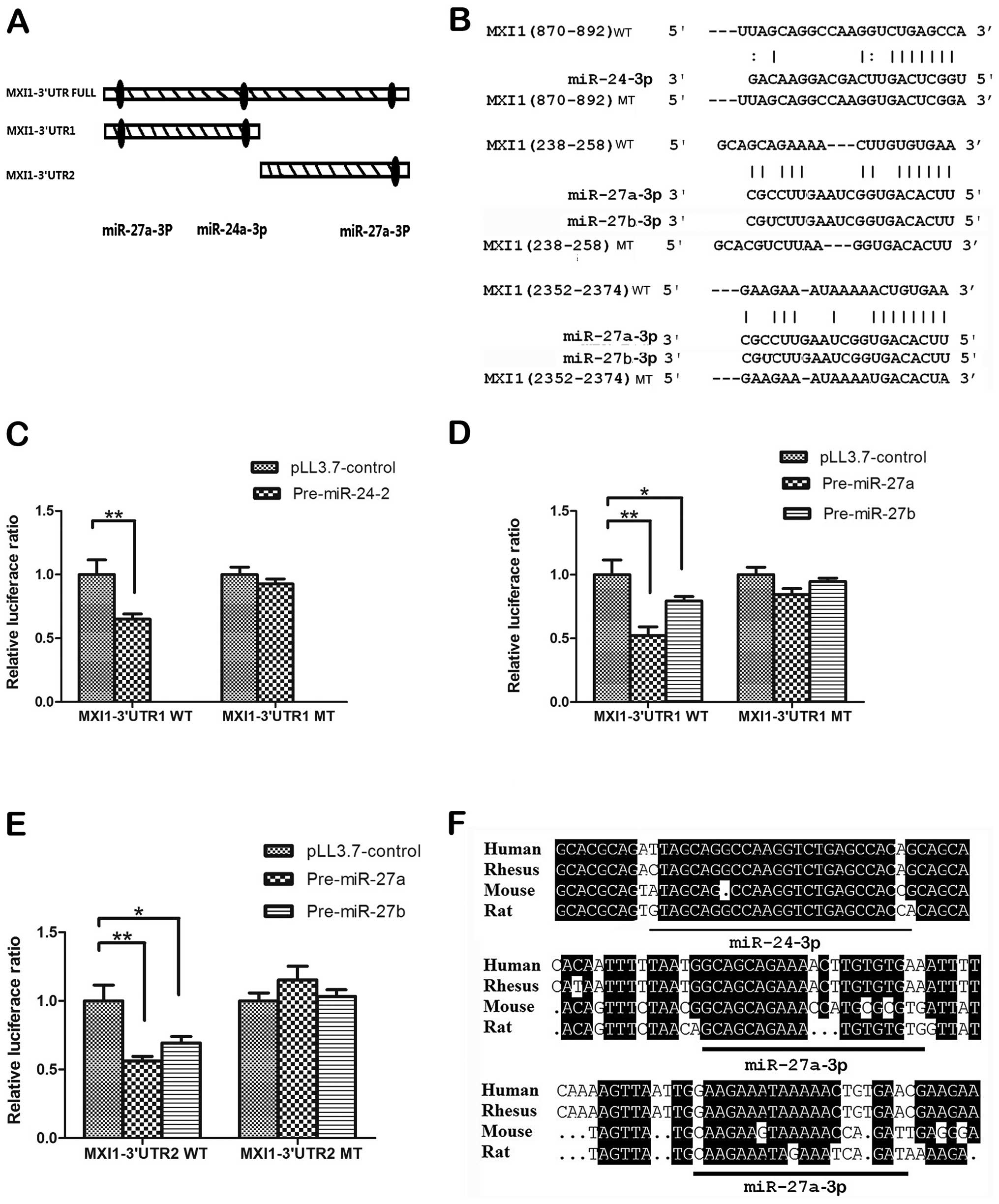
According to the prediction results, we divided MIX1 3′ UTR into
two fragments MXI1 3′ UTR1 and MXI1 3′ UTR2 containing these
predicted sites (Fig. 5A), then we
cloned these wild-type 3′ UTR fragments into the psiCHECK-2
luciferase reporter vectors separately. The mutants within binding
regions were also cloned as shown in Fig. 5B. The results of luciferase ratios
indicated that pre-miR-24-2 could downregulate MXI1 3′ UTR1,
pre-miR-27a could downregulated both MXI1 3′ UTR1 and MXI1 3′ UTR2
(Fig. 5C–E). Also, pre-miR-27b has
similar effect to the pre-miR-27a (Fig. 5D and E). By contrast, the
repressive effect of these two miRNAs on luciferase activity was
abrogated by mutations in the MXI1 3′ UTR fragments (Fig. 5C–E). In addition, we found that the
target sites are quite conserved when aligning the sequences from
human, rhesus, rat and mouse (Fig.
5F). Taken together, all these results suggest that MXI1 is a
direct downstream target of miR-24-3p, miR-27a-3p and
miR-27b-3p.
MXI1 rescues the effects of miR-24-3p and
miR-27a-3p in cell proliferation
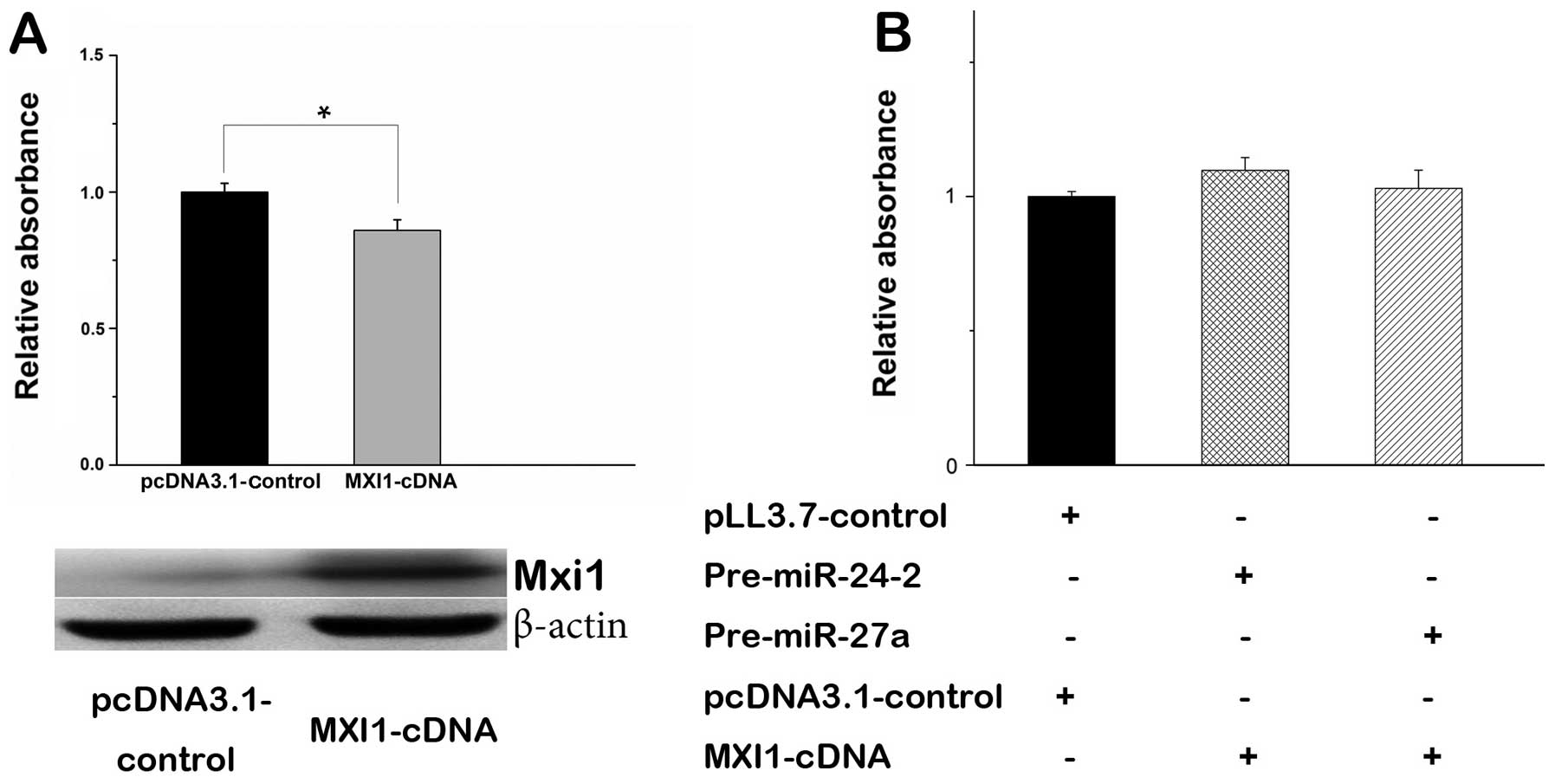
MXI1 is a tumor suppressor gene involved in
regulation of cell proliferation (34). Especially, overexpression of MXI1
in glioma cells is capable of inhibiting cell proliferation
(Fig. 6A). To better understand
the role of miR-24-3p and miR-27a-3p with MXI1 in regulating cell
proliferation, we constructed the MXI1 expression vector by
inserting MXI1-cDNA without 3′ UTR into the pcDNA3.1 vector. The
MTT assays showed that transient transfection of MXI1 expression
vector in U87 cells significantly decreased cell proliferation
(Fig. 6A). When MXI1 gene without
3′ UTR was co-transfected with pre-miR-24-2 or pre-miR-27a, the
effect of miR-24-3p or miR-27a-3p in cell proliferation was rescued
(Fig. 6B), suggesting that MXI1 is
an antagonist of miR-24-3p or miR-27a-3p.
miR-27a-3p is upregulated in human
neoplastic brain tissues
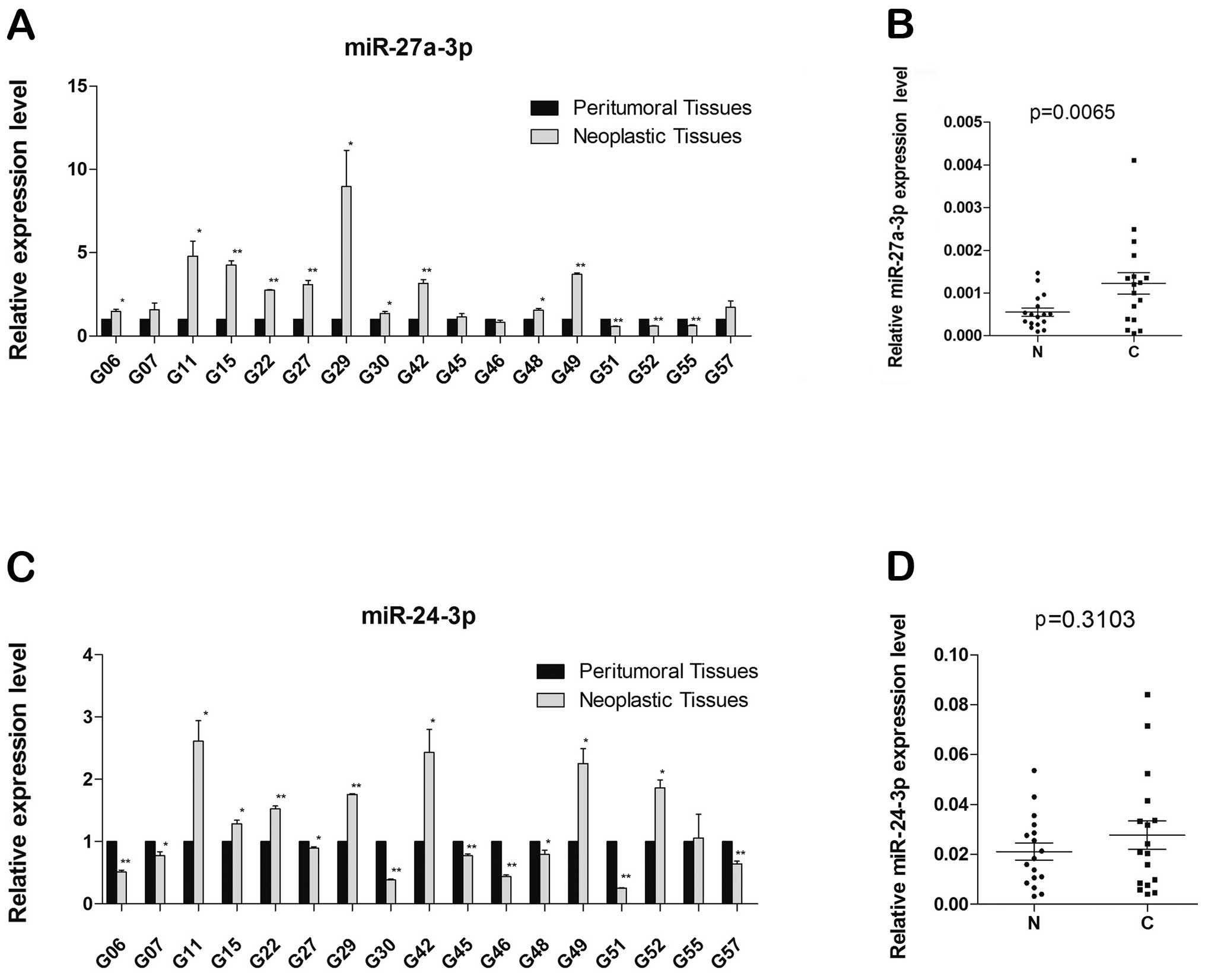
We experimentally tested miR-24-3p and miR-27a-3p
involved in neoplastic brain tissues and peritumoral brain tissues
from GBM patient samples, in which qRT-PCR were used to analyze the
expression of miR-27a-3p and miR-24-3p, and expression levels were
normalized to those of U6 RNA used as the internal control.
Compared with neoplastic brain tissues and peritumoral brain
tissues, we observed that expression of miR-27a-3p in neoplastic
brain significantly increased in 10 of 17 pairs of samples, the
expression of miR-27a-3p decreased in 3 pairs of samples and 4
pairs have no significant difference (*p<0.05;
**p<0.01) (Fig. 7A).
Densitometry of tissue samples revealed that miR-27a-3p was
significantly induced about 3-fold (p= 0.0065) in neoplastic
tissues compared with peritumoral tissues (Fig. 7B). However, while testing
miR-24-3p, the results did not show significant difference between
the two tissues (Fig. 7C), with
the densitometry of neoplastic and peritumoral brain tissue samples
equal to.1.3-fold (p=0.3103) (Fig.
7D).
Discussion
Oligonucleotides have potential to be widely used in
diagnostics and therapeutics. They have many advantages such as
easy production, high target selectivity and stability (37). Oligonucleotide-based approaches,
including miRNA and siRNA, may provide a strategy for cancer
therapy. miRNAs play an important role in cell cycle control,
differentiation, proliferation and apoptosis (38), and deregulation of miRNA expression
has been observed in glioma tumors compared with normal tissues, to
find the miRNAs involved in tumorgenesis will be an important issue
in identification of the therapeutical targets. miR-24-3p and
miR-27a-3p were upregulated in glioma tissues or cell lines
(15,39) and several studies have reported
that miR-24-3p might regulate cell proliferation in different types
of cells (11,16,17),
it is important to study the function of these miRNAs in glioma
cells. In this study, we demonstrated that both miR-24-3p and
miR-27a-3p promoted cell proliferation in glioma cells.
miR-24-3p is a master regulator in gene regulation
(11) and plays a critical role in
diverse physiological process. miR-27a-3p has also been reported to
regulate several genes including FOXO-1 in breast cancer cells
(40), MDR1/P-glycoprotein in
multidrug resistant (MDR) cancer cells lines (41) and RXRα in adipogenesis (22). However, the synergetic regulation
by the two miRNAs from this cluster is poorly characterized. In the
present study, we report for the first time that miR-24-3p and
miR-27a-3p, as well as miR-27b-3p have been found to regulate the
same gene MXI1 in 293T and glioma cells. Several lines of evidence
support a direct interaction between the three miRNAs and the MXI1
3′ UTRs. Firstly, the human MXI1 3′ UTR contains one putative
miR-24-3p binding site and two putative miR-27a/b-3p binding sites
with prominent seed matches (Fig. 5A
and B). Secondly, miR-24-3p, miR-27a-3p and miR-27b-3p can
suppress the activity of luciferase reporter genes fused to the 3′
UTR of MXI1 mRNA. While the target sites were mutated, no
significant changes have been shown in luciferase activities.
Thirdly, the miRNA precursors pre-miR-24-2 and pre-miR-27a or
synthetic mimics of miR-24-3p and miR-27a-3p are able to repress
endogenous expression of human MXI1 in 293T cells and U87 glioma
cells at the protein level. Previous studies reported that
overexpression of MXI1 in glioma cells suppressed cell
proliferation and promote cell differentiation (42). We observed that cell proliferation
increased when miR-24-3p or miR-27a-3p was overexpressed in U87 and
U251 glioma cells, which suggests that miR-24-3p or miR-27a-3p
affects cell proliferation in glioma cells via downregulation of
MXI1. This hypothesis was confirmed by the rescue experiments.
Overexpression of MXI1 without the 3′ UTR, which the miR-24-3p or
miR-27a-3p has no effect on, inhibited the ability of miR-24-3p or
miR-27a-3p to promote cell proliferation in U87 glioma cells. This
study provides the first identification and demonstration of three
miRNAs from the two miRNA clusters miR-23a∼miR-27a∼miR-24-2 and
miR-23b∼miR-27b∼miR-24-1 which directly regulate MXI1 and this in
turn enhances our understanding of the function and regulation of
MXI1 in glioma cells.
MXI1 plays an important role in oncogenesis
(43). MXI1 gene alterations could
be involved in the development and/or the progression of prostate
cancer (44). Loss of MXI1
function contributes to renal cystic disease (43). Abnormal cell proliferation and
tumorigenesis have been observed in organs of MXI1−/− mice
(34). MXI1 also plays an
important role in hepatocyte proliferation (45). MXI1 is a member of the Mad family,
the interaction between MXI1 and Max could antagonize Myc
oncoproteins and thereby suppress glioma progression (46). For cancer therapies, Myc proteins
are validated targets (47).
However, because of the difficulty to target the transcription
factors that lack clear binding domains, targeting Myc directly is
not yet a treatment option (46).
In contrast, small molecular drugs that inhibit Myc-Max
interactions are effective (46).
For this reason, we provide a clue that inhibition of miR-24-3p and
miR-27a-3p upregulated MXI1, which then inhibited Myc protein level
and finally inhibited the glioma cell proliferation (12).
It is documented that all three miRNAs of this
cluster are derived from a single primary transcript (25), however, different biological
conditions makes the expression pattern varied (11). Sometimes all three miRNAs have
similar expression patterns, but in some cases one or two of the
three miRNAs are expressed and the third is not (11). This might be the reason that
miR-27a-3p is upregulated in glioma tissues, however, there was no
significant difference for miR-24-3p between neoplastic brain
tissues and peritumoral brain tissues.
This study demonstrates the role of miR-24-3p and
miR-27a-3p in promoting cell proliferation of glioma cells. Their
function was realized partially via its downstream target MXI1. It
is likely that inhibition of miR-24-3p and miR-27a-3p could serve
in prevention of gliomas. Our studies shed light on developing
therapeutic strategies against gliomas by abrogating the level of
miR-24-3p and miR-27a-3p and increasing MXI1.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (grant no.
81272773 and 81101960); the Scientific and Technological Planning
of Guangzhou (grant no. 2012J4100082) and the Fundamental Research
Funds for the Central Universities (grant no. 101gpy23).
References
|
1
|
Surawicz TS, Davis F, Freels S, Laws ER Jr
and Menck HR: Brain tumor survival: results from the National
Cancer Data Base. J Neurooncol. 40:151–160. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Furnari FB, Fenton T, Bachoo RM, et al:
Malignant astrocytic glioma: genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu X, Xi L, Zeng J and Yao Q: A functional
+61G/A polymorphism in epidermal growth factor is associated with
glioma risk among Asians. PLoS One. 7:e414702012.
|
|
4
|
Lu J, Getz G, Miska EA, et al: MicroRNA
expression profiles classify human cancers. Nature. 435:834–838.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dong H, Siu H, Luo L, Fang X, Jin L and
Xiong M: Investigation gene and microRNA expression in
glioblastoma. BMC Genomics. 11(Suppl 3): S162010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen CZ: MicroRNAs as oncogenes and tumor
suppressors. N Engl J Med. 353:1768–1771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bushati N and Cohen SM: microRNA
functions. Annu Rev Cell Dev Biol. 23:175–205. 2007. View Article : Google Scholar
|
|
9
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang D, Qiu C, Zhang H, Wang J, Cui Q and
Yin Y: Human microRNA oncogenes and tumor suppressors show
significantly different biological patterns: from functions to
targets. PLoS One. 5:e130672010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chhabra R, Dubey R and Saini N:
Cooperative and individualistic functions of the microRNAs in the
miR-23a∼27a∼24-2 cluster and its implication in human diseases. Mol
Cancer. 9:2322010.PubMed/NCBI
|
|
12
|
Lal A, Navarro F, Maher CA, et al: miR-24
inhibits cell proliferation by targeting E2F2, MYC, and other
cell-cycle genes via binding to ‘seedless’ 3′UTR microRNA
recognition elements. Mol Cell. 35:610–625. 2009.PubMed/NCBI
|
|
13
|
Lal LS, Miller LA, Arbuckle R, et al:
Disparities in outpatient antidepressant prescribing patterns and
determinants of resource utilization at a tertiary care cancer
center. J Support Oncol. 7:237–244. 2009.PubMed/NCBI
|
|
14
|
Jones KJ, Skinner A, Xu L, Sun J and
Mueller K: The AHRQ Hospital Survey on Patient Safety Culture: A
Tool to Plan and Evaluate Patient Safety Programs. Advances in
Patient Safety: New Directions and Alternative Approaches.
Henriksen K, Battles JB, Keyes MA and Grady ML: Vol 2. Culture and
Redesign. Agency for Healthcare Research and Quality; Rockville,
MD: 2008, PubMed/NCBI
|
|
15
|
Fukuda Y, Kawasaki H and Taira K:
Exploration of human miRNA target genes in neuronal
differentiation. Nucleic Acids Symp Ser (Oxf). 341–342. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qin W, Shi Y, Zhao B, et al: miR-24
regulates apoptosis by targeting the open reading frame (ORF)
region of FAF1 in cancer cells. PLoS One. 5:e94292010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng AM, Byrom MW, Shelton J and Ford LP:
Antisense inhibition of human miRNAs and indications for an
involvement of miRNA in cell growth and apoptosis. Nucleic Acids
Res. 33:1290–1297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feng J, Iwama A, Satake M and Kohu K:
MicroRNA-27 enhances differentiation of myeloblasts into
granulocytes by post-transcriptionally downregulating Runx1. Br J
Haematol. 145:412–423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McDaneld TG, Smith TP, Doumit ME, et al:
MicroRNA transcriptome profiles during swine skeletal muscle
development. BMC Genomics. 10:772009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang T and Xu Z: miR-27 promotes
osteoblast differentiation by modulating Wnt signaling. Biochem
Biophys Res Commun. 402:186–189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mertens-Talcott SU, Chintharlapalli S, Li
X and Safe S: The oncogenic microRNA-27a targets genes that
regulate specificity protein transcription factors and the G2-M
checkpoint in MDA-MB-231 breast cancer cells. Cancer Res.
67:11001–11011. 2007. View Article : Google Scholar
|
|
22
|
Ji J, Zhang J, Huang G, Qian J, Wang X and
Mei S: Over-expressed microRNA-27a and 27b influence fat
accumulation and cell proliferation during rat hepatic stellate
cell activation. FEBS Lett. 583:759–766. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee Y, Kim M, Han J, et al: MicroRNA genes
are transcribed by RNA polymerase II. EMBO J. 23:4051–4060. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun F, Wang J, Pan Q, et al:
Characterization of function and regulation of miR-24-1 and miR-31.
Biochem Biophys Res Commun. 380:660–665. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ciafrè SA, Galardi S, Mangiola A, et al:
Extensive modulation of a set of microRNAs in primary glioblastoma.
Biochem Biophys Res Commun. 334:1351–1358. 2005.PubMed/NCBI
|
|
26
|
Landgraf P, Rusu M, Sheridan R, et al: A
mammalian microRNA expression atlas based on small RNA library
sequencing. Cell. 129:1401–1414. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Henriksson M and Lüscher B: Proteins of
the Myc network: essential regulators of cell growth and
differentiation. Adv Cancer Res. 68:109–182. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schreiber-Agus N and DePinho RA:
Repression by the Mad(Mxi1)-Sin3 complex. Bioessays. 20:808–818.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fujimoto M, Fults DW, Thomas GA, et al:
Loss of heterozygosity on chromosome 10 in human glioblastoma
multiforme. Genomics. 4:210–214. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rasheed BK, Fuller GN, Friedman AH, Bigner
DD and Bigner SH: Loss of heterozygosity for 10q loci in human
gliomas. Genes Chromosomes Cancer. 5:75–82. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ransom DT, Ritland SR, Moertel CA, et al:
Correlation of cytogenetic analysis and loss of heterozygosity
studies in human diffuse astrocytomas and mixed oligo-astrocytomas.
Genes Chromosomes Cancer. 5:357–374. 1992. View Article : Google Scholar
|
|
32
|
Carter BS, Ewing CM, Ward WS, et al:
Allelic loss of chromosomes 16q and 10q in human prostate cancer.
Proc Natl Acad Sci USA. 87:8751–8755. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lundgren R, Mandahl N, Heim S, Limon J,
Henrikson H and Mitelman F: Cytogenetic analysis of 57 primary
prostatic adenocarcinomas. Genes Chromosomes Cancer. 4:16–24. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ko JY, Yoo KH, Lee HW and Park JH: Mxi1
regulates cell proliferation through insulin-like growth factor
binding protein-3. Biochem Biophys Res Commun. 415:36–41. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tsao CC, Teh BT, Jonasch E, et al:
Inhibition of Mxi1 suppresses HIF-2alpha-dependent renal cancer
tumorigenesis. Cancer Biol Ther. 7:1619–1627. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ebert MS, Neilson JR and Sharp PA:
MicroRNA sponges: competitive inhibitors of small RNAs in mammalian
cells. Nat Methods. 4:721–726. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Catuogno S, Esposito CL, Quintavalle C,
Condorelli G, de Franciscis V and Cerchia L: Nucleic acids in human
glioma treatment: innovative approaches and recent results. J
Signal Transduct. 2012:7351352012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar
|
|
39
|
Feng SY, Dong CG, Wu WK, Wang XJ, Qiao J
and Shao JF: Lentiviral expression of anti-microRNAs targeting
miR-27a inhibits proliferation and invasiveness of U87 glioma
cells. Mol Med Rep. 6:275–281. 2012.PubMed/NCBI
|
|
40
|
Guttilla IK and White BA: Coordinate
regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast
cancer cells. J Biol Chem. 284:23204–23216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhu H, Wu H, Liu X, et al: Role of
MicroRNA miR-27a and miR-451 in the regulation of
MDR1/P-glycoprotein expression in human cancer cells. Biochem
Pharmacol. 76:582–588. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wechsler DS, Shelly CA, Petroff CA and
Dang CV: MXI1, a putative tumor suppressor gene, suppresses growth
of human glioblastoma cells. Cancer Res. 57:4905–4912.
1997.PubMed/NCBI
|
|
43
|
Schreiber-Agus N, Meng Y, Hoang T, et al:
Role of Mxi1 in ageing organ systems and the regulation of normal
and neoplastic growth. Nature. 393:483–487. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kuczyk MA, Serth J, Bokemeyer C, et al:
The MXI1 tumor suppressor gene is not mutated in primary prostate
cancer. Oncol Rep. 5:213–216. 1998.PubMed/NCBI
|
|
45
|
Mauleon I, Lombard MN, Muñoz-Alonso MJ,
Cañelles M and Leon J: Kinetics of myc-max-mad gene expression
during hepatocyte proliferation in vivo: Differential regulation of
mad family and stress-mediated induction of c-myc. Mol Carcinog.
39:85–90. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Swartling FJ: Myc proteins in brain tumor
development and maintenance. Ups J Med Sci. 117:122–131. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gustafson WC and Weiss WA: Myc proteins as
therapeutic targets. Oncogene. 29:1249–1259. 2010. View Article : Google Scholar : PubMed/NCBI
|