Introduction
Phosphatase and tensin homolog (PTEN) is one
of the most frequently lost or mutated of the tumor suppressor
genes (1–5). It has a structural motif for a dual
specificity protein phosphatase, suggesting that PTEN
functions to negatively regulate protein kinase signaling cascades,
a number of which are implicated in tumorigenesis (6). The lipid phosphatase activity of
PTEN dephosphorylates phosphatidylinositol 3,4,5
trisphosphate (PIP3), a potent activator of the protein kinase Akt
(7). Loss of PTEN function
thus results in the stimulation of cell growth and survival, which
are induced by the de-repression of the PI3K/Akt pathway (8,9).
However, recent reports suggest that PTEN also has
PI3K/Akt-independent activities (10). For example, a phosphatase-inactive
PTEN mutant still retains residual tumor suppressive
activity, leading to the hypothesis that PTEN has
phosphatase-independent tumor suppressor function (11–15).
High-grade malignant gliomas are the most common
primary brain tumor in adults. The majority of glioblastomas have
histologic characteristics of cells belonging to the astrocytic
lineage. Therefore, these tumors are thought to arise from the
transformation of astrocytes or their precursors, the neural stem
cells (16–19).
The leukemia inhibitory factor receptor β (LIFRβ)
and downstream Stat3 signaling pathway supports the self-renewal
proliferation of neural stem cells or stimulates the
differentiation of neural stem cells into astrocytes (20,21).
The enhanced expression and pro-survival activity of Stat3 in a
subset of glioblastomas supports the concept that the pleiotropic
function of Stat3 signaling contributes to glial cell
transformation (22,23). Accordingly, Stat3 associates with
the oncoprotein epidermal growth factor receptor III variant in the
nucleus to induce glial transformation, whereas it plays a key role
in the PTEN pathway to suppress malignant transformation of
astrocytes (24). These
observations indicate that Stat3 plays distinct roles in cell
transformation depending on the oncogenic environment.
Recently, there has been a dramatic advance in
research on a small subpopulation of cells identified in cancers,
which have stem cell properties. The cancer stem cell hypothesis
proposes that cancers are derived from a small fraction of cancer
stem cells that proliferate through their unique self-renewal
ability (25). Cancer stem cells
were first identified in leukemia (26,27).
Recently, many investigators have identified them in solid tumors,
including those of the breast, brain, pancreas, colon and head and
neck (28). The cancer stem cell
hypothesis established that cancer therapy must be directed against
both the resting cancer stem cells and the proliferating cancer
cells (29). Therefore, it is
essential to understand the major intracellular signaling pathways
governing the characteristics of cancer stem cells. It has become
clear that PTEN is one of the critical regulators for the
development of cancer stem cells and ultimately tumorigenesis.
In this study, we show that ectopic expression of
PTEN can disturb Akt and Stat3 phosphorylation, which leads
to reduced tumorigenicity both in vitro and in vivo.
Furthermore, as demonstrated by the loss of both CD133 expression
and neurosphere formation, we found that PTEN can suppress
the glioblastoma stem cell (GSC) population. Moreover, PTEN
expression also inhibited cell proliferation and induced senescence
in glioblastoma. The data suggest that the occurrence of GSCs, and
the proliferation and senescence of glioblastoma, which are closely
correlated with PTEN expression, are independently regulated
by Akt and Stat3 signaling.
Materials and methods
Cell culture and genetic
modification
Human glioblastomaastrocytoma, epithelial-like cell
line U-87MG was purchased from ATCC (www.atcc.org). These
cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin, and 100 μg/ml streptomycin (all from Gibco Invitrogen,
Carlsbad, CA, www.invitrogen.com). Neurospheres were
formed in DMEM containing Nutrient Mixture F-12 (DMEM/F12, Gibco
Invitrogen) supplemented with 20 ng/ml basic fibroblast growth
factor (Gibco Invitrogen), 20 ng/ml recombinant human epidermal
growth factor (R&D Systems, Minneapolis, MN, http://www.rndsystems.com), and 20 μl/ml N2-supplement
(Gibco Invitrogen). A PTEN-expressing plasmid was
constructed by cloning PCR-amplified PTEN cDNA into pLPCX
(Clontech, Mountain View, CA, http://www.clontech.com). The PTEN-expressing
plasmid was transfected into U-87MG cells with Lipofectamine 2000
(Invitrogen) according to the manufacturer’s instructions. U-87MG
cells that stably express PTEN, by chromosomal integration
of the PTEN-expressing plasmid, were selected in DMEM supplemented
with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 500
μg/ml puromycin (Gibco Invitrogen) for 2 to 3 weeks.
RNA extraction and real-time RT-PCR
Total-RNA was extracted using TRIzol (Invitrogen)
and 2–5 μg total-RNA was reverse-transcribed using the
SuperScriptII™ First-Strand Synthesis System (Invitrogen) according
to the manufacturer’s instructions. Real-time RT-PCR was carried
out on cDNAs using the QuantiTect SYBR-Green PCR Kit (Qiagen,
Valencia, CA). Reactions were conducted in triplicate using the
Exicycler™96 Real-Time Quantitative Thermal Block (Bioneer, Korea,
http://www.bioneer.co.kr). For quantification,
the expression of target genes was normalized against that of the
glyceraldehyde 3-phosphate dehydrogenase gene (GAPDH). The
PCR primer pairs (sense, antisense) were as follows: CD133
(cag agt aca acg cca aac ca, aaa tca cga tga ggg tca gc) and
GAPDH (ggg tgt gaa cca tga gaa, gtc ttc tgg gtg gca gtg
at).
Protein extraction and western blot
analysis
Cells were washed twice with cold phosphate-buffered
saline (PBS), lysed with tissue lysis buffer (20 mM Tris base pH
7.4, 137 mM NaCl, 2 mM EDTA, 1% Triton X-100, 25 mM
β-glycerophosphate, 2 mM sodium pyrophosphate, 10% glycerol, 1 mM
sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride and 1 mM
benzamidine) and clarified by centrifugation at 12,000 x g for 10
min. Whole-cell extracts were prepared and 20–50 μg protein was
resolved by SDS-PAGE and blotted using antibodies against PTEN
(sc-6817-R, Santa Cruz Biotechnology), Akt (sc-8312, Santa Cruz
Biotechnology), p-Akt (#4058, Cell Signaling Technology), p-Stat3
(#9131, Cell Signaling Technology), Stat3 (sc-482, Santa Cruz
Biotechnology) and β-actin (sc-47778, Santa Cruz Biotechnology).
Immunoreactivity was detected by enhanced chemiluminescence
(Amersham Biosciences).
Immunocytochemical staining
Cells were fixed with 4% paraformaldehyde in PBS for
20 min and permeabilized with 0.1% Triton X-100 in PBS for 15 min.
After treatment with 1% goat serum for 1 h, cells were incubated
with primary antibodies against goat anti-human p-Stat3 or CD133
(1:100 dilution) at 4°C overnight. Cells were washed with PBS and
then incubated with Alexa 488-labeled anti-goat IgG secondary
antibody (1:300; Molecular Probes, Eugene, OR) for 2 h.
Senescence analysis
The SA-β-gal staining method was used to analyze
senescence. Cells were washed twice with PBS (pH 7.2), fixed with
2% formaldehyde and 0.2% glutaraldehyde in PBS (pH 7.2), and washed
in PBS (pH 7.2) supplemented with 1 mM MgCl2. Cells were
stained in X-gal solution [1 mg/ml X-gal (Boehringer Ingelheim,
Ridgefield, CT, http://us.boehringer-ingelheim.com/)], 0.12 mM
K3Fe[CN]6, 0.12 mM
K4Fe[CN]6, 1 mM MgCl2 in PBS at pH
6.0) at 37°C overnight and then examined under microscope (CKX41,
Olympus, Japan, http://www.olympus-global.com/).
Migration assay
For scratch tests, cells were plated in 6-well
plates at 90–100% confluency. A scratch was made at the center of
the well with a 200-μl pipette tip. Cell migration was observed
under microscope 24 h after wounding. This experiment was repeated
three times.
Rat model of glioblastoma
All of the experimental animals were manipulated in
accordance with the guidelines of the CHA University Institutional
Animal Care and Use Committee (IACUC090012). Adult male
Sprague-Dawley rats (n=11) weighing 260–280 g (Orient, Seoul,
Korea) were used in this experiment. One million ordinary U-87 MG
cells (called WT thereafter) or U-87 MG cells overexpressing
PTEN (called PTEN OE thereafter) in a volume of 2 μl
were stereotaxically implanted into one hemisphere of the brain
(n=5 WT and n=6 PTEN OE) using the following coordinates:
AP, +1.0 mm; ML, −3.0 mm and DV, −5.0 mm. Animals were euthanized
at 5 weeks following implantation and brains were taken for
histological analyses. H&E staining was carried out on 10
μm-thick brain sections, whereas immunohistochemical staining was
carried out on 40 μm-thick sections using the following antibodies:
p-Stat3 (1:100, Cell Signaling), PTEN (1:100, Santa Cruz
Biotechnology), human-specific nuclei (hNu) (1:200, Chemicon), goat
anti-mouse IgG-conjugated Alexa-555 (1:200, Molecular Probes) and
goat anti-rabbit IgG-conjugated Alexa-488 (1:200, Molecular
Probes).
Statistical analysis
Graphical data are presented as means ± SD. Each
experiment was performed at least three times and subjected to
statistical analysis. Statistically significant differences between
two groups were determined using Student’s t-test. A p<0.05 was
considered significant. Statistical analysis was performed using
the SAS statistical package vs. 9.13 (SAS Inc., Cary, NC,
http://www.sas.com/).
Results and discussion
PTEN expression results in the depletion
of GSCs in glioblastoma
The PTEN pathway is known to control normal
stem cell maintenance, self-renewal and migration. PTEN loss
confers increased self-renewal capacity to normal stem
cells/progenitors, which can cause the development of cancer stem
cells and ultimately tumorigenesis. Expression of the GSC tumor
marker CD133 was used to examine the effect of PTEN
expression on the GSC population. First, the PTEN-deficient
U-87MG cells (wild-type, WT) were engineered to stably express
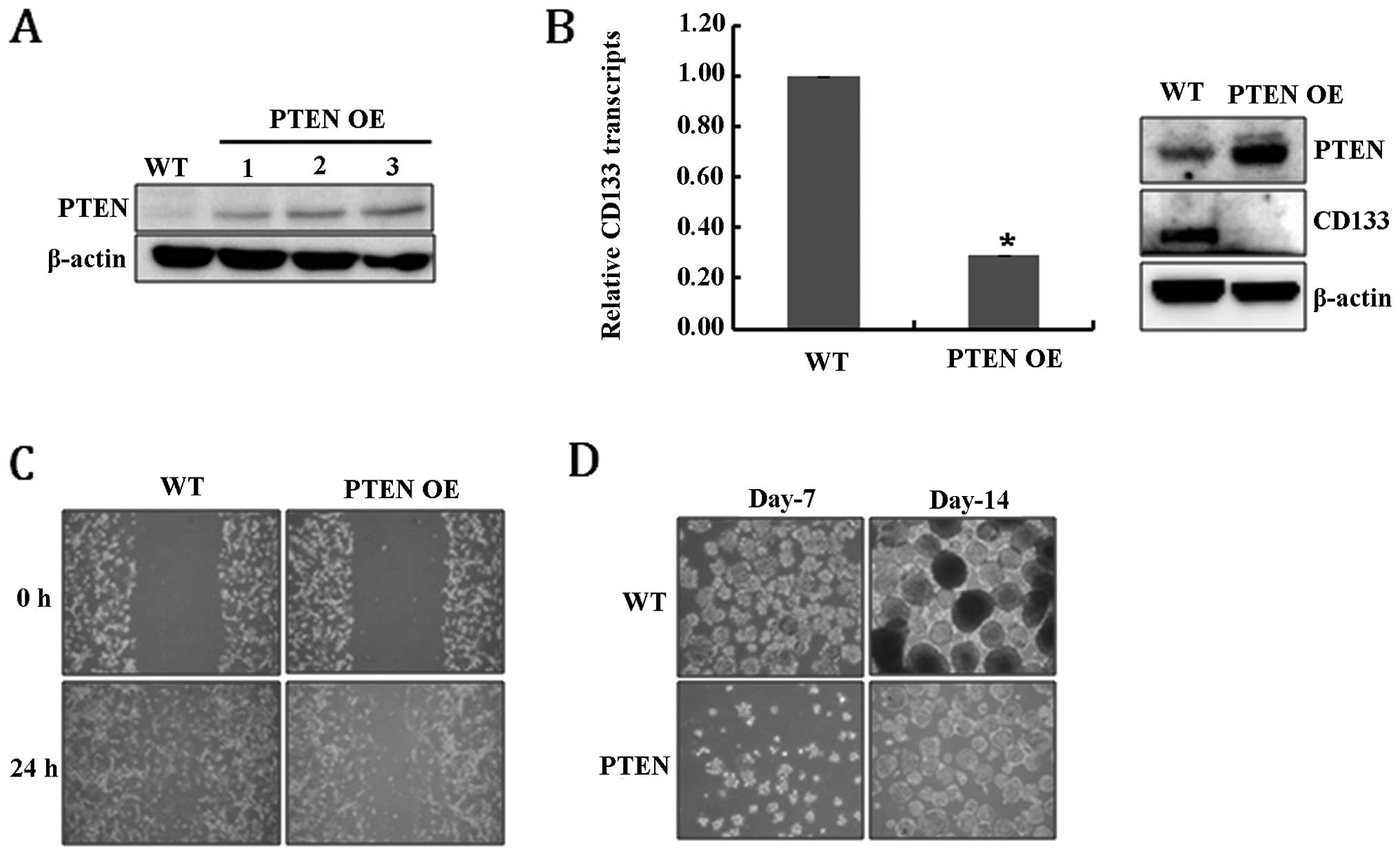
PTEN (PTEN-overexpressing, PTEN OE) (Fig. 1A). Fig. 1B shows that the overexpression of
PTEN significantly reduced the levels of CD133 RNA and protein,
suggesting that PTEN expression resulted in the depletion of
GSCs. As confirmation, the loss of further GSC characteristics was
analyzed. Cell migration ability is one of the prominent
characteristics of glioblastoma cancer stem-like cells (30), and, as expected, PTEN OE
cells showed defective migration ability compared to WT (Fig. 1C). In addition, the ability to form
neurospheres, another feature of GSCs, was also significantly
diminished in PTEN OE cells (Fig. 1D). Taken together, these results
suggest that PTEN expression clearly reduced the population
of GSCs among U-87MG cells.
PTEN expression induces growth
retardation and senescence of glioblastoma cells
Negative regulation by PTEN of PI3K activity
and the PI3K/Akt pathway is of critical importance for cell
proliferation. To further analyze the tumor suppressive mechanism
of PTEN, the effect of PTEN expression on U-87MG cell
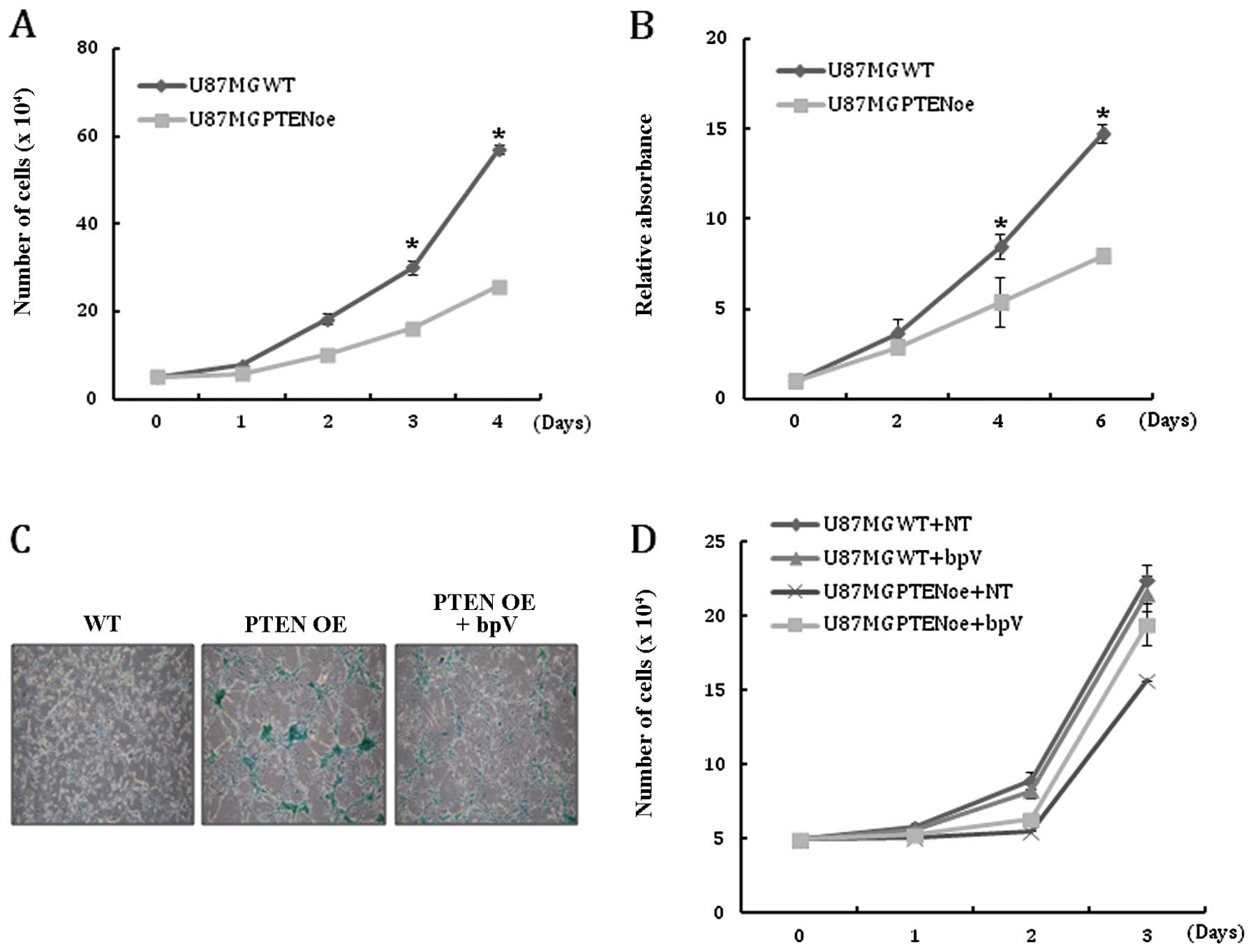
growth was examined. As expected, cell counting and MTT assay
revealed that PTEN expression reduced the proliferation of
U-87MG cells (Fig. 2A and B).
Cellular senescence, which can be induced by various stimuli, can
function as a critical barrier for cancer development and
significantly restrict cancer progression. Since proliferation is
directly linked to cellular senescence, the effect of PTEN
expression on the induction of senescence in U-87MG cells was
analyzed. Compared with the WT cells, the number of cells positive
for β-gal staining was significantly increased in PTEN OE at
each passage (Fig. 2C). Addition
of bpV to the culture medium to inhibit PTEN activity
reduced the population of β-gal-positive cells in PTEN OE,
suggesting that cellular senescence is induced by PTEN
activity (Fig. 2C). Treatment with
bpV consistently rescued the growth retardation of PTEN OE
(Fig. 2D). Taken together, these
results indicate that the restoration of PTEN activity in
glioblastoma cells markedly inhibits cell proliferation and leads
to senescence.
PTEN expression in U-87MG cells disrupts
Akt and Stat3 signaling
PTEN inhibits the PI3K/Akt signaling pathway by
catalyzing the dephosphorylation of PIP3, while the loss of PTEN
induces the activation of the PI3K/Akt cascade, which leads to the
stimulation of cell growth and survival (7,8). The
LIFRβ-Stat3 signaling pathway is also regulated by PTEN and
functions as a brain tumor suppressor in astrocytes (24). In contrast, Stat3 activation has
been described in a subset of glioblastoma, indicating that Stat3
plays distinct roles in cell transformation depending on the
oncogenic environment (23,31).
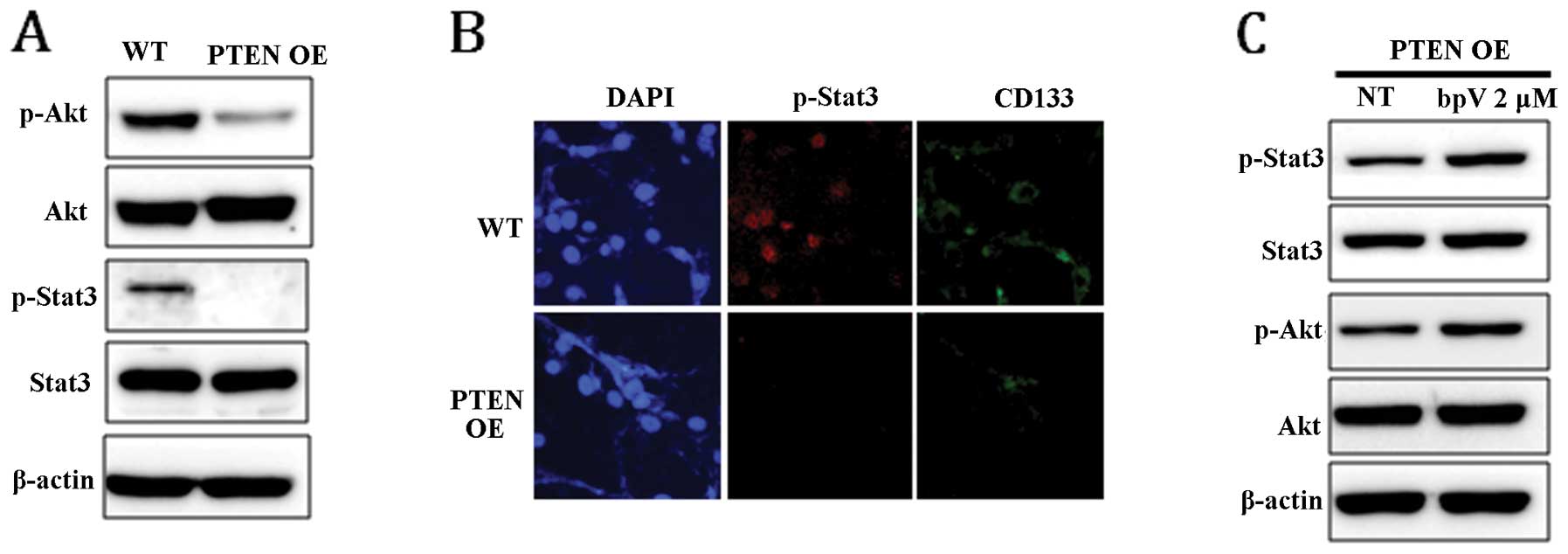
To examine whether PTEN expression can affect Stat3 and Akt
signaling pathways, the phosphorylation level of Stat3 and Akt in
PTEN OE was compared with that in WT. In response to
PTEN expression, the level of phospho-Stat3 (p-Stat3) and
phospho-Akt (p-Akt) was markedly reduced (Fig. 3A). Dephosphorylation of Stat3 in
PTEN OE was confirmed by immunocytochemical staining
(Fig. 3B). Consistent with the
result shown in Fig. 1B, the
expression level of CD133 was also significantly decreased in
PTEN-expressing cells (Fig.
3B). To confirm that the dephosphorylation of Stat3 and Akt was
induced by PTEN, the phosphorylation status of STAT3 and AKT
was examined in PTEN OE following treatment with bpV for 24
h. As shown in Fig. 3C, chemical
inhibition of PTEN activity enhanced Stat3 and Akt
phosphorylation, suggesting that Stat3 and Akt signaling is
directly regulated by PTEN.
Cell growth, senescence and the
population of GSCs are independently regulated by Akt and Stat3
signaling pathways in U-87MG cells
Since the expression of PTEN in U-87MG cells
were shown to modulate Stat3 and Akt signals, we next examined
whether Stat3 signals are regulated by Akt pathway in U-87MG. To do
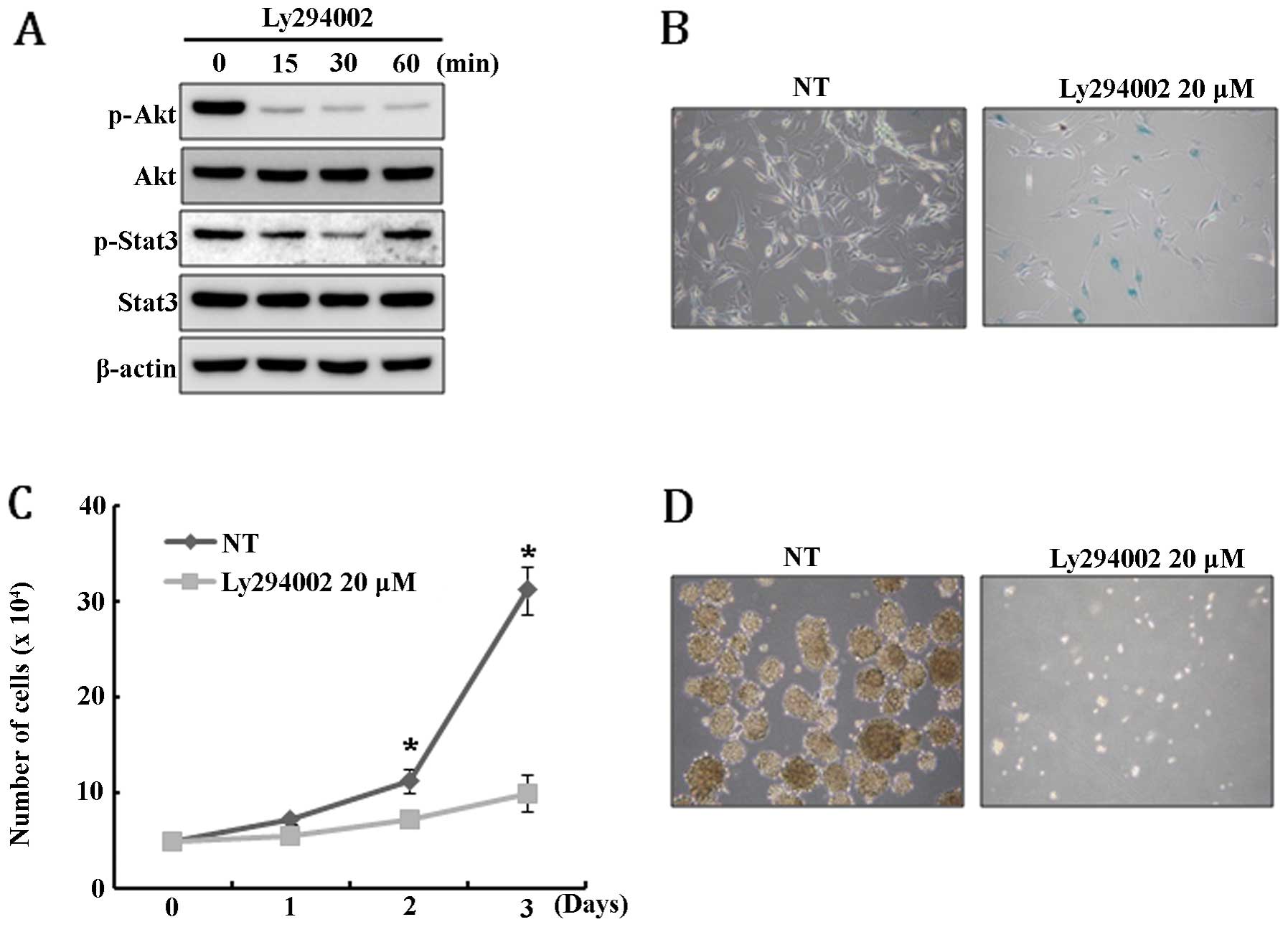
this, U-87MG cells were treated with PI3K inhibitor LY294002 and,
unexpectedly, Stat3 was only transiently dephosphorylated and
rapidly restored to its original level within 60 min after
LY294002-mediated inhibition of Akt activity (Fig. 4A). This result suggests that Stat3
signal is not a downstream target of Akt pathway in U-87MG. Since
PTEN expression resulted in the depletion of the GSC
population and caused growth retardation and senescence of U-87MG
cells, we speculate that these phenomena might be induced by the
disturbance of Akt and Stat3 signaling. To test this hypothesis,
Akt activity in U-87MG cells was inhibited by treatment with
LY294002 and then the extent of senescence, cell growth and
neurosphere formation was examined. As expected, LY294002 treatment
specifically induced senescence and severely inhibited cell growth
(Fig. 4B and C). In addition,
neurosphere formation was completely blocked by the treatment of
LY294002 (Fig. 4D). Taken
together, these results suggest that the senescence, growth
retardation, and depletion of GSCs in U-87MG cells are, at least in
part, mediated by the Akt signaling pathway.
Constitutive activation of Stat3 is frequently
detected in clinical samples from a wide range of human carcinomas
and the established cancer cell lines, suggesting that Stat3
activity is crucial to cell survival and growth (32,33).
In addition, the JAK/Stat3 signaling pathway is required for the
growth of stem-like cancer cells in various tumors (34,35).
For these reasons, we decided to examine the effect of inhibition
in Stat3 signaling on on cell growth, senescence, and neuro-sphere
formation of U-87MG cells. We found that Stat3 was completely
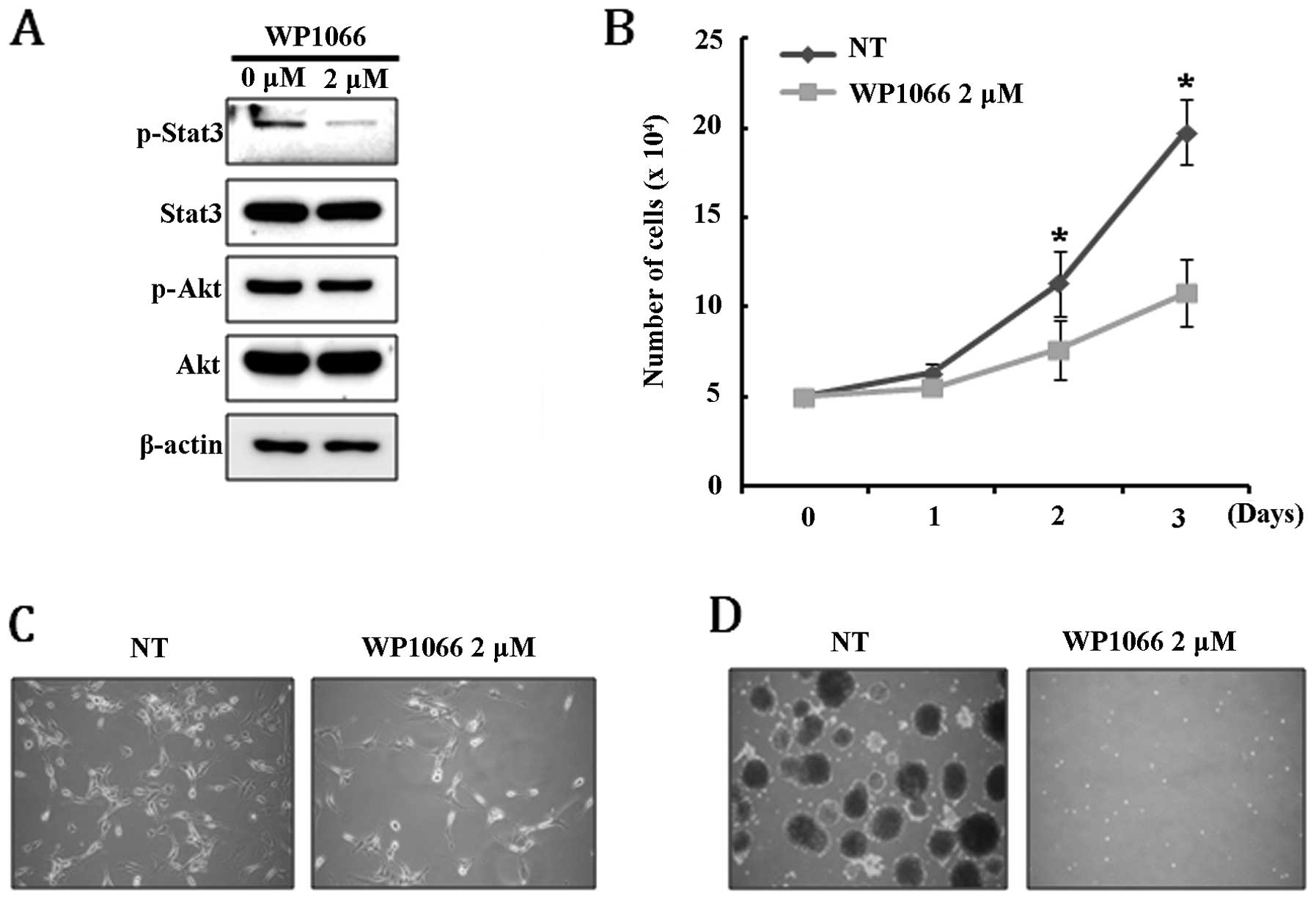
dephosphorylated by treatment with 2 μM WB1066, while
phosphorylation of AKT was not affected (Fig. 5A). Interestingly, we observed that
cellular senescence, which can be estimated by β-gal staining, was
not directly induced by inhibiting the Stat3 signaling pathway,
although it caused significant growth retardation (Fig. 5B and C). Treatment with WB1066
completely inhibited the ability of U-87MG to form neurospheres,
suggesting that the Stat3 signaling pathway is required for
maintaining the cancer stem-like cell population (Fig. 5D). These results demonstrate that
Stat3 signaling regulates proliferation and maintenance of the
cancer stem-like cell population but is not involved in the
senescence of U-87MG cells. Since PTEN expression in U-87MG
disrupts Akt and Stat3 signals, and that growth retardation,
senescence and depletion of cancer stem cells are caused by
PTEN expression in U-87MG, we speculate that PTEN
induces growth retardation and cancer stem cell depletion of U-87MG
in conjunction with both Stat3 andAkt signaling pathways, whereas
the senescence response of U-87MG cells upon PTEN expression
is mainly regulated by the Akt signaling pathway.
Overexpression of PTEN inhibits tumor
formation by U-87MG glioblastoma cells in vivo
To address whether PTEN is involved in the
proliferation of GSCs, PTEN OE or as controls, WT U-87MG
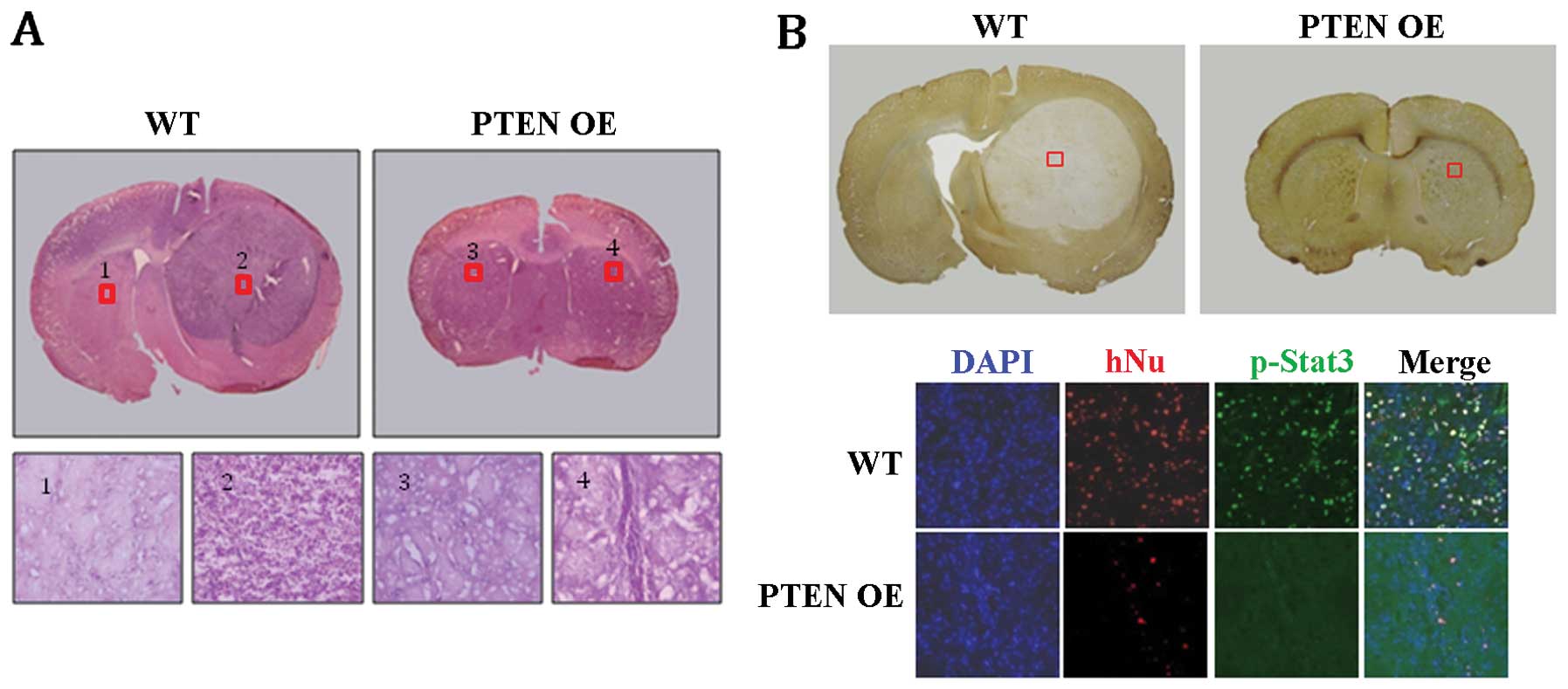
cells were implanted into the brain of 8-week-old rats. Strikingly,
no tumor-like structures were detected in any of the PTEN
OE-implanted group (n=6), whereas robust tumor-like structures were
formed in all of the WT-implanted group (n=5) (Fig. 6A and B). Histological analysis of
the WT-implanted brains indicated that the tumor-like structures
consisted mainly of undifferentiated glioblastoma cells (Fig. 6A; WT 2). In contrast, such cells
were not detected in the PTEN OE-implanted brains (Fig. 6B; PTEN OE 4). Furthermore,
immunohistochemical staining revealed that many human-specific
nuclear antigen (hNu)-positive cells were detected in the
WT-implanted brain sections, indicating that U-87MG glioblastoma
cells had proliferated extensively after implantation (Fig. 6B; WT). p-Stat3-positive cells were
also common in the area of tumor formation (Fig. 6B; WT). In contrast, in the
PTEN OE-implanted brains, only a few hNu-positive cells were
detected, suggesting that most of the PTEN OE U-87MG
glioblastoma cells were not maintained after implantation (Fig. 6B; PTEN OE in the bottom
panel). No p-Stat3-positive cells were detected either in the
PTEN OE-implanted brains (Fig.
6B; PTEN OE in the bottom panel). Taken together, these
results strongly suggest that PTEN negatively regulates the
proliferation of GSCs in U-87MG glioblastoma cells, presumably
through the inhibition of the Stat3 pathway.
From our study, we found that analysis of the
signaling pathways underlying the tumor suppressor function of
PTEN in U-87MG cells can provide experimental evidence how
the loss of PTEN promotes pathogenesis of glial tumors. In
particular, identification of the Stat3 and Akt signaling pathways
as downstream targets of PTEN provides a mechanism by which
PTEN modulates cell proliferation and senescence and
maintenance of the GSC population. Accumulating evidence supports
the concept that GSCs, which have stem cell-like properties, are
the major cause of the occurrence and maintenance of glioblastoma.
In this regard, our results will provide important experimental
basis for developing therapeutics to treat glioblastoma in the
future.
Acknowledgements
This study was supported by the Korea
Science and Engineering Foundation (KOSEF) of the Korean government
(MOST) (2010-0003254, 2011-0014084). This study was also supported
by Priority Research Centers Program through the National Research
Foundation of Korea (NRF) funded by the Ministry of Education,
Science and Technology (2009-0093821).
References
|
1
|
Cairns P, Okami K, Halachmi S, Halachmi N,
Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS and Sidransky D:
Frequent inactivation of PTEN/MMAC1 in primary prostate cancer.
Cancer Res. 57:4997–5000. 1997.PubMed/NCBI
|
|
2
|
Feilotter HE, Nagai MA, Boag AH, Eng C and
Mulligan LM: Analysis of PTEN and the 10q23 region in primary
prostate carcinomas. Oncogene. 16:1743–1748. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gray IC, Stewart LM, Phillips SM, Hamilton
JA, Gray NE, Watson GJ, Spurr NK and Snary D: Mutation and
expression analysis of the putative prostate tumour-suppressor gene
PTEN. Br J Cancer. 78:1296–1300. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li DM and Sun H: TEP1, encoded by a
candidate tumor suppressor locus, is a novel protein tyrosine
phosphatase regulated by transforming growth factor beta. Cancer
Res. 57:2124–2129. 1997.PubMed/NCBI
|
|
5
|
Steck PA, Pershouse MA, Jasser SA, Yung
WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T,
Frye C, Hu R, Swedlund B, Teng DH and Tavtigian SV: Identification
of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3
that is mutated in multiple advanced cancers. Nat Genet.
15:356–362. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kolibaba KS and Druker BJ: Protein
tyrosine kinases and cancer. Biochim Biophys Acta. 1333:F217–F248.
1997.PubMed/NCBI
|
|
7
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:13375–13378. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stambolic V, Suzuki A, de la Pompa JL,
Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM,
Siderovski DP and Mak TW: Negative regulation of PKB/Akt-dependent
cell survival by the tumor suppressor PTEN. Cell. 95:29–39. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun H, Lesche R, Li DM, Liliental J, Zhang
H, Gao J, Gavrilova N, Mueller B, Liu X and Wu H: PTEN modulates
cell cycle progression and cell survival by regulating
phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B
signaling pathway. Proc Natl Acad Sci USA. 96:6199–6204. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Salmena L, Carracedo A and Pandolfi PP:
Tenets of PTEN tumor suppression. Cell. 133:403–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Blanco-Aparicio C, Renner O, Leal JF and
Carnero A: PTEN, more than the AKT pathway. Carcinogenesis.
28:1379–1386. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Georgescu MM, Kirsch KH, Kaloudis P, Yang
H, Pavletich NP and Hanafusa H: Stabilization and productive
positioning roles of the C2 domain of PTEN tumor suppressor. Cancer
Res. 60:7033–7038. 2000.PubMed/NCBI
|
|
13
|
Gildea JJ, Herlevsen M, Harding MA,
Gulding KM, Moskaluk CA, Frierson HF and Theodorescu D: PTEN can
inhibit in vitro organotypic and in vivo orthotopic invasion of
human bladder cancer cells even in the absence of its lipid
phosphatase activity. Oncogene. 23:6788–6797. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koul D, Jasser SA, Lu Y, Davies MA, Shen
R, Shi Y, Mills GB and Yung WK: Motif analysis of the tumor
suppressor gene MMAC/PTEN identifies tyrosines critical for tumor
suppression and lipid phosphatase activity. Oncogene. 21:2357–2364.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maier D, Jones G, Li X, Schonthal AH,
Gratzl O, Van Meir EG and Merlo A: The PTEN lipid phosphatase
domain is not required to inhibit invasion of glioma cells. Cancer
Res. 59:5479–5482. 1999.PubMed/NCBI
|
|
16
|
Bachoo RM, Maher EA, Ligon KL, Sharpless
NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R,
Rowitch DH, Louis DN and DePinho RA: Epidermal growth factor
receptor and Ink4a/Arf: convergent mechanisms governing terminal
differentiation and transformation along the neural stem cell to
astrocyte axis. Cancer Cell. 1:269–277. 2002. View Article : Google Scholar
|
|
17
|
Bajenaru ML, Hernandez MR, Perry A, Zhu Y,
Parada LF, Garbow JR and Gutmann DH: Optic nerve glioma in mice
requires astrocyte Nf1 gene inactivation and Nf1 brain
heterozygosity. Cancer Res. 63:8573–8577. 2003.PubMed/NCBI
|
|
18
|
Holland EC: Gliomagenesis: genetic
alterations and mouse models. Nat Rev Genet. 2:120–129. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Uhrbom L, Dai C, Celestino JC, Rosenblum
MK, Fuller GN and Holland EC: Ink4a-Arf loss cooperates with KRas
activation in astrocytes and neural progenitors to generate
glioblastomas of various morphologies depending on activated Akt.
Cancer Res. 62:5551–5558. 2002.PubMed/NCBI
|
|
20
|
Bonni A, Sun Y, Nadal-Vicens M, Bhatt A,
Frank DA, Rozovsky I, Stahl N, Yancopoulos GD and Greenberg ME:
Regulation of gliogenesis in the central nervous system by the
JAK-STAT signaling pathway. Science. 278:477–483. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yoshimatsu T, Kawaguchi D, Oishi K, Takeda
K, Akira S, Masuyama N and Gotoh Y: Non-cell-autonomous action of
STAT3 in maintenance of neural precursor cells in the mouse
neocortex. Development. 133:2553–2563. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rahaman SO, Harbor PC, Chernova O, Barnett
GH, Vogelbaum MA and Haque SJ: Inhibition of constitutively active
Stat3 suppresses proliferation and induces apoptosis in
glioblastoma multiforme cells. Oncogene. 21:8404–8413. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weissenberger J, Loeffler S, Kappeler A,
Kopf M, Lukes A, Afanasieva TA, Aguzzi A and Weis J: IL-6 is
required for glioma development in a mouse model. Oncogene.
23:3308–3316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
de la Iglesia N, Konopka G, Puram SV, Chan
JA, Bachoo RM, You MJ, Levy DE, Depinho RA and Bonni A:
Identification of a PTEN-regulated STAT3 brain tumor suppressor
pathway. Genes Dev. 22:449–462. 2008.PubMed/NCBI
|
|
25
|
Papagiannakopoulos T and Kosik KS:
MicroRNAs: regulators of oncogenesis and stemness. BMC Med.
6:152008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dick JE: Normal and leukemic human stem
cells assayed in SCID mice. Semin Immunol. 8:197–206. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin L, Hope KJ, Zhai Q, Smadja-Joffe F and
Dick JE: Targeting of CD44 eradicates human acute myeloid leukemic
stem cells. Nat Med. 12:1167–1174. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Al-Hajj M and Clarke MF: Self-renewal and
solid tumor stem cells. Oncogene. 23:7274–7282. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rich JN: Cancer stem cells in radiation
resistance. Cancer Res. 67:8980–8984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Inoue A, Takahashi H, Harada H, Kohno S,
Ohue S, Kobayashi K, Yano H, Tanaka J and Ohnishi T: Cancer
stem-like cells of glioblastoma characteristically express MMP-13
and display highly invasive activity. Int J Oncol. 37:1121–1131.
2010.PubMed/NCBI
|
|
31
|
Wang H, Zhang W, Huang HJ, Liao WS and
Fuller GN: Analysis of the activation status of Akt, NFkappaB, and
Stat3 in human diffuse gliomas. Lab Invest. 84:941–951. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Buettner R, Mora LB and Jove R: Activated
STAT signaling in human tumors provides novel molecular targets for
therapeutic intervention. Clin Cancer Res. 8:945–954.
2002.PubMed/NCBI
|
|
33
|
Kusaba T, Nakayama T, Yamazumi K, Yakata
Y, Yoshizaki A, Nagayasu T and Sekine I: Expression of p-STAT3 in
human colorectal adenocarcinoma and adenoma; correlation with
clinicopathological factors. J Clin Pathol. 58:833–838. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Korkaya H, Liu S and Wicha MS: Regulation
of cancer stem cells by cytokine networks: attacking cancer’s
inflammatory roots. Clin Cancer Res. 17:6125–6129. 2011.PubMed/NCBI
|
|
35
|
Marotta LL, Almendro V, Marusyk A,
Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ,
Choudhury SA, Maruyama R, Wu Z, Gonen M, Mulvey LA, Bessarabova MO,
Huh SJ, Silver SJ, Kim SY, Park SY, Lee HE, Anderson KS, Richardson
AL, Nikolskaya T, Nikolsky Y, Liu XS, Root DE, Hahn WC, Frank DA
and Polyak K: The JAK2/STAT3 signaling pathway is required for
growth of CD44CD24 stem cell-like breast cancer cells in human
tumors. J Clin Invest. 121:2723–2735. 2011. View Article : Google Scholar : PubMed/NCBI
|