Introduction
The influence of the cancer microenvironment on the
behavior of tumors has received considerable attention since Poste
and Fidler (1,2) re-illuminated ‘seed and soil’
hypothesis, first postulated by the British surgeon Paget (3). The cancer microenvironment, which
consists of variety of cell types, the extracellular matrix and
signaling molecules, significantly affects the initiation,
progression and metastasis of cancers (4,5).
Among these components of the tumor microenvironment, mesenchymal
stem cells (MSCs) or mesenchymal stem-like cells (MSLCs) have been
focus of particular research interest.
With accumulating evidence suggesting a strong
association between MSCs and various tumors, including breast
cancer (6,7), lipoma (8), gastric cancer (9) and bone sarcomas (10), the veiled relationship between MSCs
and tumors has started to come into focus. Tumors of the human
brain are no exception. Lang et al first described that
cells which adhere and grow on plastic; differentiate into
osteocytes, adipocytes and chondrocytes; and express the
appropriate MSC surface markers isolated from surgical glioma
specimens [Society for Neuro-Oncology Annual Meeting, Neuro Oncol
9: abs. 596, 2007, and ASCO Annual Meeting, J Clin Oncol 26 (Suppl
15): abs. 2001, 2008].
Moreover, a series of studies reported the isolation
of cells resembling MSCs from normal mouse brain (11), glioma xenograft specimens (12) and Korean glioma specimens (13). These cells, referred to as MSLCs,
do not completely meet all the MSC-defining criteria proposed by
the International Society for Cellular Therapy (14). However, notwithstanding their
glioma origin, they have properties similar to those of MSCs, such
as adherence to plastic, expression of surface antigens
characteristic of MSCs, mesenchymal differentiation potential and a
lack of gliomagenesis potential.
Given that MSCs in the tumor microenvironment play a
critical role in determining the biological behavior of the tumor
(15), the fact that MSLCs are
present in glioma specimens and share characteristics of MSCs
suggests that these cells also likely make an important
contribution to the tumor microenvironment. Hossain et al
recently presented that tumor-associated mesenchymal stromal cells
in glioblastomas (GBMs) enhance the tumorigenic and proliferative
properties of glioma cancer stem cells (gCSCs) through the
interleukin (IL)-6/STAT3 (signal transducer and activator of
transcription 3) pathway [Society for Neuro-Oncology annual
meeting, Neuro Oncol 13 (Suppl 3): abs. iii19, 2011]. Moreover,
changes in the microenvironment have been clearly shown to affect
the biological characteristics of gCSCs (16). The exact functions of MSCs and
MSLCs, however, are a matter of controversy.
In this study, we investigated the possible role of
MSLCs in GBM by examining how glioma stroma mesenchymal stem-like
cells (GS-MSLCs) obtained from Korean glioma specimens affect
gCSCs, which are thought to be the major cells responsible for the
initiation, maintenance, and progression of glioma (17–22).
We hypothesized that GS-MSLCs favor tumor growth by altering the
biological nature of gCSCs. To verify this, we transfected gCSCs
with green fluorescent protein (GFP) and cultured them alone or
together with GS-MSLCs. After 3 weeks in culture, we compared the
numbers of GFP-emitting gCSCs (GFP-gCSCs) obtained from the two
culture groups. Finally, we prepared orthotopic xenograft mouse
models using GFP-gCSCs cultured alone and GFP-gCSCs isolated from
GS-MSLCs co-cultures, and then compared survival, tumor volume,
tumor cell proliferation and apoptosis, and microvessel density
between the two groups.
Materials and methods
Isolation and culture of human gCSCs and
GS-MSLCs
Specimens for isolation of gCSCs were collected in
the operating room from GBM patients undergoing surgery. Approval
for harvest and use was obtained from the institutional review
boards of Severance Hospital, Yonsei University College of Medicine
(4-2012-0212), and Seoul St. Mary’s Hospital, the Catholic
University of Korea College of Medicine (KC10SNS10466). Informed
consent was provided according to the Declaration of Helsinki.
Neuropathologists diagnosed each surgical specimen according to
world health organization (WHO) classifications (23). gCSCs were isolated from GBM
specimens within one hour of glioma removal using a previously
described mechanical dissociation method for isolating gCSCs from
human brain (19,22,24).
Briefly, surgical specimens were minced and dissociated with a
scalpel in Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12
(DMEM/F-12; Mediatech, Manassas, VA, USA) and then passed through a
series of 100-μm nylon mesh cell strainers (BD Falcon, Franklin
Lakes, NJ, USA). Cell suspensions were washed twice in DMEM/F-12
and cultured in gCSC complete medium (DMEM/F-12) containing 1X B27
(Invitrogen, San Diego, CA, USA) plus 20 ng/ml basic fibroblast
growth factor (bFGF; Sigma, St. Louis, MO, USA), 20 ng/ml epidermal
growth factor (EGF; Sigma), and 50 U/ml penicillin/50 mg/ml
streptomycin (Gibco, Invitrogen Korea, Seoul, Korea). One type of
gCSC (gCSC0504) from GBM was used for this study (22).
Methods shown to be reliable for isolating MSLCs
from normal brain (11),
orthotopic glioma xenografts (12)
and Korean glioma specimens (13)
were used for isolation of GS-MSLCs from GBM specimens. Cells were
isolated within one hour of glioma removal using the mechanical
dissociation method described above for gCSCs. Cell suspensions
were washed twice in minimal essential medium-α (MEMα; Mediatech,
Herndon, VA, USA), placed in a 10-cm2 cell culture dish
at a density of 2×106 cells/cm2, and cultured
in complete MSC medium consisting of MEMα (Mediatech), 10% fetal
bovine serum (FBS; Lonza, Basel, Switzerland), 2 mM L-glutamine
(Mediatech) and antibiotic-antimycotic solution (Gibco, Invitrogen
Korea, Seoul, Korea). After 24 h, non-adherent cells were removed
by washing twice with phosphate-buffered saline (PBS; Mediatech)
and the adherent cells were cultured until they reached confluence.
Cells were then trypsinized (0.25% trypsin with 0.1% EDTA) and
subcultured at a density of 5,000 cells/cm2. Cells were
cultured continuously through three passages, consistent with their
status as progenitor/stem cells. Cell morphology was examined by
observing cell cultured with inverted phase-contrast microscope
(IX71 Inverted Microscope; Olympus, Tokyo, Japan). One type of
GS-MSLCs (KGS-MSC0503) form GBM was used for this study (13).
Lentiviral vector transduction and
expression
GFP-gCSCs for cell counting were generated by
growing gCSCs in complete medium and then applying GFP-expressing
lentiviral supernatants. Polybrene (Sigma) was added to a final
concentration of 8 μg/ml and incubated with cells for 18 h.
After infection, the cells were placed in fresh growth medium and
cultured in a standad manner. Cells were treated with 1 mg/ml
puromycin (Life Technologies Korea, Seoul, Korea) to eliminate
uninfected cells and generated stable GFP-gCSC. GFP-expressing
gCSCs were isolated for use in further experiments.
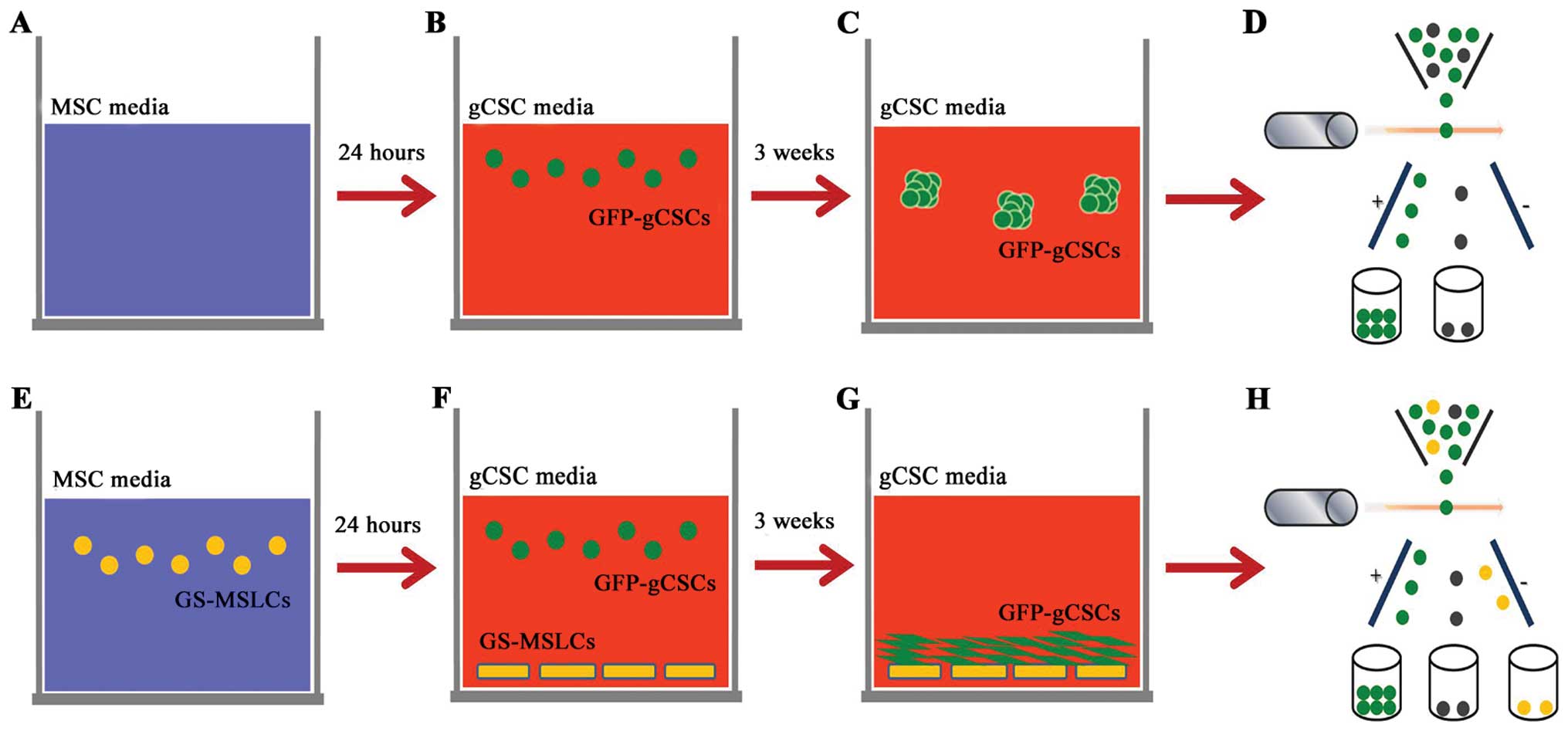
Sorting and counting of GFP-gCSCs
Increase in cell numbers was evaluated in GFP-gCSCs
cultured alone or together with GS-MSLCs for 3 weeks. For the
GFP-gCSCs monoculture group, complete MSC medium (without GS-MSLCs)
was incubated for 24 h (Fig. 1A),
after which the medium was removed, plates were washed three times
with PBS, and fresh gCSCs complete medium containing
1.25×105 GFP-gCSCs was added (Fig. 1B). The medium was replaced with
fresh gCSCs complete medium every 3 days. After 3 weeks,
gliomasphere formation by GFP-gCSCs was evident (Fig. 1C). For the GFP-gCSCs/GS-MSLC
co-culture group, GS-MSLCs (2.5×105) were first isolated
and seeded in 100-mm culture dishes containing MSC complete medium
(Fig. 1E). The next day, the
culture medium was removed and cells were washed three times with
PBS. gCSC complete medium containing 1.25×105 GFP-gCSCs
were then added (Fig. 1F), and
cells were incubated at 37°C in a humidified 5% CO2
incubator for 3 weeks. The medium was replaced with fresh gCSC
complete medium every 3 days. After 3 weeks, GFP-gCSCs had adhered
to GS-MSLCs at the bottom of the culture dish (Fig. 1G). In the last step, GFP-gCSC
monoculture (Fig. 1D) and GS-MSC
co-culture (Fig. 1H) groups were
isolated by fluorescence-activated cell sorting (FACS).
Orthotopic glioma xenograft model
Male athymic nude mice (4–8-weeks-old; Central
Laboratory Animal Inc., Seoul, Korea) were used for tumor xenograft
experiments. Mice were housed in micro-isolator cages under sterile
conditions and observed for at least 1 week before study initiation
to ensure proper health. Temperature, lighting and humidity were
controlled centrally. All experimental procedures were approved by
Yonsei University College of Medicine Institutional Animal Care and
Use Committee and the Catholic University of Korea College of
Medicine Institutional Animal Care and Use Committee. Mice were
anesthetized with a solution of Zoletil (30 mg/kg; Virbac Korea,
Seoul, Korea) and xylazine (10 mg/kg; Bayer Korea, Seoul, Korea)
delivered intraperitoneally. GFP-gCSCs (5×105) isolated
from monoculture or gCSC/GS-MSLC co-culture groups were implanted
into the right frontal lobe via a Hamilton syringe (Dongwoo Science
Co., Seoul, Korea) inserted to a depth of 4.5 mm using a
guide-screw system, as described previously (25). Cells from each culture condition
were simultaneously injected into 10 mice at a rate of 0.5
μl/min using a multiple micro-infusion syringe pump (Harvard
Apparatus, Holliston, MA, USA). The body weights of mice were
checked every other day. If body weight decreased by more than 15%
compared to the original body weight, mice were euthanized
according to the study protocol.
Tumor volume measurement
For comparison of the tumor volume, eight
GFP-gCSC-injected mice (four each from monoculture and GS-MSLC
co-culture groups) were sacrificed 40 days after injection. The
brains were carefully removed and placed in 10% buffered formalin
for fixation. The fixed brains were then cut axially every 2 mm and
embedded in paraffin. Tumor volume was calculated by measuring the
section with the largest tumor portion and applying the formula,
length × width2 × 0.5 (26,27).
Immunohistochemical analysis of PCNA,
TUNEL and CD31
Paraffin-embedded tissues were immunostained for
proliferating cell nuclear antigen (PCNA) and the endothelial
cell-specific marker CD31 to evaluate tumor cell proliferation and
microvessel density, respectively. Apoptosis was assessed using
deoxynucleotidyl transferase-mediated dUTP nick-end labeling
(TUNEL) assays. Tissue sections (4–6 μm thick) were mounted
on silanized glass slides and dried overnight. The sections were
deparaffinized using xylene, then treated with graded series of
alcohol [100, 95 and 80% ethanol/ddH2O (vol/vol)] and
rehydrated in PBS (pH 7.5). For PCNA immunostaining, sections were
microwaved for 5 min in water followed by treatment with 3%
H2O2 in methanol for 5 min to block
endogenous peroxidase. Slide-mounted slices were then incubated at
4°C overnight with mouse, anti-PCNA primary antibody (1:100;
Invitrogen Korea, Seoul, Korea) in a 5% normal horse serum, 1%
normal goat serum phosphate buffer solution. Peroxidase-conjugated
secondary antibodies were used and visualized by incubating the
slides with 3, 3′-diaminobenzidine (DAB) stable substrate solution
for 10–20 min. The sections were rinsed with distilled water,
counterstained with Gill’s hematoxylin for 1 min, and mounted with
Universal Mount (Research Genetics, Huntsville, AL, USA). Twenty
different areas of a single slide (one brain section) were scored.
Expression of CD31 (1:50; BD Pharmigen, San Diego, CA, USA) was
determined by similar method. Apoptosis in tissues was detected
using a commercially available TUNEL kit (Apop Tag Peroxidase in
situ Apoptosis Detection Kit; Chemicon, Danvers, MA, USA).
Detection of PCNA, CD31 and TUNEL was done as described previously
(27,28).
Quantification of PCNA, TUNEL and
CD31
PCNA and CD31 expression, and TUNEL staining were
quantified by counting the numbers of positive cells in 20 randomly
chosen fields at ×400 (PCNA, TUNEL) or ×200 (CD31) magnification. A
single microvessel was defined as an individual cell or discrete
cluster of CD31-positive cells. The presence of a lumen was not
required for being scored as a microvessel.
Statistics
Data are expressed as means ± standard deviations.
Survival of GFP-gCSC-implanted mice (from monoculture and GS-MSLC
co-culture groups) was evaluated using the Kaplan-Meier method.
Comparisons of cell counts, tumor volume and PCNA, TUNEL and CD31
staining were analyzed by Kruskal-Wallis test. All statistical
analyses and graphing were performed using SPSS version 18.0KO
software (SPSS Korea, Seoul, Korea).
Results
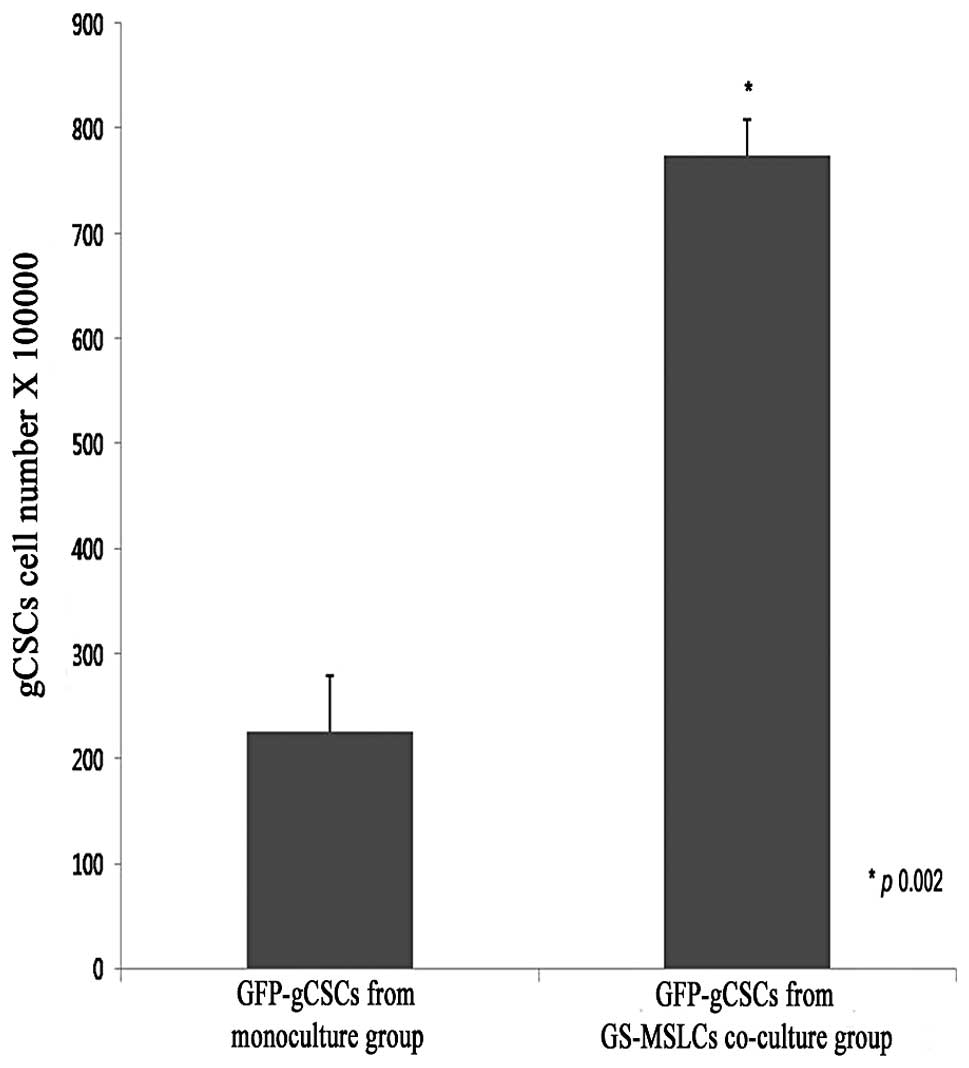
Sorting and counting GFP-gCSCs in
vitro
To analyze the effects of GS-MSLCs on the growth of
gCSCs in vitro, we cultured GFP-gCSCs alone and together
with GS-MSLCs for 3 weeks and counted the numbers of GFP-gCSCs
under a fluorescence microscope using a hemocytometer. As shown in
Fig. 2, the number of GFP-gCSCs in
the group co-cultured with GS-MSLCs (7.76±0.39×105) was
significantly increased (∼3-fold) compared to that in the GFP-gCSC
monoculture group (2.62±0.44×105; p=0.002).
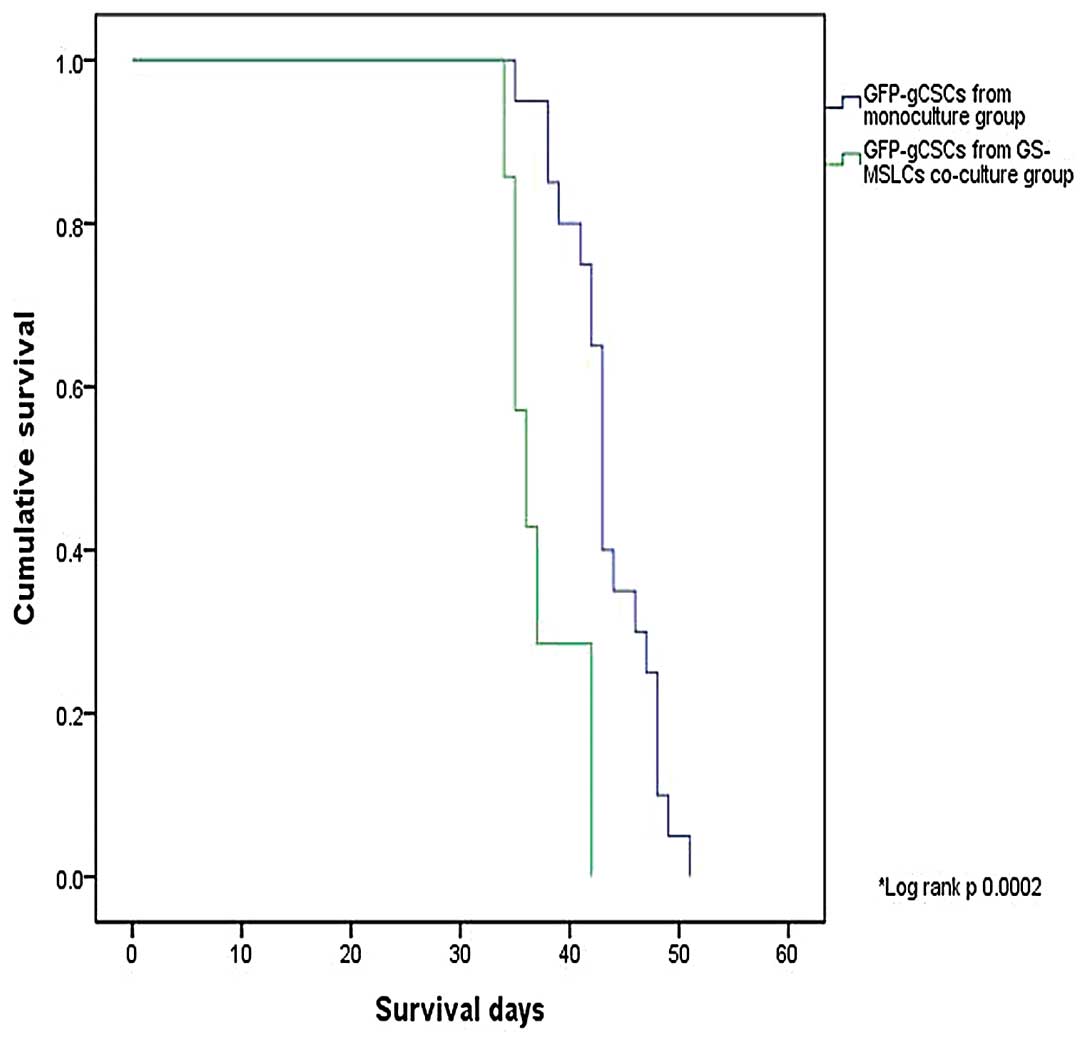
Survival of orthotopic xenograft model
mice
To evaluate the effect of GS-MSLCs on in vivo
tumor growth, we used an orthotopic xenograft mouse model of
glioma. We first compared survival of nude mice cranially implanted
with monocultured GFP-gCSCs (n=20) or GFP-gCSCs co-cultured with
GS-MSLCs (n=7). As shown in Fig.
3, average survival was significantly decreased in mice
injected with GFP-gCSCs co-cultured with GS-MSLCs compared with
mice injected with monocultured GFP-gCSCs (37.3±1.3 vs. 43.5±1.0
days; p=0.002, log-rank test). No mice implanted with mono-cultured
GFP-gCSCs showed a shorter survival than mice implanted with
GFP-gCSCs co-cultured with GS-MSLCs.
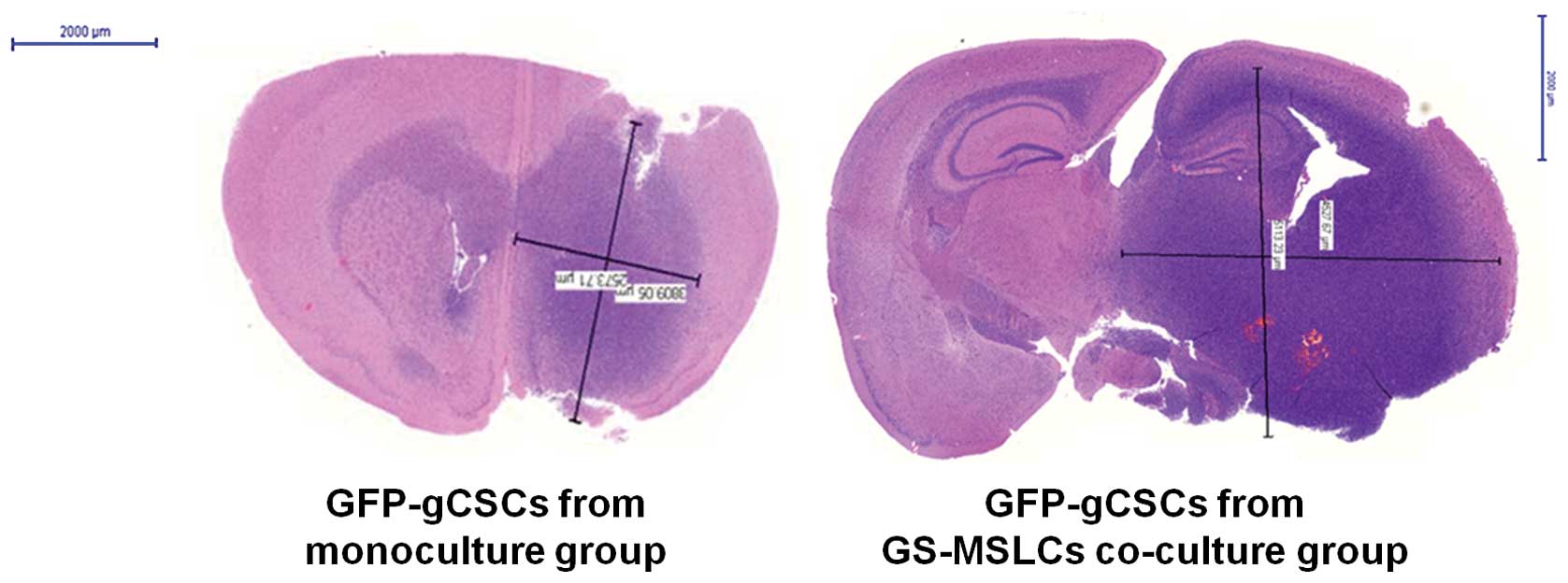
Tumor volumes in orthotopic xenograft
model mice
Four mice from each GFP-gCSC-implanted group
(monoculture and GS-MSLC co-culture) were used to study the effects
of GS-MSLCs on the rate and size of gCSC tumor growth in
vivo. On day 40 after GFP-gCSCs injection, mouse brains were
carefully removed and tumor volumes were measured. Consistent with
the survival data, tumors in mice implanted with GFP-gCSCs from
GS-MSLC co-cultures were larger in both dimensions (length and
width) than those from mice implanted with monocultured GFP-gCSCs
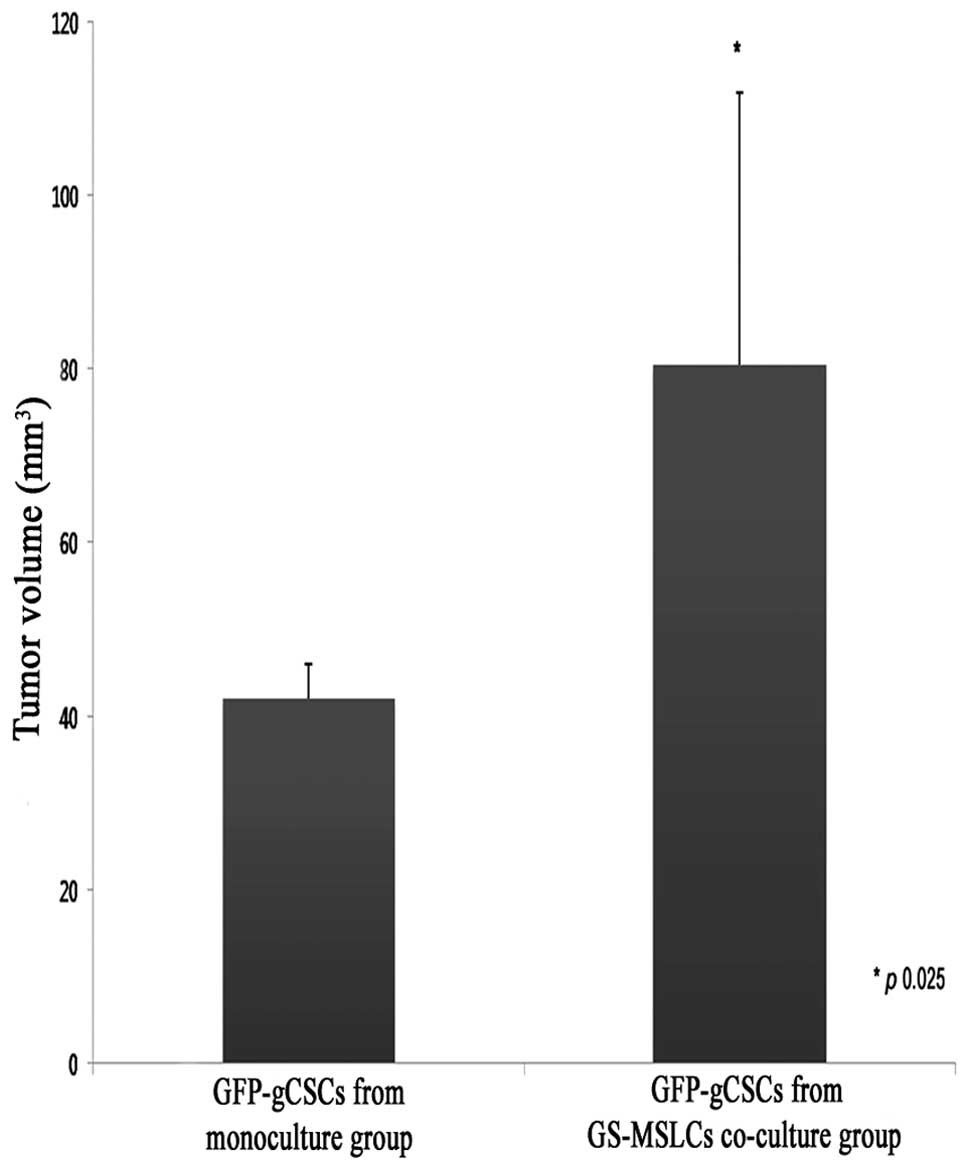
(Fig. 4). As shown in Fig. 5, average brain tumor volumes,
calculated as described in Materials and methods, were
significantly greater in mice implanted with GFP-gCSCs co-cultured
with GS-MSLCs (80.58±31.29 mm3) than in those implanted
with GFP-gCSCs cultured alone (42±4 mm3; p=0.025). These
data indicate that tumor formation and growth were more rapid in
mice injected with GFP-gCSCs co-cultured with GS-MSLCs.
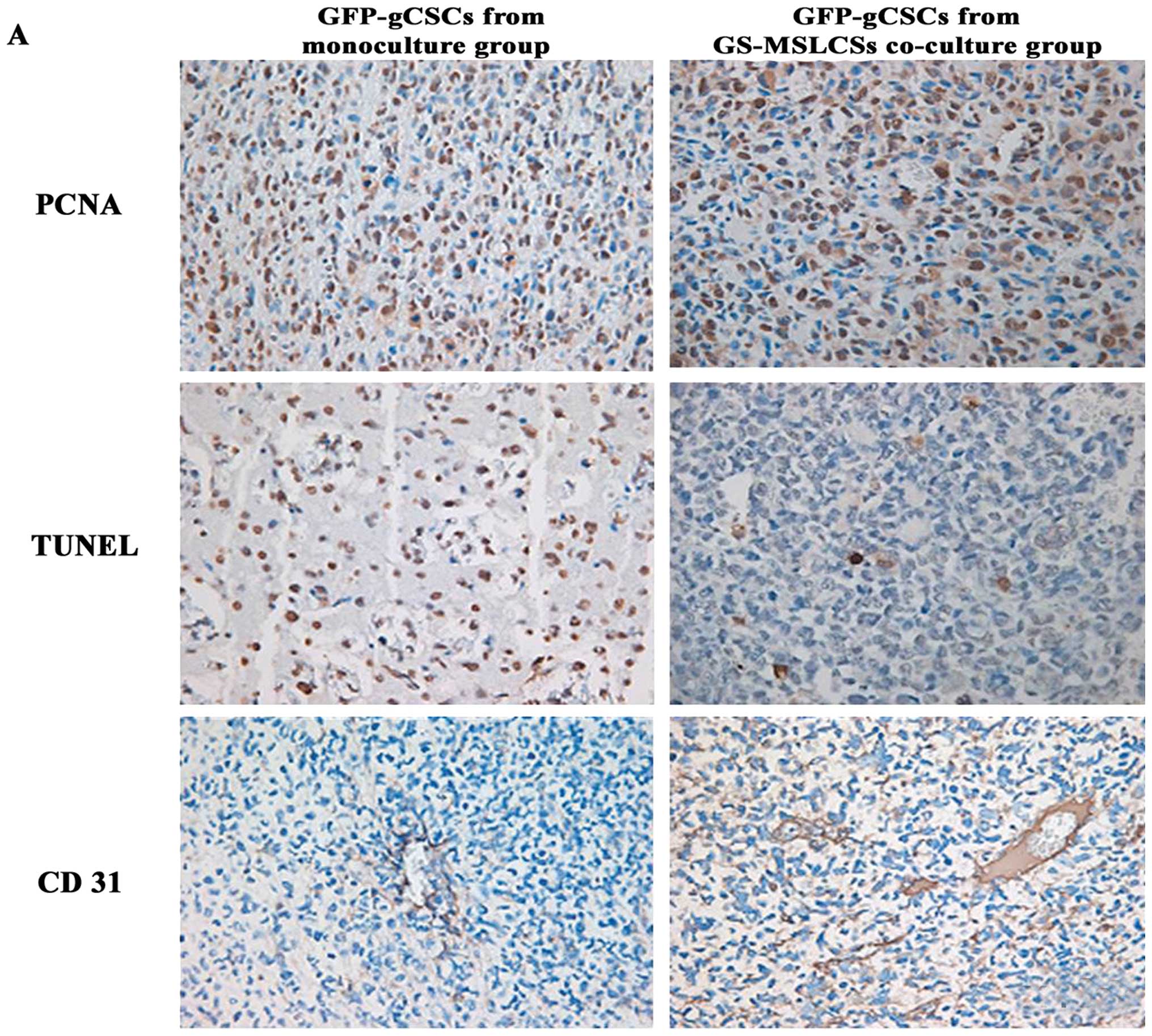
Immunohistochemical analysis of PCNA,
TUNEL and CD31
To investigate the biochemical mechanism underlying
the in vivo effects of gCSC/GS-MSLC co-culture, we examined
tumor cell proliferation (PCNA), apoptosis (TUNEL) and microvessel
formation (CD31) using immunohistochemistry (Fig. 6A). The eight mice used for
measuring tumor volume were used for these analyses. The number of
PCNA-positive cells, an indicator of the rate of proliferation, was
not significantly different between tumors formed by GFP-gCSCs
cultured alone (410±120) and GFP-gCSCs co-cultured with GS-MSLC
(380±40; Fig. 6B). The number of
TUNEL-positive tumor cells, indicative of apoptosis, was also
statistically indistinguishable between GFP-gCSCs cultured alone
(151±36.2) and those co-cultured with GS-MSLCs (222±84.6; Fig. 6C). In contrast, the expression of
CD31, an indicator of tumor microvessels, was approximately
2.5-fold higher in tumors formed by GFP-gCSCs from GS-MSLC
co-cultures (350±50) than in those formed from GFP-gCSCs
mono-cultures (140±20; p=0.016; Fig.
6D), suggesting increased tumor microvessel density.
Collectively, these data imply that co-culture with GS-MSLCs
increases the angiogenesis capacity of gCSCs.
Discussion
In this study, we tested the effects of GS-MSLCs on
tumor growth by specifically examining how GS-MSLCs in the tumor
microenvironment influence the biological properties of gCSCs. We
found that co-culture with GS-MSLCs increased the aggressiveness of
gCSCs, as evidenced by their increased cell numbers in
vitro, reduced survival of mice orthotopically xenografted, and
increased microvessel density in tumors formed from them. Our
findings suggest that GS-MSLCs influence the properties of gCSCs,
shifting them towards a more aggressive state. Moreover,
angiogenesis may be a pivotal part of this mechanism.
Our immunohistochemistry data showed increased CD31
expression in GFP-gCSCs after co-culturing with GS-MSLCs,
suggesting increased vessel formation. Since angiogenesis is a
hallmark of tumor progression, we believe that, by enhancing the
angiogenic capacity of gCSCs, GS-MSLCs might have contributed to
the more rapid and extensive tumor growth in mice implanted with
GFP-gCSCs co-cultured with GS-MSLCs (Figs. 5 and 6). The increased number of GFP-gCSCs in
co-cultures of gCSCs and GS-MSLCs in vitro (Fig. 2) suggested the possibility that a
mechanism involving increased cell proliferation and/or decreased
cell death also contributed to the more aggressive tumor phenotype.
Although the level of proliferation (PCNA expression) showed a
higher trend in tumors formed by GFP-gCSCs obtained from GS-MSLC
co-cultures, and apoptosis (TUNEL staining) lower trends that would
be consistent with the in vitro data-these differences did
not reach statistical significance (Fig. 6B and C). Thus, angiogenesis appears
to play a critical role in creating a supportive environment for
gCSCs and tumor growth (Fig. 6D),
other mechanisms may also contribute. Whether a different
experimental design and larger sample size might establish a
significant role for proliferation and/or apoptosis, or whether
some entirely different mechanism is involved, are questions
currently under investigation in our laboratory.
Our data strongly argue that GS-MSLCs favorably
influence the growth of glioma; however, not all literature reports
are in agreement on this point. Qiao et al reported that
human MSCs inhibit the proliferation and colony-forming ability of
human cancer cell lines (29); a
similar antitumoral role of MSCs was also observed in Kaposi’s
sarcomas (30) and colon carcinoma
in rats (31). Furthermore,
contrary to our data, Nakamura et al reported that rat MSCs
inhibit the growth of 9L glioma cells both in vitro and
in vivo and increase the survival of 9L glioma-bearing rats
(32). An important point that
sets our experiments apart from these data is that we used GS-MSLCs
isolated from that GBM stroma, not normal MSCs from a normal organ.
We think that it is possible that MSCs in a tumor may be
re-programmed or otherwise altered by the tumor microenvironment,
changing their characteristics to those that favor tumor
growth.
While some researchers have argued that MSCs
negatively impact tumor growth, numerous researchers have reported
experiments supporting the tumor growth-enhancing role of MSCs. It
has been speculated that, by secreting cytokines that mediate
angiogenesis and immunosuppressive effects in tumor sites, MSCs
provide a stem cell niche that facilitates tumor progression
(33–36). In a paper on the suppressive role
of MSCs in hematopoietic and non-hematopoietic tumor cells in
vitro, Ramasamy et al also demonstrated that
co-injection of MSCs with tumor cells resulted in more rapid in
vivo tumor growth (37). Tumor
cell-protecting and growth-promoting roles of MSCs have also been
observed in many other cancers, including melanoma (15,38),
breast cancer (39), myeloid
leukemia (40) and colon cancer
(41). Furthermore, MSCs have also
been shown to stimulate metastasis of breast cancer (6) and play a role in the drug resistance
of leukemia cells (42–44).
Despite our data and other evidence suggesting tumor
progression-enhancing role for the MSCs and MSLCs, negative results
on this phenomenon are not readily dismissed. The reasons for these
discrepancies are not fully understood, but Sasser et
al(45) and Karnoub et
al(6) have proposed that the
genetic background of tumor cells may be a key determinant in the
MSCs-tumor relationship. Specifically, Sasser et al reported
that an MSC paracrine factor enhanced the growth of estrogen
receptor-α (ERα)-positive breast cancer, but had no or a
substantially diminished effect on ERα-negative breast cancer
(45). Similarly, Karnoub et
al found that MSCs only accelerated the growth of ERα-positive,
but not ERα-negative, tumor xenografts in mouse skin (6). Although the mechanism underlying this
apparent duality of MSL/MSLC effects remains to be elucidated, it
is clear that the MSCs or MSLCs microenvironment is a double-edged
sword capable of exacerbating cancer by stimulating its
progression, metastasis, and resistance to drugs (46).
Mesenchymal fibroblasts within solid tumors,
referred to as activated fibroblasts or carcinoma-associated
fibroblasts (CAFs), serve as indirect evidence supporting
tumor-promoting role of MSCs or MSLCs. Although the origin and
biology of CAFs are not completely defined, the fact that CAFs and
MSCs share common surface markers and exhibit similar functions
implies that they may originate from the same progenitor cell
(47,48) or that MSCs may differentiate into
CAFs in a tumor context (41,49–51).
The hypothesis that MSCs transition into CAFs within a tumor
environment is supported by a study by Shinagawa et
al(41). These authors
observed MSCs moving towards the stroma of colon cancer tumors and
differentiating into CAFs, and thereby promoting angiogenesis,
tumor growth, migration, invasion, and metastasis. Considerable
additional experimental evidence has definitively shown that CAFs
play a role in the progression towards an aggressive phenotype in
various cancers (52–56). Although these results do not
unambiguously establish that MSCs differentiate into CAFs, they
strongly suggest that MSCs and CAFs are closely associated and
indicate that CAFs might participate in the mechanism by which MSCs
modulate the tumor microenvironment. The possibility that GS-MSLCs
may also interact with CAFs in glioma microenvironment suggested by
these reports is currently under investigation in our
laboratory.
In our experiments, gCSCs and GS-MSLCs were in
direct contact when co-cultured, allowing bidirectional crosstalk
between them. Karnoub et al have also pointed out that the
increase in the metastatic potency of tumor cells induced by MSCs
requires that the two cell types be in direct contact (6). This phenomenon is consistent with the
current understanding that MSCs interact with tumor cells by direct
contact and though paracrine factors. Tumor cells secrete cytokines
that attract MSCs to the tumor stroma and MSCs release cytokines,
matrix-degrading enzymes, and immunomodulatory factors that
regulate tumor growth, invasion and metastasis (5,6,57).
We believe that the direct contact between MSCs or MSLCs and gCSCs
in our experimentational setting mimics that of the in vivo
tumor microenvironment. In fact gCSCs (22,58)
and GS-MSLCs (12,13) both reside within vascular niches in
gliomas. Although we are not yet certain whether GS-MSLCs and
normal MSCs share the same functions and interact with gCSCs in the
same manner, the fact that they reside in the same site strengthens
the possibility for cross-talk between gCSCs and GS-MSLCs through
direct contact and paracrine factors.
The tumor-homing characteristics of MSCs have led
many researchers to investigate the potential of MSCs as targeted
delivery vehicles for cancer therapy (59,60).
The results of our experiments, however, offer a cautionary tale.
Although normal MSCs and GS-MSLCs are not identical, our
demonstration that co-culture with GS-MSLCs increases gCSC cell
numbers in vitro, reduces the survival of mice injected with
gCSCs co-cultured with GS-MSLCs, and increases tumor volume and
vessel formation in the tumors of these mice warns of the potential
dangers of MSC-gCSC interactions. These concerns notwithstanding,
MSCs still have great potential as a therapeutic tool in cancer,
but more likely as a therapeutic target rather than as a
targeted-delivery vehicle. MSCs are known to affects the biological
characteristics of cancer by three mechanisms: i) production of
growth factors, cytokines, and immunomodulatory factors that favor
tumor progression; ii) formation of a cancer stem cell niche that
facilitates the growth of cancer stem cells; and iii)
differentiation into other stromal cells, such as CAFs. Our data
further suggest that GS-MSLCs may even shift the biological
property of cancer stem cells toward a more malignant phenotype.
Although it is clear that considerable additional study will be
required before this mechanism is completely understood and can be
applied clinically, GS-MSLCs and their interactions with tumor
cells are a promising new target in cancer therapy.
In conclusion, we found that GS-MSLCs, as a
component of the tumor microenvironment, significantly change the
biological properties of gCSCs. Co-culture with GS-MSLCs increased
the aggressiveness of gCSCs, as evidenced by elevated cell counts
in vitro, shortened survival time in orthotopic xenograft
mouse models, and increased tumor volume and vessel formation in
vivo. Moreover, from the increased expression of the
endothelial cell marker CD31, we infer that GS-MSLCs positively
influence tumor growth by enhancing the angiogenic capacity of
gCSCs. The next step is to clarify the exact mechanism underlying
the supportive relationship between GS-MSLCs and gCSCs. These
experiments are currently underway in our laboratory.
Acknowledgements
This research was supported by the
Basic Science Research Program through the National Research
Foundation of Korea (NRF) funded by the Ministry of Education,
Science and Technology (2010-0004506) and a grant from the National
R&D Program for Cancer Control, Ministry for Health, Welfare
and Family Affairs, Republic of Korea (1020340).
References
|
1.
|
Fidler IJ and Poste G: The ‘seed and soil’
hypothesis revisited. Lancet Oncol. 9:8082008.
|
|
2.
|
Poste G and Fidler IJ: The pathogenesis of
cancer metastasis. Nature. 283:139–146. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Paget S: The distribution of secondary
growths in cancer of the breast. Lancet. 133:571–573. 1889.
View Article : Google Scholar
|
|
4.
|
Albini A and Sporn MB: The tumour
microenvironment as a target for chemoprevention. Nat Rev Cancer.
7:139–147. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Zhang W, Remenyik E, Zelterman D, Brash DE
and Wikonkal NM: Escaping the stem cell compartment: sustained UVB
exposure allows p53-mutant keratinocytes to colonize adjacent
epidermal proliferating units without incurring additional
mutations. Proc Natl Acad Sci USA. 98:13948–13953. 2001. View Article : Google Scholar
|
|
6.
|
Karnoub AE, Dash AB, Vo AP, et al:
Mesenchymal stem cells within tumour stroma promote breast cancer
metastasis. Nature. 449:557–563. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
El-Haibi CP and Karnoub AE: Mesenchymal
stem cells in the pathogenesis and therapy of breast cancer. J
Mammary Gland Biol Neoplasia. 15:399–409. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Lin TM, Chang HW, Wang KH, et al:
Isolation and identification of mesenchymal stem cells from human
lipoma tissue. Biochem Biophys Res Commun. 361:883–889. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Houghton J, Stoicov C, Nomura S, et al:
Gastric cancer originating from bone marrow-derived cells. Science.
306:1568–1571. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Gibbs CP, Kukekov VG, Reith JD, et al:
Stem-like cells in bone sarcomas: implications for tumorigenesis.
Neoplasia. 7:967–976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Kang SG, Shinojima N, Hossain A, et al:
Isolation and perivascular localization of mesenchymal stem cells
from mouse brain. Neurosurgery. 67:711–720. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kim SM, Kang SG, Park NR, et al: Presence
of glioma stroma mesenchymal stem cells in a murine orthotopic
glioma model. Childs Nerv Syst. 27:911–922. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kim YG, Jeon S, Sin GY, et al: Existence
of glioma stroma mesenchymal stem-like cells in Korean glioma
specimens. Childs Nerv Syst. Dec 29–2012.(Epub ahead of print).
View Article : Google Scholar
|
|
14.
|
Dominici M, Le Blanc K, Mueller I, et al:
Minimal criteria for defining multipotent mesenchymal stromal
cells. The International Society for Cellular Therapy position
statement. Cytotherapy. 8:315–317. 2006. View Article : Google Scholar
|
|
15.
|
Kucerova L, Matuskova M, Hlubinova K,
Altanerova V and Altaner C: Tumor cell behaviour modulation by
mesenchymal stromal cells. Mol Cancer. 9:1292010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Shin GY, Shim JK, Lee JH, et al: Changes
in the biological characteristics of glioma cancer stem cells after
serial in vivo subtransplantation. Childs Nerv Syst. 29:55–64.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Dirks PB: Brain tumor stem cells: bringing
order to the chaos of brain cancer. J Clin Oncol. 26:2916–2924.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Galli R, Binda E, Orfanelli U, et al:
Isolation and characterization of tumorigenic, stem-like neural
precursors from human glioblastoma. Cancer Res. 64:7011–7021. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Singh SK, Hawkins C, Clarke ID, et al:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Sulman E, Aldape K and Colman H: Brain
tumor stem cells. Curr Probl Cancer. 32:124–142. 2008. View Article : Google Scholar
|
|
21.
|
Yuan X, Curtin J, Xiong Y, et al:
Isolation of cancer stem cells from adult glioblastoma multiforme.
Oncogene. 23:9392–9400. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kong BH, Park NR, Shim JK, et al:
Isolation of glioma cancer stem cells in relation to histological
grades in glioma specimens. Childs Nerv Syst. 29:217–219. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Louis DN, Ohgaki H, Wiestler OD, et al:
The 2007 WHO classification of tumours of the central nervous
system. Acta Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Singh SK, Clarke ID, Terasaki M, et al:
Identification of a cancer stem cell in human brain tumors. Cancer
Res. 63:5821–5828. 2003.PubMed/NCBI
|
|
25.
|
Lal S, Lacroix M, Tofilon P, Fuller GN,
Sawaya R and Lang FF: An implantable guide-screw system for brain
tumor studies in small animals. J Neurosurg. 92:326–333. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Nam DH, Park K, Park C, et al:
Intracranial inhibition of glioma cell growth by cyclooxygenase-2
inhibitor celecoxib. Oncol Rep. 11:263–268. 2004.PubMed/NCBI
|
|
27.
|
Kang SG, Kim JS, Park K, Kim JS, Groves MD
and Nam DH: Combination of celecoxib and temozolomide in C6 rat
glioma orthotopic model. Oncol Rep. 15:7–13. 2006.PubMed/NCBI
|
|
28.
|
Kim SJ, Uehara H, Karashima T, Shepherd
DL, Killion JJ and Fidler IJ: Blockade of epidermal growth factor
receptor signaling in tumor cells and tumor-associated endothelial
cells for therapy of androgen-independent human prostate cancer
growing in the bone of nude mice. Clin Cancer Res. 9:1200–1210.
2003.
|
|
29.
|
Qiao L, Xu Z, Zhao T, et al: Suppression
of tumorigenesis by human mesenchymal stem cells in a hepatoma
model. Cell Res. 18:500–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Khakoo AY, Pati S, Anderson SA, et al:
Human mesenchymal stem cells exert potent antitumorigenic effects
in a model of Kaposi’s sarcoma. J Exp Med. 203:1235–1247.
2006.PubMed/NCBI
|
|
31.
|
Ohlsson LB, Varas L, Kjellman C, Edvardsen
K and Lindvall M: Mesenchymal progenitor cell-mediated inhibition
of tumor growth in vivo and in vitro in gelatin matrix. Exp Mol
Pathol. 75:248–255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Nakamura K, Ito Y, Kawano Y, et al:
Antitumor effect of genetically engineered mesenchymal stem cells
in a rat glioma model. Gene Ther. 11:1155–1164. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Roorda BD, ter Elst A, Kamps WA and de
Bont ES: Bone marrow-derived cells and tumor growth: contribution
of bone marrow-derived cells to tumor micro-environments with
special focus on mesenchymal stem cells. Crit Rev Oncol Hematol.
69:187–198. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Djouad F, Plence P, Bony C, et al:
Immunosuppressive effect of mesenchymal stem cells favors tumor
growth in allogeneic animals. Blood. 102:3837–3844. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Le Blanc K and Ringden O: Immunobiology of
human mesenchymal stem cells and future use in hematopoietic stem
cell transplantation. Biol Blood Marrow Transplant. 11:321–334.
2005.PubMed/NCBI
|
|
36.
|
Zhu W, Xu W, Jiang R, et al: Mesenchymal
stem cells derived from bone marrow favor tumor cell growth in
vivo. Exp Mol Pathol. 80:267–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Ramasamy R, Lam EW, Soeiro I, Tisato V,
Bonnet D and Dazzi F: Mesenchymal stem cells inhibit proliferation
and apoptosis of tumor cells: impact on in vivo tumor growth.
Leukemia. 21:304–310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Wang X, Zhang Z and Yao C: Survivin is
upregulated in myeloma cell lines cocultured with mesenchymal stem
cells. Leuk Res. 34:1325–1329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Patel SA, Meyer JR, Greco SJ, Corcoran KE,
Bryan M and Rameshwar P: Mesenchymal stem cells protect breast
cancer cells through regulatory T cells: role of mesenchymal stem
cell-derived TGF-beta. J Immunol. 184:5885–5894. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Konopleva M, Konoplev S, Hu W, Zaritskey
AY, Afanasiev BV and Andreeff M: Stromal cells prevent apoptosis of
AML cells by up-regulation of anti-apoptotic proteins. Leukemia.
16:1713–1724. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Shinagawa K, Kitadai Y, Tanaka M, et al:
Mesenchymal stem cells enhance growth and metastasis of colon
cancer. Int J Cancer. 127:2323–2333. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Lin YM, Zhang GZ, Leng ZX, et al: Study on
the bone marrow mesenchymal stem cells induced drug resistance in
the U937 cells and its mechanism. Chin Med J (Engl). 119:905–910.
2006.PubMed/NCBI
|
|
43.
|
Kurtova AV, Balakrishnan K, Chen R, et al:
Diverse marrow stromal cells protect CLL cells from spontaneous and
drug-induced apoptosis: development of a reliable and reproducible
system to assess stromal cell adhesion-mediated drug resistance.
Blood. 114:4441–4450. 2009. View Article : Google Scholar
|
|
44.
|
Vianello F, Villanova F, Tisato V, et al:
Bone marrow mesenchymal stromal cells non-selectively protect
chronic myeloid leukemia cells from imatinib-induced apoptosis via
the CXCR4/CXCL12 axis. Haematologica. 95:1081–1089. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Sasser AK, Mundy BL, Smith KM, et al:
Human bone marrow stromal cells enhance breast cancer cell growth
rates in a cell line-dependent manner when evaluated in 3D tumor
environments. Cancer Lett. 254:255–264. 2007. View Article : Google Scholar
|
|
46.
|
Zhang W: Mesenchymal stem cells in cancer:
friends or foes. Cancer Biol Ther. 7:252–254. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Haniffa MA, Wang XN, Holtick U, et al:
Adult human fibroblasts are potent immunoregulatory cells and
functionally equivalent to mesenchymal stem cells. J Immunol.
179:1595–1604. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Paunescu V BF, Tatu CA, Gavriliuc OI,
Rosca A, Gruia AT, Tanasie G, Bunu C, Crisnic D, Gherghiceanu M,
Tatu FR, Tatu CS and Vermesan S: Tumour-associated fibroblasts and
mesenchymal stem cells: more similarities than differences. J Cell
Mol Med. 15:635–646. 2011.PubMed/NCBI
|
|
49.
|
Spaeth EL, Dembinski JL, Sasser AK, et al:
Mesenchymal stem cell transition to tumor-associated fibroblasts
contributes to fibrovascular network expansion and tumor
progression. PLoS One. 4:e49922009. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Mishra PJ, Humeniuk R, Medina DJ, et al:
Carcinoma-associated fibroblast-like differentiation of human
mesenchymal stem cells. Cancer Res. 68:4331–4339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Hall B, Dembinski J, Sasser AK, Studeny M,
Andreeff M and Marini F: Mesenchymal stem cells in cancer:
tumor-associated fibroblasts and cell-based delivery vehicles. Int
J Hematol. 86:8–16. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Brentnall TA, Lai LA, Coleman J, Bronner
MP, Pan S and Chen R: Arousal of cancer-associated stroma:
overexpression of palladin activates fibroblasts to promote tumor
invasion. PLoS One. 7:e302192012. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Zhi K, Shen X, Zhang H and Bi J:
Cancer-associated fibroblasts are positively correlated with
metastatic potential of human gastric cancers. J Exp Clin Cancer
Res. 29:662010. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Liao D, Luo Y, Markowitz D, Xiang R and
Reisfeld RA: Cancer associated fibroblasts promote tumor growth and
metastasis by modulating the tumor immune microenvironment in a 4T1
murine breast cancer model. PLoS One. 4:e79652009. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Casey TM, Eneman J, Crocker A, et al:
Cancer associated fibroblasts stimulated by transforming growth
factor beta1 (TGF-beta 1) increase invasion rate of tumor cells: a
population study. Breast Cancer Res Treat. 110:39–49. 2008.
View Article : Google Scholar
|
|
56.
|
Ostman A and Augsten M: Cancer-associated
fibroblasts and tumor growth - bystanders turning into key players.
Curr Opin Genet Dev. 19:67–73. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Honczarenko M, Le Y, Swierkowski M, Ghiran
I, Glodek AM and Silberstein LE: Human bone marrow stromal cells
express a distinct set of biologically functional chemokine
receptors. Stem Cells. 24:1030–1041. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Gilbertson RJ and Rich JN: Making a
tumour’s bed: glioblastoma stem cells and the vascular niche. Nat
Rev Cancer. 7:733–736. 2007.
|
|
59.
|
Ciavarella S, Dominici M, Dammacco F and
Silvestris F: Mesenchymal stem cells: a new promise in anticancer
therapy. Stem Cells Dev. 20:1–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Dai LJ, Moniri MR, Zeng ZR, Zhou JX, Rayat
J and Warnock GL: Potential implications of mesenchymal stem cells
in cancer therapy. Cancer Lett. 305:8–20. 2011. View Article : Google Scholar : PubMed/NCBI
|