Introduction
Heat shock protein 90 (HSP90) is a molecular
chaperone, which in complex with co-chaperones (such as HSP70)
forms the HSP90 chaperone machine. This complex has been
demonstrated to play an important role in stabilization and
activation of numerous key oncogenic client proteins including Akt,
MEK, EGFR, ErbB2 and IGF-IR (1,2). The
HSP90 monomer consists of 3 domains: the amino terminal domain,
which contains the ATP-binding site, the middle domain with docking
sites for client proteins and the carboxy terminal region, which
resembles a dimerization motif (2). Co-chaperone binding sites are present
in all 3 domains whereas drug-binding regions are present in the
amino and carboxy terminal domains (2). Normal chaperone function requires
dimerization of two HSP90 monomers and recruitment of co-chaperones
which regulate the conformational dynamics and activity of the
chaperone (2). HSP90 is
over-expressed in cancer cells and associated with decreased
survival in breast cancer, gastrointestinal stromal tumors and
non-small cell lung cancer (3).
Given the potential to block multiple oncogenic signaling pathways
simultaneously and thus possibly counteract escape mechanisms and
resistance to targeted monotherapies, several HSP90 inhibitors are
currently undergoing clinical trials in multiple indications as
single agents or in combination therapy (2–4).
Neuroendocrine tumors (NETs) of the
gastroenteropancreatic (GEP) system comprise a rare group of tumors
which accounts for ∼2% of all gastrointestinal tumors (5,6).
Over the last decades the incidence of GEP NETs has increased
considerably (6,7). Around 25% of all NETs present with
distant metastasis at the time of first diagnosis and despite
advances in surgical and medical therapy the overall 5-year
survival rate remains rather low (∼60%) (6,8,9).
Thus, novel therapeutic tools are needed for this heterogeneous
group of tumors. Due to high activity of Akt and Erk signaling in
NETs and compensatory activation of Akt in response to mTOR and Raf
inhibitors, targeting HSP90 could provide a tool to simultaneously
suppress both survival pathways (10,11).
Furthermore, HSP90 overexpression has recently been reported in
NETs and the HSP90 inhibitors 17-AAG and IPI-504 have demonstrated
antiproliferative efficacy in several NET cell lines in
vitro (12,13).
Here we report antiproliferative effects of the
novel small molecule HSP90 inhibitors AUY922 and HSP990 and
characterize HSP90 downstream signaling in neuroendocrine tumor
cells of pancreatic, midgut and bronchopulmonary origin.
Materials and methods
Materials
DMEM/F12 media, penicillin and streptomycin were
purchased from Gibco/Invitrogen (Karlsruhe, Germany) and RPMI
medium was from PAA Laboratories (Pasching, Austria). Fetal bovine
serum (FBS) and amphotericin B were from Biochrom (Berlin,
Germany), and AUY922 and HSP990 were kindly provided from Novartis
Pharma (Basel, Switzerland).
Cell cultures
All human neuroendocrine cell lines were received
and cultured as described (14).
Briefly, pancreatic neuroendocrine BON1 tumor cells (kindly
provided by R. Göke, Marburg) were cultured in DMEM/F12 (1:1)
medium supplemented with 10% FBS, 1% penicillin/streptomycin and
0.4% amphotericin B. Human midgut carcinoid GOT1 cells (kindly
provided by Professor Ola Nilsson, Sahlgrenska University Hospital,
Gothenburg, Sweden) and human broncho-pulmonary neuroendocrine
NCI-H727 tumor cells (purchased from ATCC, Manassas, VA, USA) were
both cultured in RPMI medium supplemented with 10% FBS, 1%
penicillin/streptomycin and 0.4% amphotericin B. Additional
supplements in GOT1 culture medium were 0.135 IU/ml insulin and 5
mg/dl apo-transferrin.
Assessment of cell viability
Cell viability was assessed as described (14). Briefly, cells were seeded into
96-well plates at densities of 3,000 (BON1), 50,000 (GOT1) and
4,000 (NCIH727) cells per well, respectively, and grown for 24 h.
The next day, medium was replaced by serum rich medium (10% FBS)
containing various concentrations of AUY922 and HSP990 (0.1, 0.5,
1, 5, 10, 50, 100 nM) and the cells were further incubated for
indicated time intervals. Cell viability expressed by metabolic
activity was measured with Cell Titer 96 aqueous One Solution Cell
Proliferation assay (Promega, Madison, WI, USA) according to the
manufacturer’s instructions. Following 3 h of incubation with Cell
Titer 96 solution, absorbance at 492 nm was determined using an
ELISA plate reader.
SYBR-DNA-labeling assay
The SYBR-DNA-labeling experiment was performed
identically to that described for the Cell Titer 96 aqueous One
Solution Cell Proliferation assay. Assays were stopped after
indicated time intervals by flicking off the medium and freezing
the plate. Cells were stained with 200 μl/well of
SYBR-Green® I (Lonza, Basel, Switzerland) 1:4,000 in
aqua destillata for 30 min in the dark and then quantified
by flourimetry at 530 nm with 485 nm excitation, measured using a
CytoFluor® Multi-Well Plate Reader Series 4000
(PerSeptive Biosystems, Framingham, MA, USA).
Cell cycle analysis
Apoptosis and cell cycle distribution were analyzed
using flow cytometry as described (14). Briefly, cells were scraped with a
rubber policeman, washed with PBS and incubated in staining buffer
containing 0.1% sodium citrate, 0.1% Triton X-100 (Sigma) and 50
μg/ml propidium iodide overnight. Sub-G1 events and cell
cycle distribution were measured in a fluorescence-activated cell
sorter (FACSCalibur, Becton-Dickinson, Franklin Lakes, NJ, USA).
Nuclei to the left of the G1-peak containing hypodiploid DNA were
considered apoptotic.
Caspase assay
Activity of effector caspases 3 and 7 was measured
with Caspase-Glo 3/7 assay (Promega) according to the
manufacturer’s instructions. Following 1 h of incubation with
Caspase-Glo 3/7 reagent, luminescence was determined using a
plate-reading luminometer.
Protein extraction and western blot
analysis
Protein extraction and western blot analysis were
performed as described (14).
Briefly, cells were lysed in 500 μl lysis buffer. The
lysates were centrifuged for 10 min at 4°C and 13,000 × g and
supernatans were adjusted to equal protein loads and diluted 1:1
with SDS sample buffer. Samples were boiled for 5 min and separated
on an SDS polyacrylamide gel. Proteins were electrotransferred for
60 min onto PVDF membranes (Immobilone; Millipore, Eschborn,
Germany) using a semi-dry western blot technique. After blocking in
2% non-fat dried milk, the membranes were incubated overnight in
appropriate dilutions of antibodies against pAkt (Ser 473)
(1:20,000), Akt (1:5,000), pErk 1/2 (1:10,000), Erk 1/2 (1:20,000),
PARP (1:1,000), IGFR (1:5,000), p70S6K (1:1,000), pp70S6K
(1:2,000), 4EBP1 (1:2,000) p4EBP1 (1:1,000) (all from Cell
Signaling, Danvers, MA, USA), HSP70 (1:10,000) (Biomol Stressgen,
Hamburg, Germany), HSP90 (1:5,000), EGFR (1:1,000), ErbB2 (1:500),
ErbB3 (1:1,000) and STAT3 (1:10,000) (all from Santa Cruz,
Heidelberg, Germany). After washing with PBS, the membranes were
incubated with peroxidise-conjugated secondary antibody (1:25,000)
for 2 h. The blots were washed and immersed in the chemiluminescent
substrate SuperSignal West Dura (Thermo Scientific, Rockford, IL,
USA) and exposed to Super RX Fujifilm (Fujifilm Corporation, Tokyo,
Japan).
Statistical analysis
IC50 inhibition values were determined
with the use of Prism 6 for May OS X software (www.graphpad.com). Cell cycle phases were analyzed by
Cell Quest Software (Becton-Dickinson) and comparisons evaluated
using 2-tailed Student’s t-test. Results are expressed as mean ± SD
of independently performed experiments. Statistical significance
was set at p<0.05.
Results
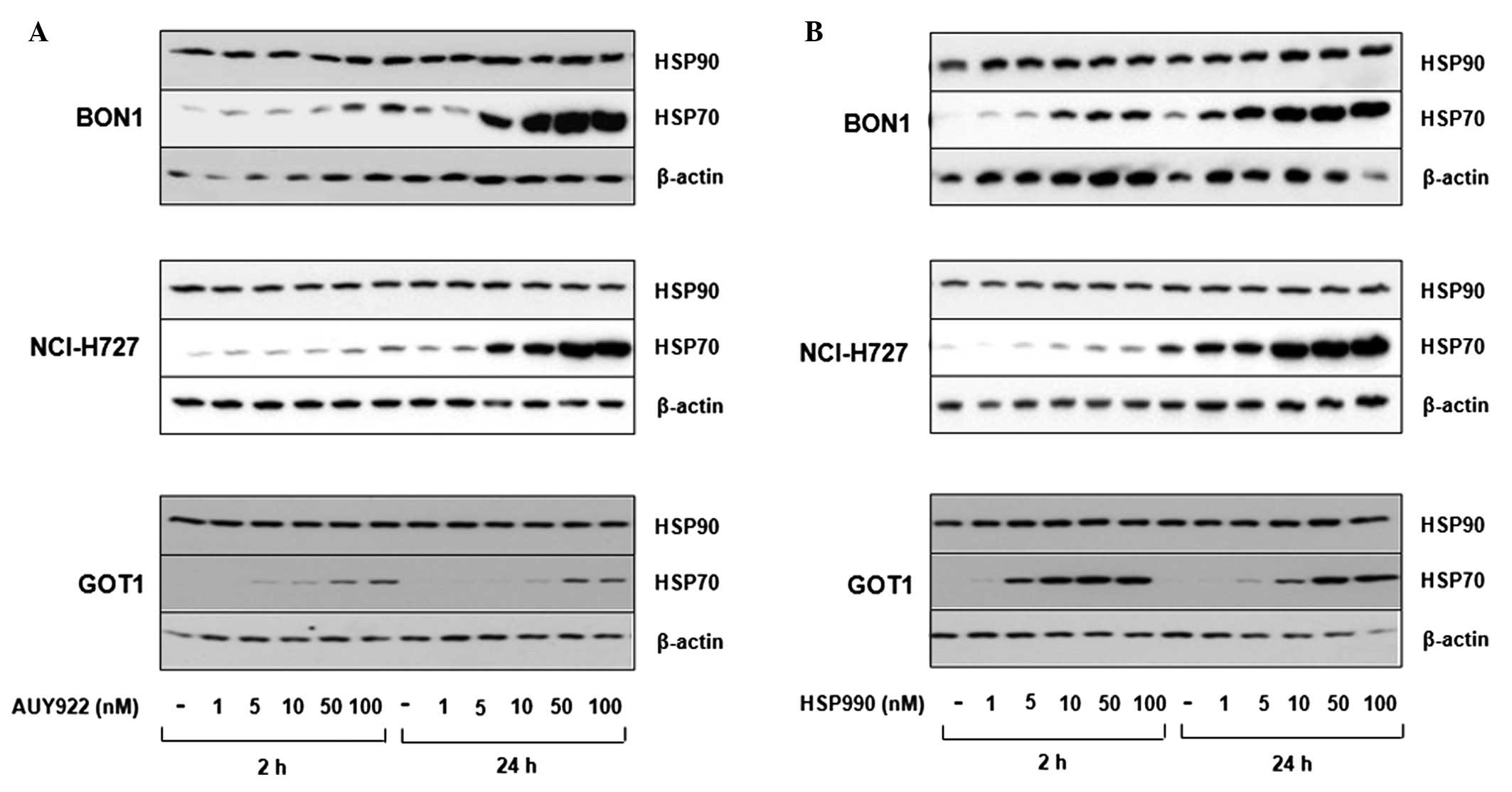
HSP90 expression and inhibitor
specificity
Western blot analysis revealed BON1, NCI-H727 and
GOT1 cells to express easily detectable levels of HSP90, while
HSP70 was poorly expressed under baseline conditions. As increased
HSP70 expression is a hallmark of specific HSP90 inhibition, we
analyzed HSP70 expression after treatment with the HSP90 inhibitors
AUY922 and HSP990. Treatment with increasing concentrations (10–100
nM) of both inhibitors induced HSP70 expression in a dose-dependent
manner (Fig. 1).
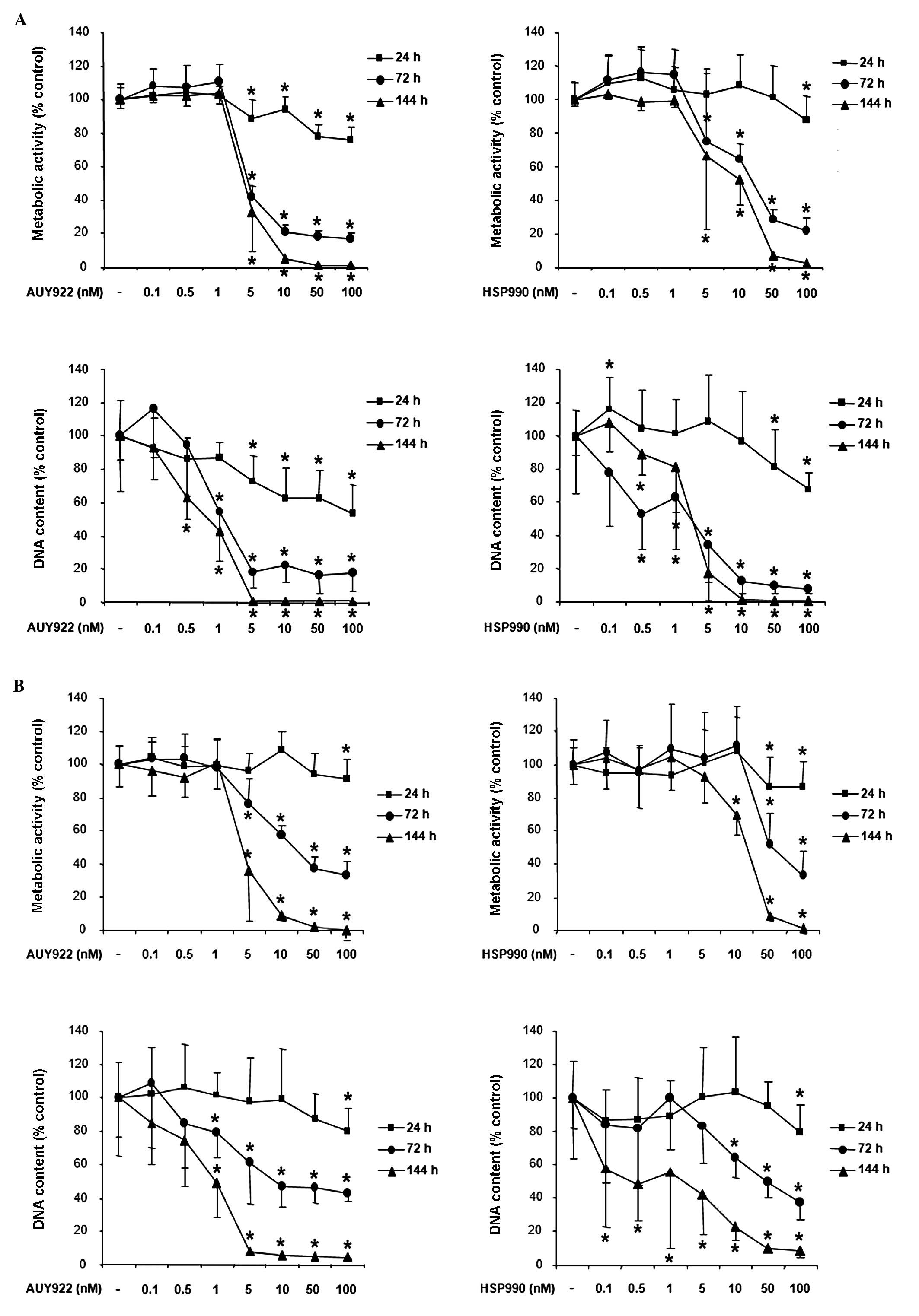
Inhibition of neuroendocrine cell
viability by HSP90 inhibitors
Treatment of human pancreatic neuroendocrine BON1
tumor cells with the HSP90 inhibitor AUY922 dose-dependently
suppressed cell viability as assessed by measurement of metabolic
activity and DNA content (Fig. 2A,
left panel). Significant effects were observed at all time
points tested (24, 72 and 144 h) beginning at AUY922 concentrations
as low as 5 nM (suppression of metabolic activity to ∼89, 42 and
36% compared to non-treated controls, respectively; p<0.05) and
peaked at the highest dose tested (100 nM; suppression of metabolic
activity to ∼75, 17 and 1%, respectively; p<0.05). Similar to
AUY922, the HSP90 inhibitor HSP990 also suppressed BON1 cell
viability (Fig. 2A, right panel).
At 24 h, significant effects were observed at the highest HSP990
dose tested (100 nM; reduction of metabolic activity to ∼75%
compared to controls, p<0.05). At 72 h, significant effects were
observed at a HSP990 concentration as low as 5 nM (reduction of
metabolic activity to ∼71%, p<0.05) with a maximum effect at 100
nM (reduction of metabolic activity to ∼21%, p<0.05). At 144 h,
5 nM HSP990 suppressed BON1 cell metabolic activity to ∼66%
(p<0.05), 10 nM to ∼52% (p<0.05), 50 nM to ∼7% (p<0.05)
and 100 nM to ∼3% (p<0.05). For all concentrations and time
points the decrease of metabolic activity (Fig. 2A, upper panels) correlated with the
decrease of DNA content (Fig. 2A,
lower panels).
Treatment of human bronchopulmonary neuroendocrine
NCI-H727 tumor cells with AUY922 also suppressed cell viability in
a dose-dependent manner (Fig. 2B, left
panel). Significant effects were observed at all times points,
beginning at a treatment concentration of 5 nM and peaking at the
highest concentration tested. Similar to the observed effects of
AUY922, treatment with HSP990 dose-dependently suppressed NCI-H727
cell viability (Fig. 2B, right
panel). Significant effects were observed at 24, 72 and 144 h
with HSP990 concentrations as low as 10 nM.
Due to their slow growth rate, cell proliferation
experiments with human midgut carcinoid GOT1 cells were performed
for 72 and 144 h. Treatment with AUY922 dose-dependently suppressed
GOT1 cell viability. Significant effects at both time points were
observed with AUY922 concentrations as low as 5 nM and peaked at
the highest dose tested (100 nM; Fig.
2C, left panel). HSP990 also suppressed GOT1 cell viability
with a similar potency (Fig. 2C, right
panel).
Table I summarizes
the IC50 inhibitory values of AUY922 and HSP990 on
proliferation of BON1, NCI-H727 and GOT1 cells (based on metabolic
activity data determined by Cell Titer 96 aqueous One Solution
Proliferation assay). Lowest IC50 values were observed
for AUY922-mediated BON1 and HSP990-mediated GOT1 metabolic
activity (at 144 h).
 | Table I.IC50 values (nM) of AUY922
and HSP990-mediated inhibition of NET cell proliferation at 72 and
144 h. |
Table I.
IC50 values (nM) of AUY922
and HSP990-mediated inhibition of NET cell proliferation at 72 and
144 h.
| AUY922
| HSP990
|
|---|
| 72 h | 144 h | 72 h | 144 h |
|---|
| BON1 | 4.4 | 4.4 | 22.1 | 11.8 |
| NCI-H727 | 12 | 4.6 | 50.9 | 19 |
| GOT1 | 30.3 | 5.1 | 26.2 | 9.4 |
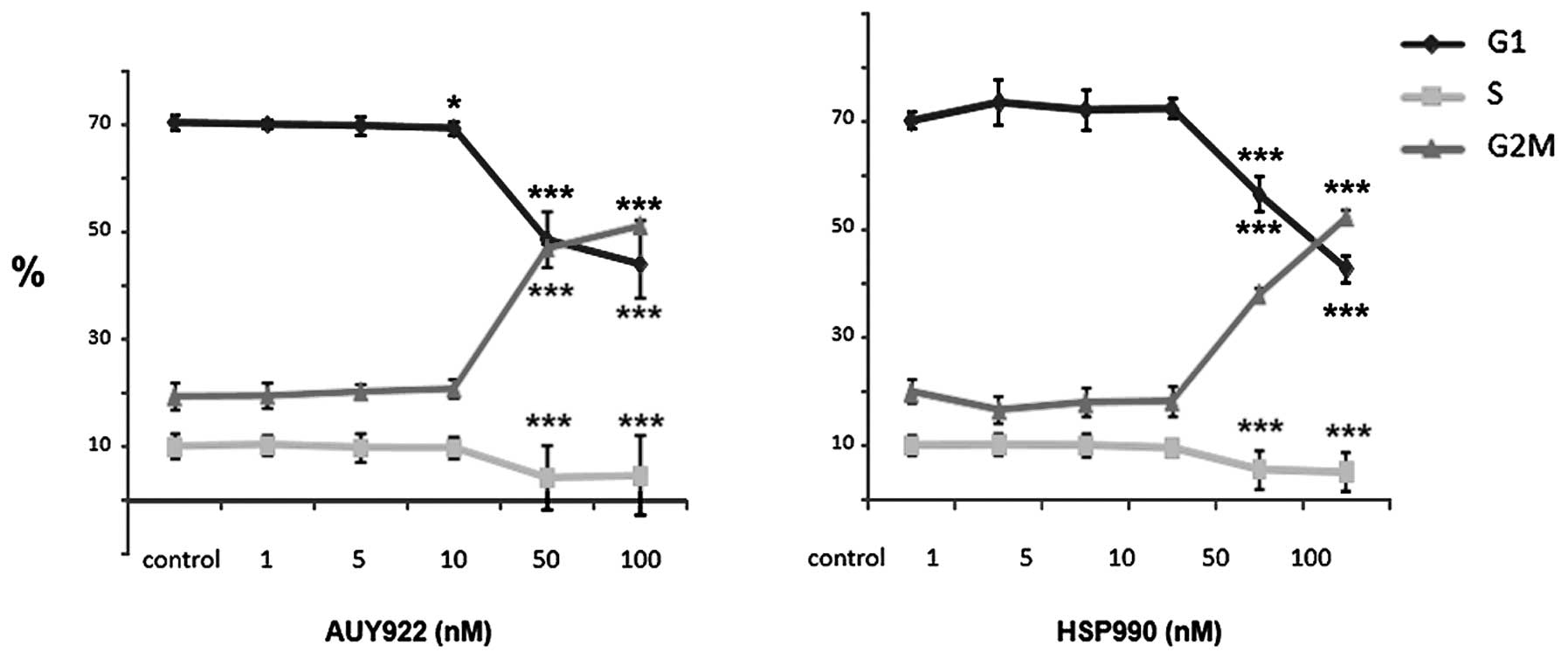
Effect of HSP90 inhibition on cell cycle
distribution of neuroendocrine tumor cells
To further explore mechanisms for the observed
inhibition of neuroendocrine tumor cell viability by HSP90
inhibition, we performed cell cycle analysis of BON1, NCI-H727 and
GOT1 cells treated for 24 h with AUY922 and HSP990, respectively.
BON1 cells responded to AUY922 treatment with a significant
reduction of cells in S phase from ∼10 to ∼5%, while the number of
cells in G2/M phase was dose-dependently increased (at 50 and 100
nM; p<0.001) (Fig. 3, left
panel). HSP990 also decreased the number of BON1 cells in S
phase and increased the number of cells in G2/M phase with a
similar potency (Fig. 3, right
panel). In contrast, cell cycle phase distribution of NCI-H727
and GOT1 cells was not altered by overnight HSP inhibition (data
not shown).
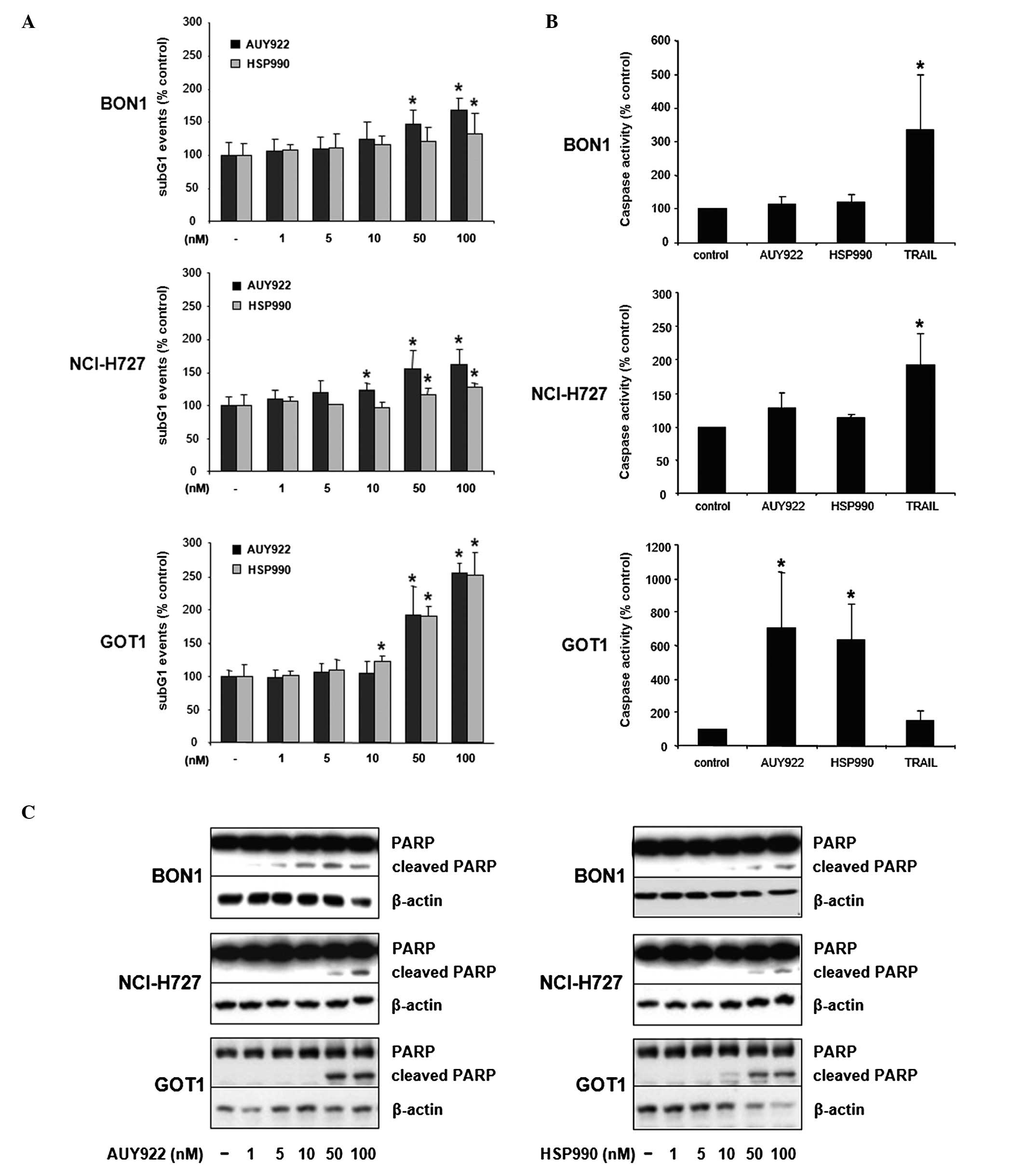
HSP90 inhibition induces apoptosis in
neuroendocrine tumor cells
Twenty-four hour treatment of BON1 cells with AUY922
dose-dependently increased the number of cells in sub-G1 phase up
to ∼1.7-fold (100 nM, p<0.05; Fig.
4A). HSP990 also increased the number of sub-G1 events up to
∼1.4-fold (100 nM, p<0.05; Fig.
4A). Furthermore, AUY922 and HSP990 treatment resulted in a
significant increase of NCI-H727 cells in sub-G1 phase up to
∼1.6-fold (100 nM AUY922, p<0.05; Fig. 4A) and ∼1.3-fold (100 nM HSP990,
p<0.05; Fig. 4A), respectively.
GOT1 cells showed the strongest increase of DNA fragmentation in
response to HSP90 inhibition (up to ∼2.5-fold at 100 nM AUY922 or
HSP990, p<0.05; Fig. 4A).
To further specify the observed HSP90
inhibition-mediated increase of the sub-G1 fraction, cells were
additionally assayed for the activity of effector caspases 3 and 7.
While inducing only slight increases of caspase 3/7 activity in
BON1 and NCI-H727 cells, both HSP90 inhibitors induced a massive
increase of caspase 3/7 activity in GOT1 cells up to ∼7.0-fold (100
nM AUY922, p<0.05; Fig. 4B).
The induction of PARP cleavage confirmed the results obtained by
measurement of caspase 3/7 activity, demonstrating more potent
induction of PARP cleavage in GOT1 compared to BON1 and NCI-H727
cells (Fig. 4C).
Mechanisms for HSP90 inhibition in
neuroendocrine tumor cells: effects on downstream signaling
As the HSP90 inhibitor 17-AAG has recently been
reported to reduce EGFR and IGF-IR expression in the
bronchopulmonary typical carcinoid cell line NCI-H727 (13,15),
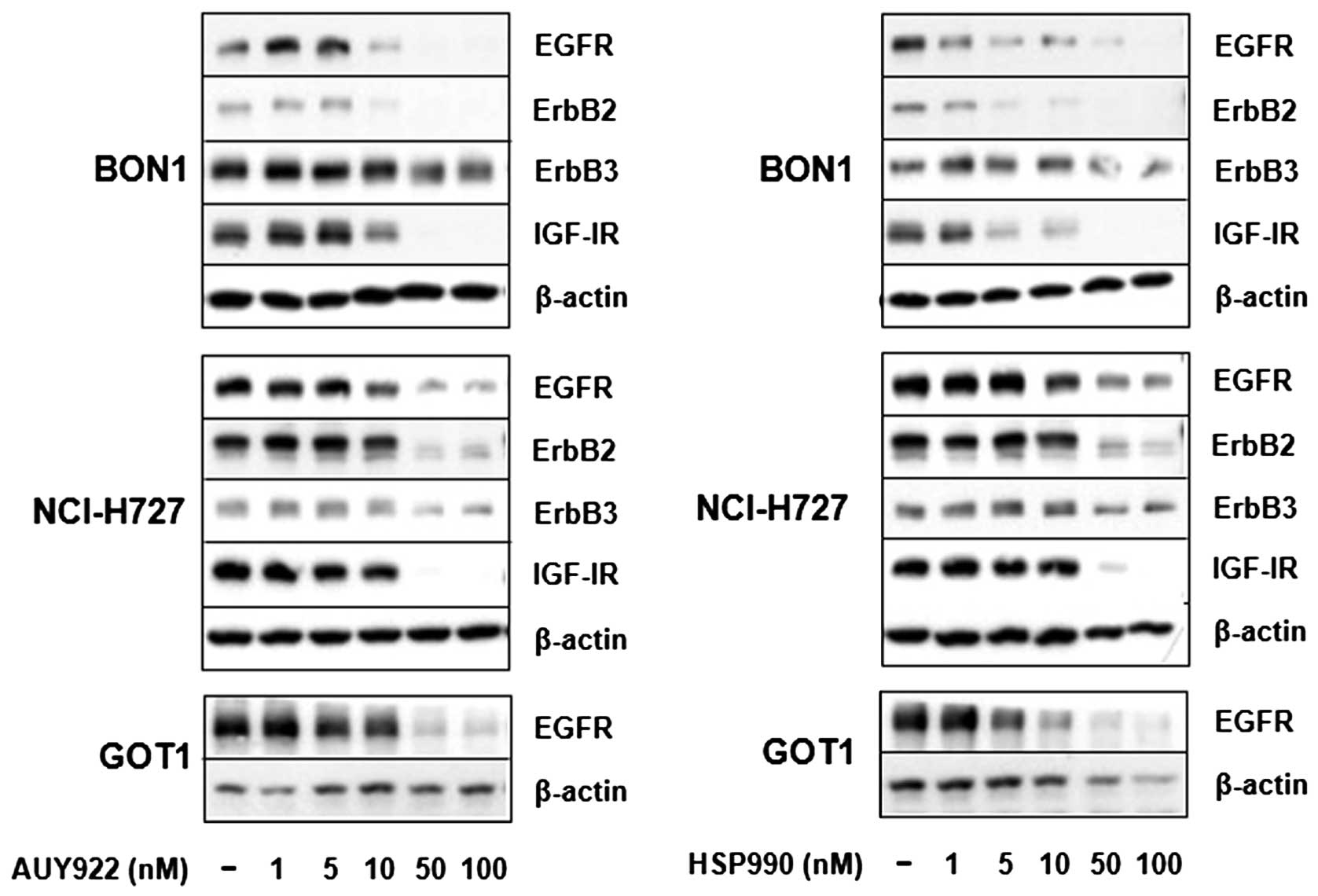
we examined the effect of AUY922 and HSP990 on ErbB and IGF-I
receptor expression. Treatment of BON1 cells with AUY922 and HSP990
for 24 h suppressed both ErbB2 and EGF receptor expression starting
at concentrations of 5 to 10 nM with minor inhibitory effects
observed on ErbB3 expression (Fig.
5). In addition, strong inhibitory effects were observed on
IGF-I receptor expression (Fig.
5). In NCI-H727 cells, HSP90 inhibition abolished ErbB2 and
IGF-I receptor expression, while inhibitory effects were observed
on ErbB3 and EGF receptor expression (Fig. 5). In contrast to BON1 and NCI-H727
cells, ErbB2, ErbB3 and IGF-I receptor are not detectable in GOT1
cells. However, treatment of GOT1 cells with AUY922 and HSP990 for
24 h strongly suppressed EGF receptor expression (Fig. 5).
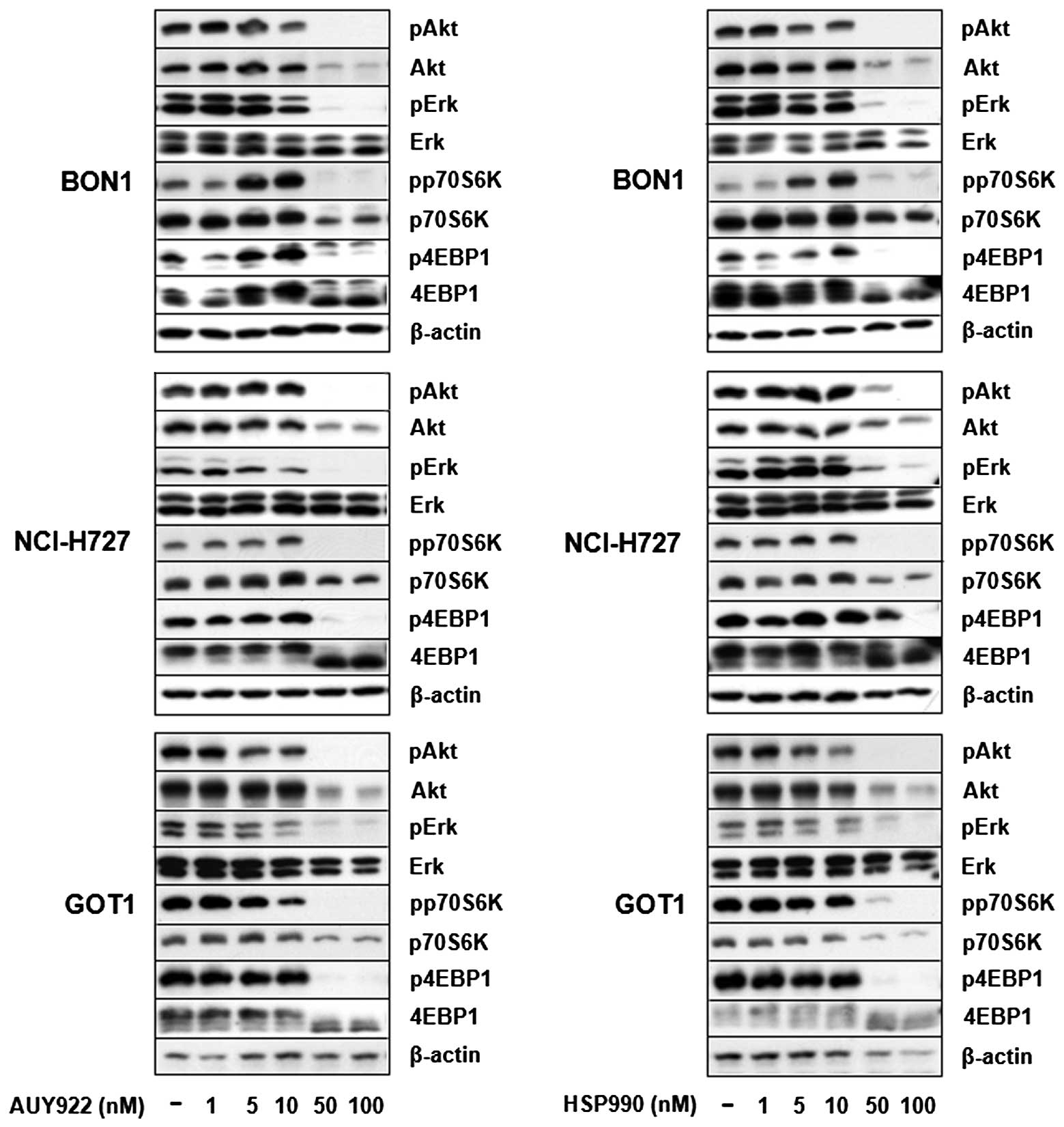
Untreated BON1, NCI-H727 and GOT1 cells cultured in
complete medium, exhibited baseline activation of Akt and Erk
signaling pathways (Fig. 6).
Treatment of all cells with AUY922 and HSP990 dose-dependently
suppressed Erk, Akt, p70S6K and 4EBP1 phosphorylation (Fig. 6). Potent inhibition on PI3K/Akt
signaling was also demonstrated by suppression of Akt and p70S6K
protein expression. Total Erk and β-actin protein expression
remained unaffected at all concentrations tested (Fig. 6).
Discussion
Due to increased incidence and relatively poor
prognosis of GEP-NETs alternative treatment options are required
for this heterogeneous group of tumors (5,6,9).
High Akt and Erk activity in NETs as well as compensatory Akt
activation in response to mTOR and Raf inhibitors, suggest that
simultaneous blockade of multiple oncogenic neuroendocrine
signaling cascades could be a more effective therapeutic approach
(10,11). As HSP90 is overexpressed in NETs
and controls the function of multiple oncogenic proteins (2,13),
we examined the effect of HSP90 inhibition on neuroendocrine cell
proliferation and signaling.
HSP90 inhibition was performed with novel low
molecular weight ATP-competitive non-geldamycin HSP90 inhibitors
AUY922 and the novel oral inhibitor HSP990. These compounds have
been speculated to offer advantages over ansamycin benzoquinone
HSP90 inhibitors such as 17-allylamino-17-demethoxygeldanamycin
(17-AAG) based on the independence from NAD(P)H:quinone
oxidoreductase 1 (NQO1) metabolism, P-glycoprotein expression and
favorable aqueous solubility (16,17).
All three compounds are currently in clinical trials in different
solid tumor entities.
Treatment of human pancreatic BON1, bronchopulmonary
NCI-H727 and midgut GOT1 carcinoid cells with increasing
concentrations of AUY922 and HSP990 dose-dependently decreased cell
viability. Recently, also the HSP90 inhibitors 17-AAG and IPI504
have been reported to inhibit cell viability in NCI-H727 cells with
an IC50 value of 70.4 and 192 nM, respectively (12,13).
In the current study, significant inhibition of cell viability was
observed with inhibitor concentrations as low as 5 nM particularly
after prolonged treatment (72–144 h). Inhibition of neuroendocrine
cell viability was associated with the induction of apoptosis
(especially in GOT1 cells) as demonstrated by increased number of
sub-G1 events, as well as the induction of caspase 3/7 and PARP
cleavage. Consistently, AUY922 has recently been demonstrated to
induce apoptosis in multiple myeloma and glioblastoma cell lines
(18,19).
In addition to apoptosis induction, HSP90 inhibition
in human pancreatic BON1 cells was also associated with a
pronounced increase of cells in G2/M phase, while no effect on cell
cycle distribution was observed in NCI-H727 and GOT1 cells.
Induction of apoptosis combined with inhibition of cell cycle
progression (arrest in G2M phase) could explain the more potent
inhibition of cell viability in BON1 compared to NCI-H727 cells.
However, different growth rates of the cell lines tested may also
have an impact on cell cycle analysis of non-synchronized cells at
a given time point (overnight treatment).
In addition to HSP90, members of the ErbB receptor
family EGFR and ErbB2, as well as IGF-IR are also expressed in NETs
and could present alternative treatment targets (13,20).
Here we show AUY922 and HSP90-mediated inhibition of ErbB receptor
(EGFR, ErbB2 and ErbB3), and IGF-I receptor expression in BON1 and
NCI-H727 cells, likely as a result of dissociation from HSP90 and
subsequent increase in ubiquitinylation and proteosomal degradation
(1). NETs are characterized by
high activity of Erk and Akt signalling (10). Consistently, we detected baseline
activity of Erk and Akt signalling pathways (phosphorylation of
Erk, Akt, p70S6K, 4EBP1) in all NET cell lines tested.
Neuroendocrine HSP90 inhibition suppressed both signalling
pathways, possibly at least in part through depletion of upstream
signalling RTKs. Interestingly, HSP90 inhibitors at lower
concentrations (5–10 nM) increased p70S6K and 4EBP1 signalling in
BON1 cells possibly due to incomplete HSP90 inhibition at this
concentration and compensatory activation of alternate stimulatory
feedback. HSP90 inhibition was not a toxic effect, since no effect
was observed on Erk, or β-actin expression. Furthermore, HSP90
inhibition was associated with HSP70 upregulation, a hallmark of
HSP90 inhibition.
Considering the complexity of neuroendocrine tumors,
targeting a single pathway by inhibiting the activity of one
component is unlikely to be effective in the long term due to
development of resistance and activation of compensatory mechanisms
(11). We report simultaneous
suppression of neuroendocrine Erk and Akt signalling associated
with induction of apoptosis and inhibition of cell viability by the
novel HSP90 inhibitors AUY922 and HSP990. As HSP90 overexpression
was recently reported in NETs (13) inhibition of HSP90 alone or in
combination with other molecular targets could be useful for the
treatment of aggressive neuroendocrine tumors resistant to
conventional therapy. Further preclinical and clinical studies are
needed to evaluate the potential role of HSP90 inhibitors in
neuroendocrine tumors.
Acknowledgements
C.J.A. has received research
contracts, lecture honorarium and advisory board honorarium from
Novartis. F.B. has received research contracts from Novartis. This
study contains parts of the unpublished doctoral theses of G.
Ailer. This study has been supported in part by a grant from the
German Federal Ministry of Education and Research [01EX1021B,
Spitzencluster M4, Verbund Personalisierte Medizin: Teilprojekt
NeoExNET (PM1)] to C.J.A. and F.B. and a restricted research grant
from Novartis Oncology Germany to C.J.A.
References
|
1.
|
Peterson LB and Blagg BS: To fold or not
to fold: modulation and consequences of Hsp90 inhibition. Future
Med Chem. 1:267–283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Trepel J, Mollapour M, Giaccone G and
Neckers L: Targeting the dynamic HSP90 complex in cancer. Nat Rev
Cancer. 10:537–549. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Porter JR, Fritz CC and Depew KM:
Discovery and development of Hsp90 inhibitors: a promising pathway
for cancer therapy. Curr Opin Chem Biol. 14:412–420. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Kim YS, Alarcon SV, Lee S, Lee MJ,
Giaccone G, Neckers L and Trepel JB: Update on Hsp90 inhibitors in
clinical trial. Curr Top Med Chem. 9:1479–1492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Massironi S, Sciola V, Peracchi M,
Ciafardini C, Spampatti MP and Conte D: Neuroendocrine tumors of
the gastro-enteropancreatic system. World J Gastroenterol.
14:5377–5384. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Modlin IM, Oberg K, Chung DC, Jensen RT,
de Herder WW, Thakker RV, Caplin M, Delle Fave G, Kaltsas GA,
Krenning EP, Moss SF, Nilsson O, Rindi G, Salazar R, Ruszniewski P
and Sundin A: Gastroenteropancreatic neuroendocrine tumours. Lancet
Oncol. 9:61–72. 2008. View Article : Google Scholar
|
|
7.
|
Janson ET, Sørbye H, Welin S, Federspiel
B, Grønbaek H, Hellman P, Mathisen O, Mortensen J, Sundin A,
Thiis-Evensen E, Välimäki MJ, Oberg K and Knigge U: Nordic
Guidelines 2010 for diagnosis and treatment of
gastroenteropancreatic neuroendocrine tumours. Acta Oncol.
49:740–756. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Auernhammer CJ and Göke B: Therapeutic
strategies for advanced neuroendocrine carcinomas of jejunum/ileum
and pancreatic origin. Gut. 60:1009–1021. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Yao JC, Hassan M, Phan A, Dagohoy C, Leary
C, Mares JE, Abdalla EK, Fleming JB, Vauthey JN, Rashid A and Evans
DB: One hundred years after ‘carcinoid’: epidemiology of and
prognostic factors for neuroendocrine tumors in 35,825 cases in the
United States. J Clin Oncol. 26:3063–3072. 2008.
|
|
10.
|
Shah T, Hochhauser D, Frow R, Quaglia A,
Dhillon AP and Caplin ME: Epidermal growth factor receptor
expression and activation in neuroendocrine tumours. J
Neuroendocrinol. 18:355–360. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Zitzmann K, Rüden Jv, Brand S, Göke B,
Lichtl J, Spöttl G and Auernhammer CJ: Compensatory activation of
Akt in response to mTOR and Raf inhibitors - a rationale for
dual-targeted therapy approaches in neuroendocrine tumor disease.
Cancer Lett. 295:100–109. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Gloesenkamp C, Nitzsche B, Lim AR, Normant
E, Vosburgh E, Schrader M, Ocker M, Scherübl H and Höpfner M: Heat
shock protein 90 is a promising target for effective growth
inhibition of gastrointestinal neuroendocrine tumors. Int J Oncol.
40:1659–1667. 2012.PubMed/NCBI
|
|
13.
|
Gilbert JA, Adhikari LJ, Lloyd RV, Rubin
J, Haluska P, Carboni JM, Gottardis MM and Ames MM: Molecular
markers for novel therapies in neuroendocrine (carcinoid) tumors.
Endocr Relat Cancer. 17:623–636. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Zitzmann K, de Toni E, von Rüden J, Brand
S, Göke B, Laubender RP and Auernhammer CJ: The novel Raf inhibitor
Raf265 decreases Bcl-2 levels and confers TRAIL-sensitivity to
neuroendocrine tumour cells. Endocr Relat Cancer. 18:277–285. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Cakir M and Grossman A: The molecular
pathogenesis and management of bronchial carcinoids. Expert Opin
Ther Targets. 15:457–491. 2011.PubMed/NCBI
|
|
16.
|
Eccles SA, Massey A, Raynaud FI, Sharp SY,
Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall
F, Aherne W, Rowlands M, Hayes A, Martins V, Urban F, Boxall K,
Prodromou C, Pearl L, James K, Matthews TP, Cheung KM, Kalusa A,
Jones K, McDonald E, Barril X, Brough PA, Cansfield JE, Dymock B,
Drysdale MJ, Finch H, Howes R, Hubbard RE, Surgenor A, Webb P, Wood
M, Wright L and Workman P: NVP-AUY922: a novel heat shock protein
90 inhibitor active against xenograft tumor growth, angiogenesis,
and metastasis. Cancer Res. 68:2850–2860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Sharp SY, Prodromou C, Boxall K, Powers
MV, Holmes JL, Box G, Matthews TP, Cheung KM, Kalusa A, James K,
Hayes A, Hardcastle A, Dymock B, Brough PA, Barril X, Cansfield JE,
Wright L, Surgenor A, Foloppe N, Hubbard RE, Aherne W, Pearl L,
Jones K, McDonald E, Raynaud F, Eccles S, Drysdale M and Workman P:
Inhibition of the heat shock protein 90 molecular chaperone in
vitro and in vivo by novel, synthetic, potent resorcinylic
pyrazole/isoxazole amide analogues. Mol Cancer Ther. 6:1198–1211.
2007. View Article : Google Scholar
|
|
18.
|
Gaspar N, Sharp SY, Eccles SA, Gowan S,
Popov S, Jones C, Pearson A, Vassal G and Workman P: Mechanistic
evaluation of the novel HSP90 inhibitor NVP-AUY922 in adult and
pediatric glioblastoma. Mol Cancer Ther. 9:1219–1233. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Stühmer T, Zöllinger A, Siegmund D,
Chatterjee M, Grella E, Knop S, Kortüm M, Unzicker C, Jensen MR,
Quadt C, Chène P, Schoepfer J, García-Echeverría C, Einsele H,
Wajant H and Bargou RC: Signalling profile and antitumour activity
of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma.
Leukemia. 22:1604–1612. 2008.PubMed/NCBI
|
|
20.
|
Papouchado B, Erickson LA, Rohlinger AL,
Hobday TJ, Erlichman C, Ames MM and Lloyd RV: Epidermal growth
factor receptor and activated epidermal growth factor receptor
expression in gastrointestinal carcinoids and pancreatic endocrine
carcinomas. Mod Pathol. 18:1329–1335. 2005. View Article : Google Scholar
|