Introduction
Vitamin A is obtained through the diet in the form
of retinol, retinyl ester or β-carotene (1). Retinoic acid (RA) is one of the
principal active metabolites of vitamin A which plays a critical
role in cell proliferation, differentiation and apoptosis in normal
tissues during embryonic development (2). RA induces differentiation in many
cell types and is the most widely used differentiating therapeutic
agent (3,4). Retinol has 6 biologically active
isoforms that among others includes all-trans (ATRA, tretinoin) and
9-cis RA (alitretinoin); ATRA is the predominant physiological form
(5). RA mediates the
transcriptional regulation of several genes by binding to the
nuclear retinoic acid receptors (RARs), namely RARα, RARβ and RARγ
(6,7). Like other nuclear receptors, RARs
contain a domain that mediates interaction with ATRA, a zinc
finger-containing DNA binding domain that binds to RA response
elements (RAREs) in target genes, and a dimerization domain that
engages members of the retinoid X receptor (RXR) subfamily in
RXR/RAR heterodimers (8).
Different isomers activate different receptors and thus lead to
different biological effects. RARs can be activated by both
all-trans (ATRA) and 9-cis-RA, while RXR are exclusively activated
by 9-cis RA; however, due to the conversion of ATRA to 9-cis RA,
high concentrations (10−5 M) of ATRA can also activate
gene transcription in cells transfected with RXRs (9). It has also been shown that retinoids
exert their effects via the nuclear receptor independent pathway
(5).
RA and its derivatives are promising anti-neoplastic
agents endowed with both therapeutic and chemopreventive potential
because they are able to regulate cell growth, differentiation and
apoptosis (10,11). It is believed that the
anti-neoplastic pathways induced by RA are regulated predominantly
by RAR-β, which is known to induce apoptosis; thus it has been
suggested that RAR-β plays a critical role in mediating the growth
arrest and differentiation in several breast cancer cell types
(12–14).
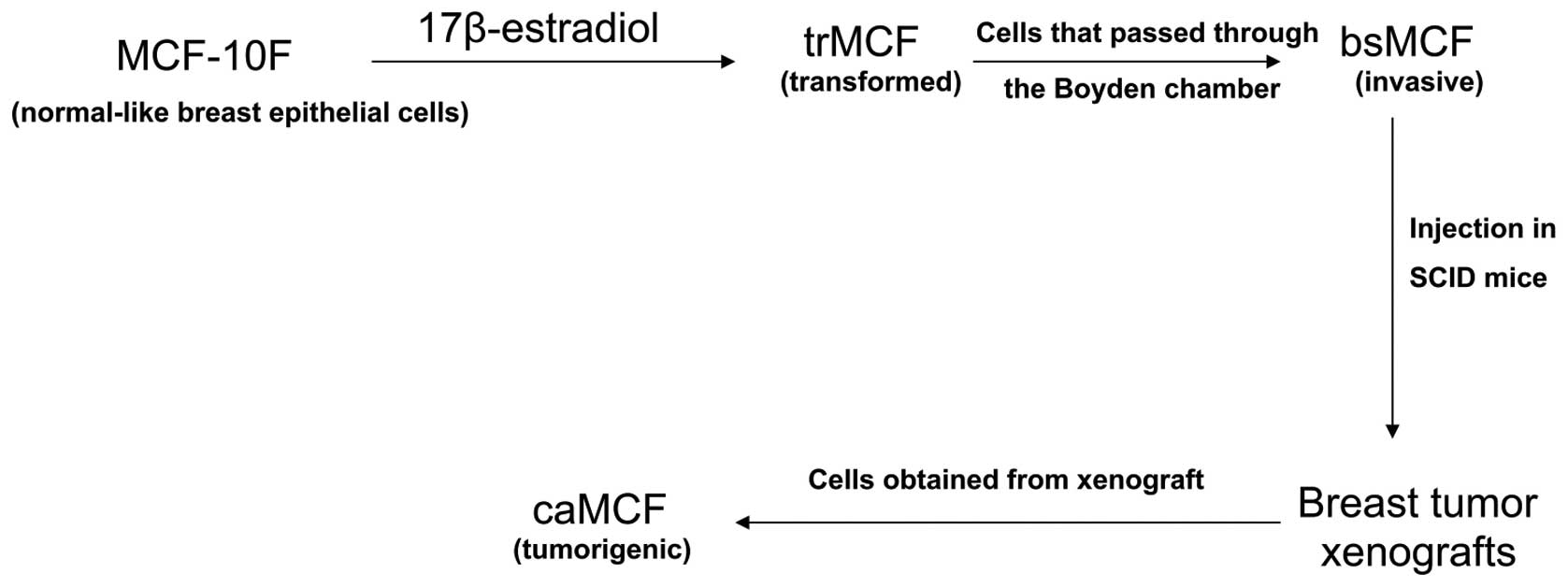
We have developed an in vitro-in vivo
model of breast cancer progression by treating the human
normal-like breast epithelial cells MCF-10F with a high dose of
estradiol (70 nM) (Fig. 1)
(15,16). This model consists of four cell
lines: i) the spontaneously immortalized cell line MCF-10F, which
is considered to be a normal-like breast epithelial cell line; ii)
the transformed trMCF cells; iii) the invasive bsMCF cells; and iv)
cells isolated from xenografts, caMCFs, which show all
characteristics of fully malignant breast cancer cells (Fig. 1). Gene expression studies showed
the highest number of deregulated genes in caMCF, being slightly
lower in bsMCF, and lowest in trMCF and, this order was consistent
with the extent of chromosome aberrations
(caMCF>bsMCF>>trMCF) (16). This model of breast cancer
progression resembles the different steps of neoplastic
transformation of the mammary gland; it is widely held that breast
cancer initiates as the premalignant stage of atypical ductal
hyperplasia (ADH), progresses into the pre-invasive stage of ductal
carcinoma in situ (DCIS), and culminates in the potentially
lethal stage of invasive ductal carcinoma (IDC) (17). In collagen, the normal-like MCF-10F
cells form tubules resembling the structures observed in the normal
mammary gland although after treatment with estradiol, the
transformed trMCF cells form tubules and spherical masses, which
are indicative of cell transformation (8,19).
The spherical masses showed a partial filling of the lumen that
would result from decreased central apoptosis, enhanced cellular
proliferation or both (18). The
filling of the lumen of the tubular structures of the breast is the
earliest morphologic alteration and is common in atypical ductal
hyperplasia and ductal carcinoma in situ (DCIS) (18). In the presented study, we studied
the effect of all trans-RA (ATRA) using this model of breast cancer
progression. Our results showed that ATRA was able to re-program
early transformed cells to a normal stage.
Materials and methods
Cells and media
The human normal-like breast epithelial cells
MCF-10F are estrogen receptor (ER) negative, progesterone receptor
(PR) negative and HER2 negative. Cells were cultured in Dulbecco’s
modified Eagle’s medium [DMEM/F-12, Gibco, Carlsbad, CA; formula
90–5212 EF: containing DMEM/F12 (1:1) with L-glutamine and phenol
red, D-glucose 315 mg/l, sodium pyruvate 55 mg/l] with 5% horse
serum, 2.43 g/l sodium bicarbonate, 20 mg/l epidermal growth factor
(EGF), 100 mg/l Vibrio cholerae toxin, 10 mg/l insulin, 0.5
mg/l hydrocortisone, 1.05 mM calcium, antibiotics and antimicotic
(100 U/ml penicillin, 100 mg/ml streptomycin, 0.25 mg/ml
amphotericin). A 10-mM solution of all trans-retinoic acid (ATRA,
Cat# R2625, Sigma, St. Louis, MO) was prepared as a stock solution
by dissolving ATRA in dimethylsulphoxide (DMSO).
The trMCF clone 11 was isolated by seeding 100–1,000
trMCF cells in a 100-mm cell culture plate and after 1 day in
culture, several colonies were isolated by ring cloning. The trMCF
clone 11 cells were generated by expanding the cells from one of
these colonies; trMCF clone 11 cells only formed spherical masses
on collagen. To study the effect of ATRA, trMCF clone 11 cells were
treated continuously for 26 days with 10−5 M (10
μM) to 10−8 M (0.01 μM) ATRA (media was
replaced daily). As control, the cells were treated with 0.1% DMSO
(vehicle). The bsMCF and caMCF cells were treated with
10−5 to 10−8 M ATRA alone or in combination
with 2.5 μM 5-aza-dC.
Collagen assays
The cells were resuspended at a final density of
1.5×104 cells/ml in collagen matrix consisting of 2.68
mg/ml (89.3%) type I collagen (PureCol, Inamed Biomaterials Co.,
Fremont, CA), 8% 12.5X DMEM-F12 with antibiotics, 0.1 mg/ml
insulin, 14 mM NaHCO3 and 0.01 N NaOH. A total of 400
μl (3,000 cells) were plated on the top four 24-well
chambers pre-coated with 400 μl of 89.3% collagen mix. Per
each treatment, cells were plated in 4 wells and fed daily with the
medium described before. The structures in collagen matrix were
observed daily under an inverted microscope and at the end of the
observation period (8 days), the structures (spherical masses,
tubules and intermediate structures) were counted, photographed and
fixed in 10% neutral buffered formalin and processed for
histological examination. Results were expressed as the total
number of structures per well (spherical masses, tubules and
intermediate structures) and percentage of the different structures
per treatment. The t-test was used to determine if the differences
were significant.
Invasion assays
Cell invasion in real-time were performed using
xCELLigence RTCA DP device from Roche Diagnostics (Mannheim,
Germany). For this purpose, each well of the upper chamber of the
CIM-Plate 16 was covered with Matrigel (BD Biosciences, Franklin
Lakes, NJ) basement membrane matrix (1:20 in cell culture media)
and 10% fetal bovine serum (chemo-attractant) was added in the
lower chamber. A total of 40,000 cells suspended in 100 μl
serum free media were seeded per well in CIM-Plates 16 (Roche
Diagnostics). Data acquisition and analysis was performed with the
RTCA software (version 1.2, Roche Diagnostics). Changes in
impedance from cells that invade and migrate to the underside of
wells were recorded and monitored for a total of 24 h.
Gene expression profiling
RNA was isolated from the cells using RiboPure™ kit
(Life Technologies, Frederick, MD) and RNA quality was controlled
using the Agilent 2100 Bioanalyzer. Gene expression studies were
performed using Affymetrix U133 Plus 2.0 (Affymetrix, Santa Clara,
CA) human oligonucleotide microarrays containing over 47,000
transcripts and variants, including 38,500 well characterized human
genes. After hybridization, the chips were scanned using GeneChip
Scanner 3000. The data were analyzed with Microarray Suite version
5.0 (MAS 5.0) using Affymetrix default analysis settings and global
scaling as normalization method. The trimmed mean target intensity
of each array was arbitrarily set to 100. Background correction and
normalization was done using Iterative Plier 16 with GeneSpring
V11.5 software (Agilent, Palo Alto, CA). The criteria for
differentially expressed genes was set at ≥2-fold changes (p-value
<0.05). The differentially expressed gene list was loaded into
Ingenuity Pathway Analysis (IPA) 8.0 software (Ingenuity Systems,
Redwood City, CA) to perform biological network and functional
analyses.
Results
Treatment with ATRA induced branching
morphogenesis in early transformed breast epithelial cells
MCF-10F cells are normal-like breast epithelial
cells that form tubules in collagen matrix (3D culture); when these
cells were treated with high dose of estradiol (70 nM), the cells
(trMCF) formed tubules and spherical masses. To isolate transformed
cells that only form spherical masses, trMCF cells were seeded at
low density in cell culture dishes and several clones were isolated
by ring cloning. One of these clones, trMCF clone 11, did not form
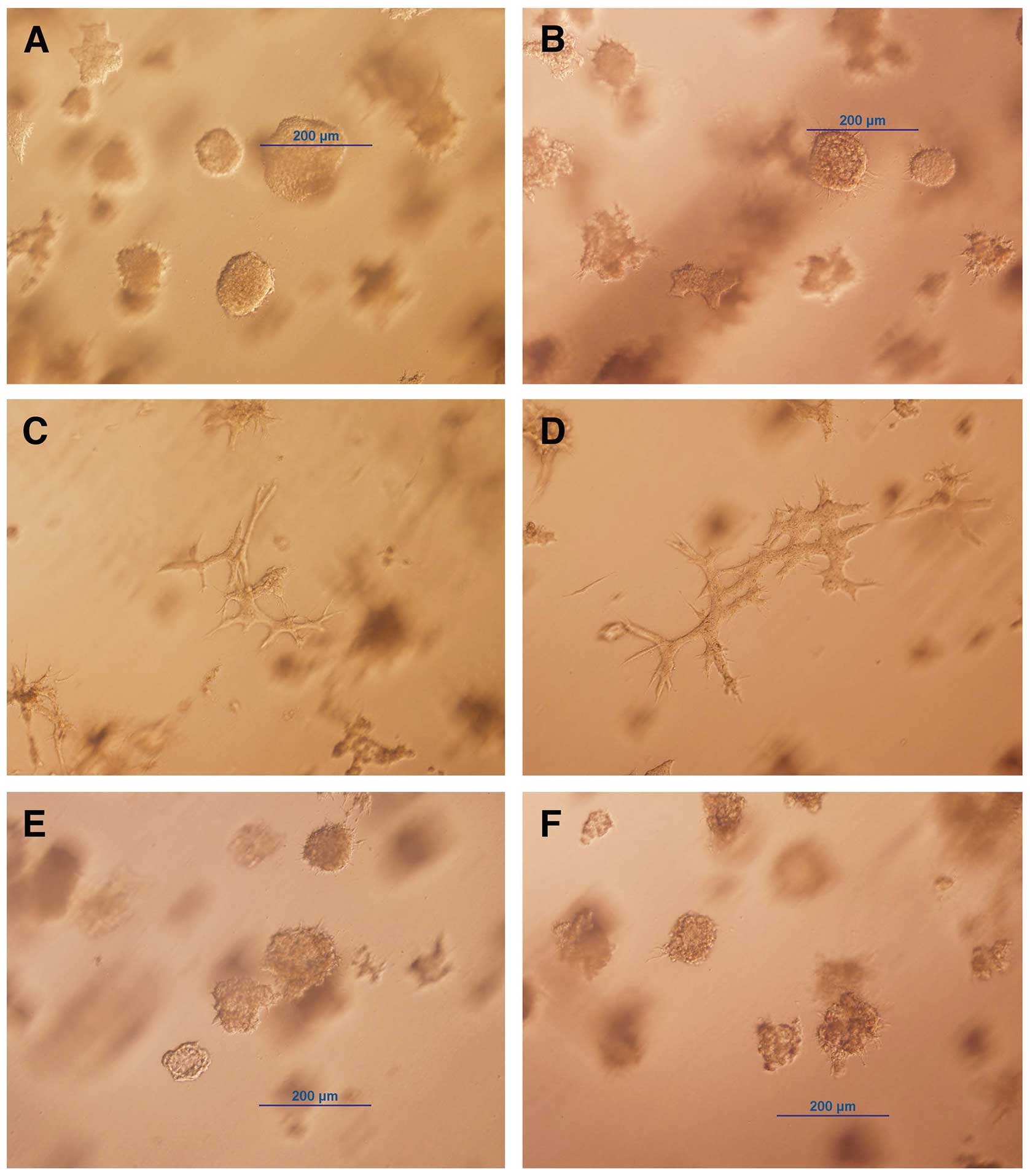
tubules in collagen; instead these cells formed spherical masses
and intermediate structures (Fig. 2A
and B). The trMCF clone 11 cells were treated continuously for
26 days with 10−5 to 10−8 M all
trans-retinoic acid (ATRA) and, we found that cells treated with
10−5 and 10−6 M ATRA were able to form
tubules in collagen (Fig. 2C and
D). Furthermore, the spherical masses formed by trMCF clone 11
treated with 10−5 and 10−6 M ATRA (Fig. 2C and D) were smaller compared to
the ones formed by the controls (Fig.
2A and B) or cells treated with 10−7 and
10−8 M ATRA (Fig. 2E and
F). The trMCF clone 11 cells treated with 10−7 and
10−8 M ATRA (Fig. 2E and
F) did not show any difference in morphology when compared to
the controls (Fig. 2A and B). The
number of spherical masses, intermediate structures and tubules for
trMCF clone 11 cells treated with different concentrations of ATRA
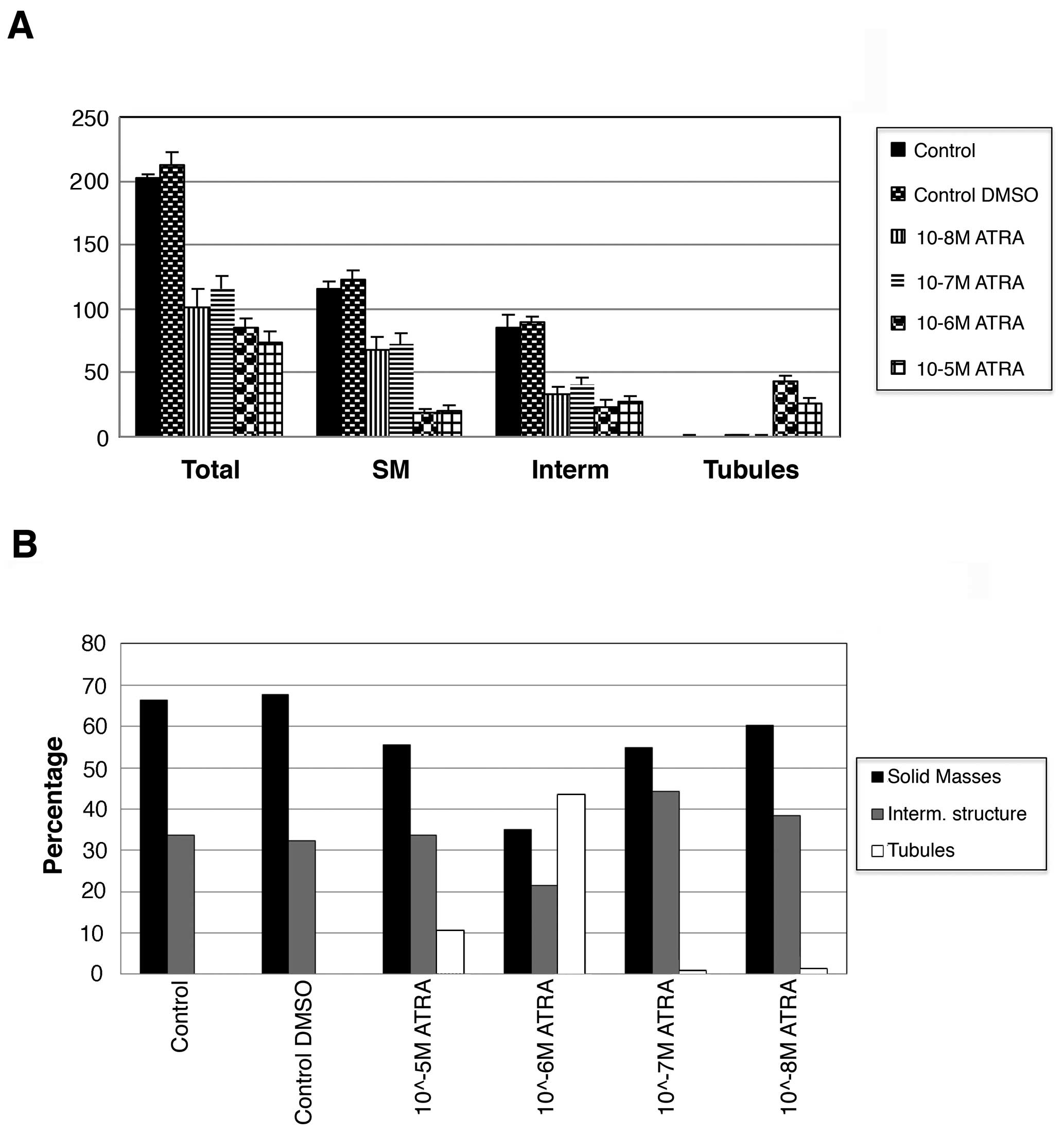
was counted (Fig. 3). The total
number of structures in collagen was significantly lower in cells
treated with ATRA compared with the controls suggesting that ATRA
treatment decrease the proliferation rate of the cells (p<0.01)
(Fig. 3A). The control trMCF clone
11 showed spherical masses and intermediate structures but no
tubules in collagen while cells treated with 10−6 and
10−5 M ATRA formed tubules and less spherical masses
(Fig. 3A). The cells treated with
10−5 or 10−6 M ATRA formed significantly less
spherical masses than the cells treated with 10−7 or
10−8 M ATRA (p<0.01) (Fig. 3A). A total of 43% of the structures
were tubules in the wells containing cells treated with
10−6 M ATRA and 10% tubules in wells with
10−5 M ATRA-treated cells (Fig. 3B).
The invasion capacity of trMCF clone 11 was studied
before and after ATRA treatment but, no differences were observed
(Fig. 4). The bsMCF and caMCF
cells did not show any changes in their morphology or invasion
capacity after treatment with ATRA alone or in combination with the
demethylating agent 5-aza-cytidine (data not shown).
Treatment with ATRA re-programmed gene
expression of early transformed cells
As trMCF clone 11 cells that only formed spherical
masses on collagen were able to form tubules after treatment with
10−5 or 10−6 M ATRA, gene expression studies
were performed on these cells. The microarray data have been
deposited into the NCBI gene expression omnibus (GEO) datasets
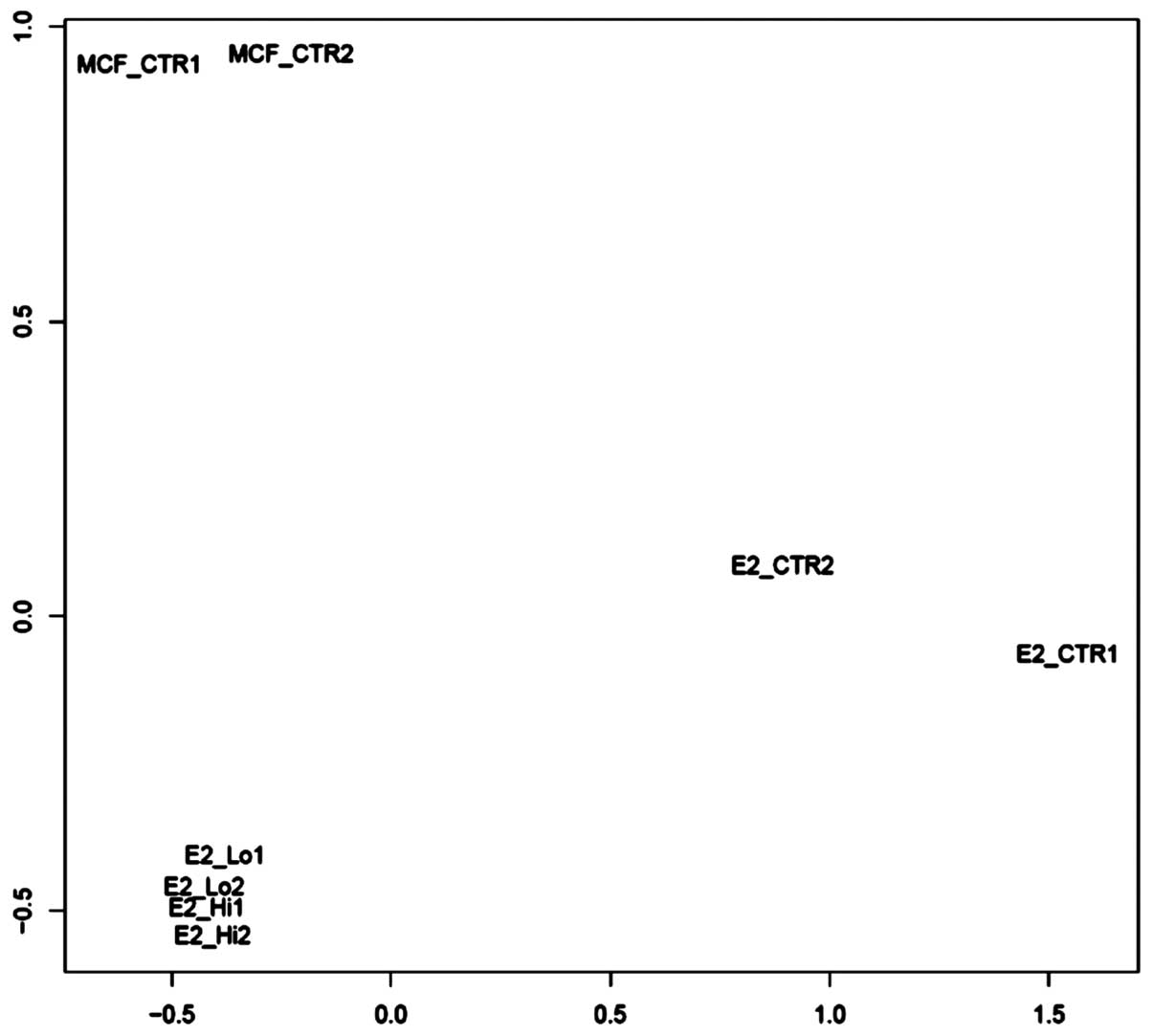
(GSE51549). The unsupervised sample classification by PCoA
(principle coordinate analysis) revealed that trMCF clone 11 cells
treated with 10−5 or 10−6 M ATRA demonstrated
a major difference with trMCF clone 11 cells, and a minor
difference with MCF-10F; also sample differences between
10−5 M ATRA and 10−6 M ATRA were weak
(Fig. 5). Although, trMCF clone 11
cells treated with 10−5 M ATRA and 10−6 M
ATRA showed minor differences at the expression level, we
considered trMCF clone 11 treated with 10−6 M ATRA for
the expression analysis since the number of tubules in collagen
matrix was higher for this concentration (43% tubules with
10−6 M ATRA vs. 10% tubules with 10−5 M
ATRA). For gene expression studies, three experimental groups were
compared using empirical Bayesian-moderated t-test implemented in R
package ‘limma’: the normal breast epithelial cells MCF-10F, the
cells transformed by treatment with estradiol trMCF clone 11 (that
only formed spherical masses on collagen) and the trMCF clone 11
after treatment with 10−6 M ATRA. We generated three
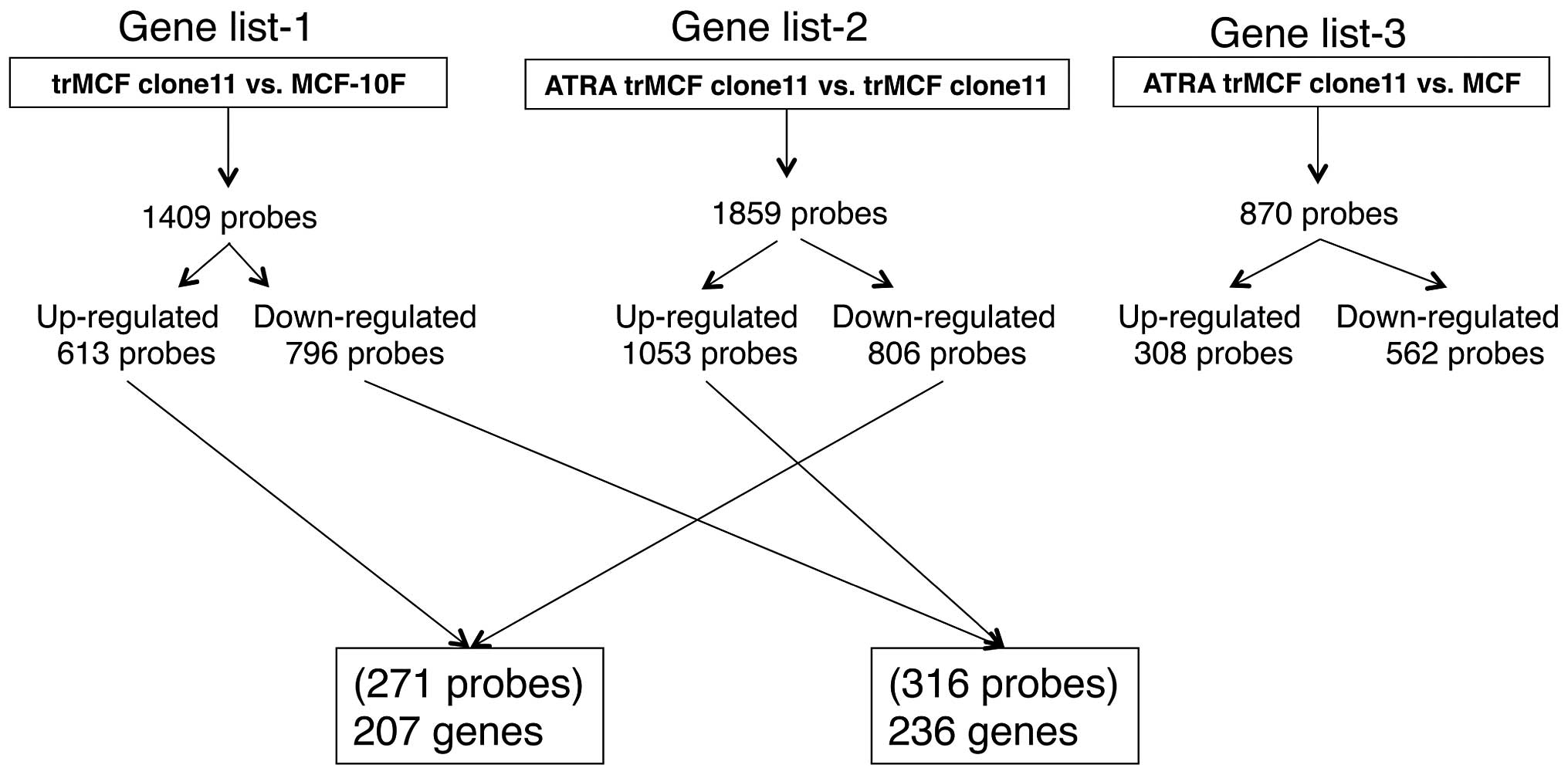
gene lists at criteria of fold change ≥2 and p≤0.05: gene list-1
(trMCF clone11 vs. MCF-10F) with 1,409 probes (613 probes
upregulated; 796 probes downregulated), gene list-2 (ATRA trMCF
clone 11 vs. trMCF clone 11) with 1,859 probe sets (1,053 probes
upregulated; 806 probes downregulated) and gene list-3 (ATRA trMCF
clone 11 vs. MCF-10F) with 870 probe sets (308 probes upregulated;
562 probes downregulated) (Fig.
6). Most importantly, 207 genes (271 probes) upregulated in the
transformed trMCF clone 11 (compared to the normal MCF-10F) were
downregulated after treatment with 10−6 M ATRA (Fig. 6 and Table IA) and 236 genes (316 probes) that
were downregulated in trMCF clone 11 (compared to MCF-10F) were
upregulated by 10−6 M ATRA treatment (Fig. 6 and Table IB). These 443 genes defined a gene
signature programming the reverse-transformation effect by ATRA
(Table I). The relatively smaller
number of significant probe sets in gene list-3 compared with other
gene lists (Fig. 6) further
supported the findings that ATRA-treatment reprograms the gene
expression status of trMCF clone 11 cells to MCF-10F.
 | A, ATRA-downregulated genes (207
genes). |
A, ATRA-downregulated genes (207
genes).
| ACSS3 | DNAJB9 | KLF11 |
PL-5283/SLC13A4 | TIMP3a |
| ALDH3A2 | DSC3 | KLHDC8B | PLAG1 | TMEM167B |
| ALDOC | DUSP5P | LCA5 | PLD1 | TMEM27 |
| ALPK1 | EFHC1 | LOC100288092 | PLD6 | TMEM40 |
| ANKRD37 | EFNB3 | LOC100289187 | PLK1S1 | TMEM59 |
| AQPEP | EPB41L4B | LOC100505894 | POFUT2 | TNFRSF25 |
| ARG2 | ERCC1a |
LOC100506057/STK32C | POLR1D | TNFSF11 |
| ARHGAP19 | ETS2a | LOC100507303 | PPIL6 | TNKS |
| ARHGEF10 | FABP6 | LOC100507547 | PPOX | TP63 |
| ATF2 | FAM117A | LOC100507644 | PPP1R13L | TPD52L1a |
| ATG14 | FAM168A | LOC439938 | PPP1R3C | TPRG1 |
| ATP2C2 | FAM46C | LOC642587 | PRKAB2 | TRAF3IP2 |
| ATP5C1 | FAT2 | LOX | PRMT2 | TRAPPC6A |
| BCAS4 | FBXO2 | LRIG1 | PROCR | TSC22D3 |
| BFSP1 | FEM1B | LYST | PTEN | TTBK2 |
| BLNK | FLCN | MAP2K5 | PTEN/PTENP1 | TTC39B |
| BTBD3 | FLJ37644 | MAPT | PTPN14 | TXNIP |
| C11orf80 | FLJ45244 | MGEA5 | RAB11FIP4 | UFM1 |
| C16orf46 | FNBP1L | MGPa | RAB38 |
UGT1A1/1A4/1A6/1A8/1A9/1A10 |
| C17orf39 | FNTA | MLF1 | RAB40C | USP3 |
| C17orf48 | FNTB | MRAP2 | RAB4A | USP32 |
| C1orf133 | FSIP1 | MXD1 | RAB7L1 | VPS8 |
| C1orf161 | FXYD2 | MYLIP | RASSF6 | WAC |
| C20orf111 | GBAS | N4BP2L1 | RMND1 | WDR59 |
| C21orf7 | GGNBP2 | NDE1 | RNF169 | WDR91 |
| C5orf41 | GGTA1 | NDUFB4 | SCARA3 | WWOXa |
| C7orf68 | GIT2 | NEK2 | SCRG1 | YOD1 |
| C9orf9 | GJA3 | NEURL1B | SEMA6A | ZBTB34 |
| CCDC28A | GKAP1 | NFKBIL1 | SFT2D1 | ZFAND5 |
| CD44a | GNA13 | NGLY1 | SHOX2 | ZNF836 |
| CELSR2 | GNAI1 | NMNAT3 | SLC25A37 | ZNRF1 |
| CLCA2 | GOSR2 | NPL | SLC2A12 | |
| CMBL | GPM6A | OGFRL1 | SLC2A9 | |
| COBL | GPNMB | PALMD | SLC5A3 | |
| CRIP2 | H19 | PDCD4a | SOCS3 | |
| CSNK2A2 | HAS3 | PDCD5 | SORL1 | |
| CYP1B1 | HBP1 | PDE7A | SPATA17 | |
| CYP39A1 | HERPUD1 | PDZD2 | STAU2 | |
| DBP | HMGCL | PER1 | STMN3 | |
| DCD | IFNAR1 | PER3 | STX6 | |
| DDAH2 | IRF6 | PHF21A | SUSD4 | |
| DDCa | IRX2 | PHLDB3 | TESK2 | |
| DDIT3 | KCMF1 | PHTF2 | THBS2 | |
| DHX40 | KDM5B | PIK3CD |
THSD1///THSD1P1 | |
| B, ATRA-upregulated genes (236
genes). |
B, ATRA-upregulated genes (236
genes).
| ABHD13 | COX7B | HIATL1 | OSTM1 | SLC43A3 | TSPAN2 |
| ACP2 | CRELD2 | HIGD1A | P2RY2 | SMPDL3A | UBE2Na |
| ADAM12 | CST6a | HOXA11 | PAPPA | SNRNP25 | UBE2Q1 |
| ALDH1A3 | CSTF2 | HPGD | PARVA | SNX19 | UBP1 |
| ANO1 | CYB561D2 | HS3ST1 | PCSK5 | SOAT1 | UNK |
| AOX1 | DCBLD2 | HS6ST2 | PDE12 | SPAG1 | VARS2 |
| APOL6 | DHRS9 | IFI44 | PHACTR3 | SPATA13 | VGLL3 |
| ARGLU1 | DHX9 | IFIT3 | PHLDA1 | SRPX2 | VSIG10 |
| ARHGAP26 | DNAJA1 | IFIT5 | PHLDA2 | SRSF10 | ZADH2 |
| ARHGAP42 | DOLK | IFNAR1 | PITPNC1 | SRSF2IP | ZBED4 |
| ARHGDIB | DPH3 | KHNYN | PKIB | SSPN | ZDHHC2 |
| ARIH2 | EFCAB2 | KLHL18 | PLCXD2 | STK39 | ZMPSTE24 |
| ASPHD2 | EFNB2 | KLHL23 |
PLGLA/PLGLB1/PLGLB2 | STS | ZNF252 |
| ATP6V0A2 | EHD4 | KRT80 | PNO1 | STYK1 | ZNF271 |
| B3GALNT1 | EIF2AK1 | LOC100131993 | PNPLA3 | SUPT7L | ZNF326 |
| BRI3BP | EIF5B | LOC100505759 | PODXL | SUSD5 | ZNF35 |
| BTG1 | ELOVL6 | LOC100507192 | POLR3K | SYNCRIP | |
| C12orf26 | ENC1a | LOC283278 | PPP2R1B | SYNJ2BP | |
| C12orf5 | ENY2 | LOC728903 | PRPS1 | SYTL2 | |
| C1GALT1C1 | ERLIN2 | MACC1 | PRR15 | SYTL5 | |
| C1orf116 | EXOG | MARCKS | PSCA | TBC1D30 | |
| C1orf135 | FADS1a | MAT2A | PSME3 | TFDP1 | |
| C1orf212 | FAIM | MCFD2 | PTGR1 | TFRC | |
| C1orf226 | FAM118B | MEIS3P1 | PTP4A2 | TGFB2 | |
| C6orf223 | FAM119A | MFAP3L | PTPRB | TGFBR2a | |
| CALM1a | FAM83A | MFI2 | PTPRJ | TGM2 | |
| CCDC68 | FBXW2 | MFSD1 | RABIF | THSD4 | |
| CCDC88A | FDX1 | MICALL1 | RBM25 | TIMM23 | |
| CCND1a | FN1a | MMACHC | RBM45 | TIMM8A | |
| CDA | FNIP2 | MRPL35 | RGS17 | TIMM8B | |
| CDC42EP2 | FRMD3 | MST1R | RHOBTB1 | TLCD1 | |
| CDH2 | FUCA1 | MTERFD3 | RHOF | TLR3 | |
| CEP78 | FXN | MYEOV | RPL27A | TLR4 | |
| CFH/CFHR1 | FZD8 | MYO5C | RPS6KA2 | TMC5 | |
| CFI | GALNT7 | NAA40 | S1PR3 | TMEM133 | |
| CHAC2 | GATAD2A | NAV3 | SAMHD1 | TMEM177 | |
| CHML | GBP1 | NECAP1 | SCEL | TMEM9B | |
| CHRNA5 | GDA | NIPAL1 | SGK223 | TP53I3 | |
| CLDN23 | GGCX | NMI | SH3TC2 | TPCN2 | |
| CMAH | GPATCH2 | NRP2 | SLC16A5 | TRAK2 | |
| CNPY2 | GPX8 | NSD1 | SLC1A1 | TRIM45 | |
| COL4A3 | GXYLT1 | OLAH | SLC35B4 | TRIOBP | |
| COL4A4 | HAS2 | OR7E14P | SLC35C1 | TRNT1 | |
| COX7A1 | HERC6 | OR7E47P | SLC37A1 | TSPAN12 | |
Ingenuity pathway analysis (IPA) revealed 4
canonical pathways significantly dysregulated in the transformed
cells trMCF clone 11: aryl hydrocarbon receptor signaling, retinoic
acid activation, xenobiotic metabolism signaling and molecular
mechanism of cancer (Table II).
Several genes of these pathways that were up- or downregulated in
trMCF clone 11 show similar levels of expression to MCF-10F after
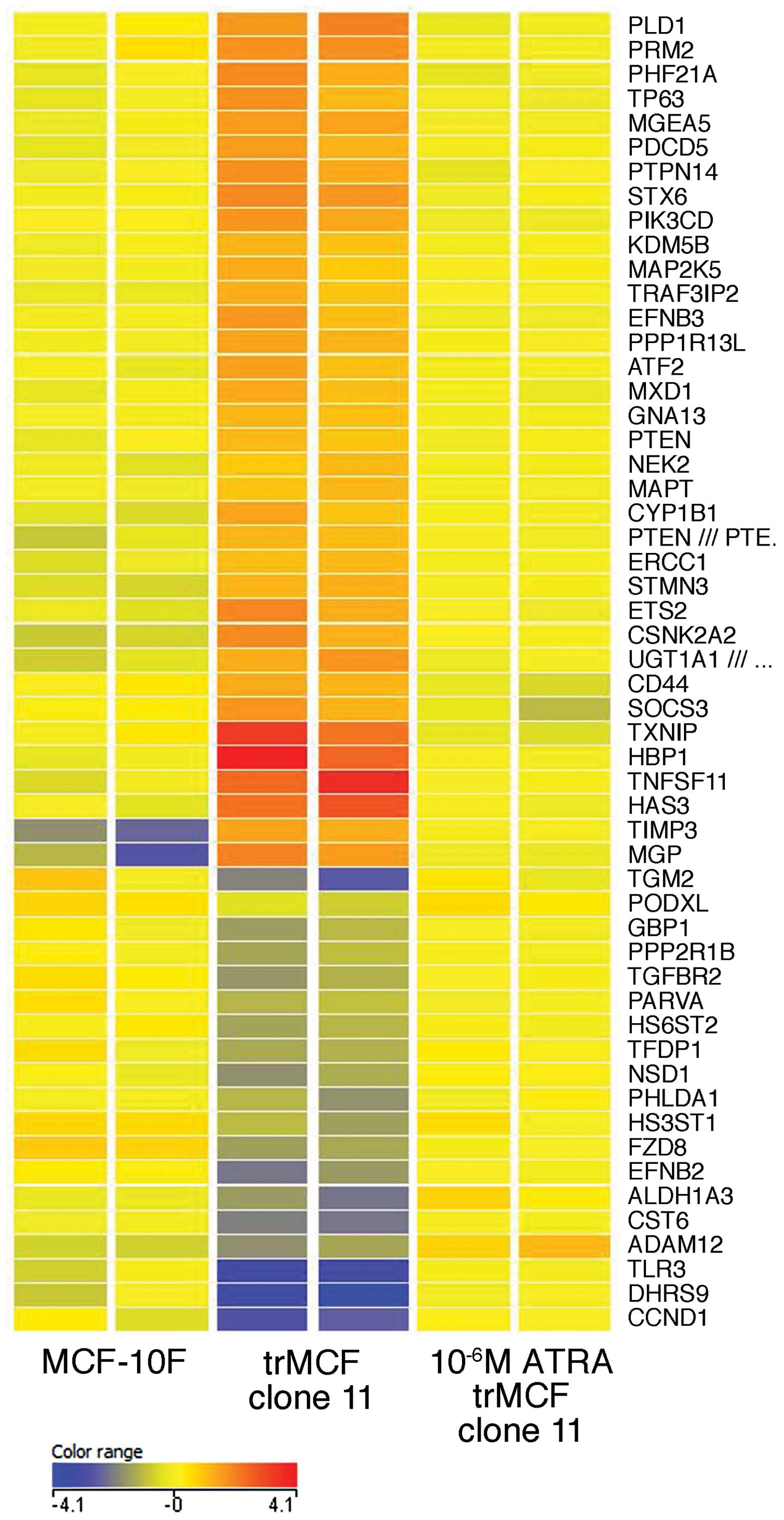
trMCF clone 11 was treated with 10−6 M ATRA (Table II and Fig. 7). Genes from the aryl hydrocarbon
receptor signaling ALDH1A3, CCND1, TGFBR2, TGM2 and TFDP1 were
downregulated in the transformed cells trMCF clone 11 when compared
to their expression in the normal breast epithelial cells MCF-10F
and, the expression of these genes was upregulated after these
cells were treated with 10−6 M ATRA reaching similar
levels to the expression in MCF-10F (Table II and Fig. 7). One of the functions that show
enrichment of dysregulated genes in the transformed trMCF clone 11
cells is cell morphology and the expression of most of these genes
reached similar levels to MCF-10F after trMCF cells were treated
with 10−6 M ATRA. The expression of some genes related
to cell morphology such as PLD1, CD44, STX6, STMN3, ATF2, ETS2,
NEK2, HAS3, MGP, GNA13 were upregulated in the transformed trMCF
clone 11 and their expression reached normal levels after
10−6 M ATRA treatment (Fig.
7); other genes related to cell morphology such as PHLDA1,
GBP1, HS6ST2 and TLR3 were downregulated in trMCF clone 11 and
their expressions increased after 10−6 M ATRA treatment
reaching similar levels to those found in the normal MCF-10F breast
epithelial cells (Fig. 7). Also,
the expression of several genes that encode enzymes involved in
chromatin modifications such as MGEA5, ATF-2, KDM5B, PRMT2 (PRM2),
PHF21A and NSD1, were dysregulated in trMCF clone 11, reaching
normal levels after 10−6 M ATRA treatment (Fig. 7).
| Table II.Canonical pathways enriched with
differentially expressed genes. |
Table II.
Canonical pathways enriched with
differentially expressed genes.
| trMCF clone 11 vs.
MCF-10F | ATRA trMCF clone 11
vs. trMCF clone 11 |
|---|
| Aryl hydrocarbon
receptor signaling | ALDH1A3↓, CCND1↓,
TGFBR2↓, TGM2 ↓, TFDP1↓, ALDH3A2↑, CYP1B1↑ | ALDH1A3↑, CCND1↑,
TGFB2↑, TGM2↑,TFDP1↑, ALDH3A2↓, CYP1B1↓ |
| Other genes:
CDKN1A↓, JUN↓ ALDH7A1↑, CSNK2A1↑, TGFB1↑, MAPK1↑, NFE2L2↑ | Other genes:
CCNE1↑, CCNE2↑, CDK6↑,DHFR↑, IL1B↑, IL6↑, NR2F1↑, NRIP1↑, POLA1↑
ALDH3B2↓, ARNT↓, NCOA3↓, HSPB2↓, ALDH6A1↓ |
| RAR activation | ALDH1A3↓, DHRS9↓,
NSD1↓, TGFB2↓, CSNK2A2↑, PIK3CD↑, PRMT2↑, PTEN↑ | ALDH1A3↑, DHRS9↑,
NSD1↑, TGFB2↑, CSNK2A2↓, PIK3CD↓, PRMT2↓, PTEN↓ |
| Other genes: JUN↓,
NR2F2↓, RBP1↓, CSNK2A1↑, CSNK2A2↑, MAPK1↑, MAPK14↑, TGFB1↑,
GNAS↑ | Other genes:
GTF2H2↑, IGFBP3↑, MAP2K1↑, MAPK13↑, NR2F1↑, NRIP1↑, RKAR2B↑, DH10↑,
CITED2↓, PNRC1↓, PRKAR1A↓, SMARCD2↓ |
| Xenobiotic
metabolism signaling | ALDH1A3↓, HS3ST1↓,
HS6ST2↓, PPP2R1B↓, ALDH3A2↑, CYP1B1↑, MAP2K5↑, PIK3CD↑, UGT1A1 (and
others UGT)↑ | ALDH1A3↑, HS3ST1↑,
HS6ST2↑, PPP2R1B↑, ALDH3A2↓, CYP1B1↓, MAP2K5↓, PIK3CD↓, UGT1A1 (and
others UGT)↓ |
| Other genes:
CHST15↓, ALDH7A1↑, CAMK1D↑, HDAC4↑, MAPK1↑, MAPK14↑, MGMT↑, UGT8↑,
NFE2L2↑ | Other genes: ILIB↑,
IL6↑, NRIP1↑, MAP2K1↑, MAPK13↑, ALDH3B2↓, ARNT↓, CAMK2D↓, CITED2↓,
MAP3K8↓, PPP2R3A↓, ALDH6A1↓, MAP3K2↓ |
| Molecular
mechanisms of cancer | TGFB2↓, TGFBR2↓,
CCND1↓, FZD8↓, PLCB4↓, RABIF↓, RHOF↓, GNA13↑, GNAI1↑, CD44↑ | TGFB2↑, TGFBR2↑,
CCND1↑, FZD8↑, PLCB4↑, RABIF↑, RHOF↑, GNA13↓, GNAI1↓, CD44↓ |
| Other genes:
CDKN1A↓, CTNNB1↓, FYN↓, IRS1↓, JUN↓, SMAD4↓, TCF4↓, XIAP↓, PIK3CD↑,
TCF3↑, TGFB1↑, GNAL↑, MAPK1↑, MAPK14↑ | Other genes: APC↑,
CCNE1↑, CCNE2↑, CDC25A↑, CDK6↑, CYCS↑, E2F2↑, MAP2K1↑, MAPK13↑,
PRKAR2B↑, RAPGEF3↑, RBL1↑, TFDP1↑, ARHGEF10↓, FOXO1↓, HHAT↓, IRS1↓,
NF1↓, PAK3↓, PIK3CD↓, PRKAR1A↓, RALGDS↓, RHOV↓ |
Discussion
In this study we showed that all trans-retinoic acid
(ATRA) induced branching of early transformed human breast
epithelial cells. The transformed trMCF clone 11 cells form
spherical masses in collagen (3D culture) and treatment with
10−6 M ATRA produced a significant decrease in spherical
masses and an increased number of tubules. Cells at an advanced
stage of transformation (bsMCF and caMCF) did not show any change
in morphology after being treated with ATRA. Our previous results
showed that RARβ (retinoic acid receptor β) was unmethylated in
MCF-10F and trMCF cells and became hypermethylated at the invasive
(bsMCF) and tumorigenic (caMCF) stages (19); although bsMCF and caMCF were
treated with 5-aza-dC to reactivate the expression of RARβ in
combination with ATRA, no changes in the phenotype of these cells
in collagen were observed. Our results indicate that ATRA is able
to re-differentiate early transformed cells to a normal stage but,
not tumor cells at later stages of the neoplastic process. We
previously showed that bsMCF and caMCF had important chromosomal
gains and losses and the earlier transformed cells trMCF showed
small genomic changes (16); this
could explain why ATRA was only effective as a re-differentiation
agent in the early transformed breast epithelial cells. Different
studies indicate that epigenetic modifications play important roles
in RA transcriptional regulation (20–24).
Histones have a long N-terminal tail extending outside the
nucleosome that is subject to acetylation, phosphorylation, and
methylation (25). In the absence
of RA, co-repressive elements (SMRT, NCoR and SIN3A) inhibit
transcription; the presence of RA releases co-repressors and
histone deacetylases allowing chromatin remodeling and access to
specific RAREs (20,24). RA treatment leads to acetylation of
histones H3 and H4 that lead to a more open stage of the chromatin
allowing the transcription of ATRA regulated genes. However, only a
limited number of information is currently available on the
epigenetic dynamics of RA response.
Recently, analysis of gene expression array datasets
of different FDA approved drugs revealed that ATRA (tretinoin) is a
drug that is negatively associated with cancer stem cell (CSC)
enriched gene expression signature (26). We found that ATRA treatment reduced
the expression of the stem cell marker CD44 in early transformed
cells. ATRA exerts effects on stem cell differentiation in part via
the modulation of the epigenome. Numerous enzymes that alter the
modifications on histones are involved in transcriptional
activation of specific genes in stem cells, and many of these
enzymes are modulated by RA treatment of stem cells (27). The expression of several genes
encoding enzymes involved in chromatin modifications such as MGEA5,
ATF-2, KDM5B, PRMT2, PHF21A and NSD1 were dysregulated in trMCF
clone 11, reaching normal levels after ATRA treatment. Others have
shown that in breast cancer, retinoids are effective inhibitors of
breast cancer cells at early stages of tumor progression, but their
effectiveness diminishes as the tumors become more aggressive
(28). Our results support these
findings.
Our results show that the RA concentration is
important to induced re-differentiation of early transformed breast
epithelial cells. The treatment of transformed cells with either
10−7 or 10−8 M ATRA did not induced any
change in morphology although, cells were able to form tubules
after treatment with 10−5 and 10−6 M ATRA,
more tubules being developed after treatment with 10−6 M
(1 μM) ATRA.
Little is known about the genomic targets and
effects of the different isoforms of the RARs and mechanism or
extent of crosstalk between RA signaling and other signaling
pathways. It has been recently shown that RAR binding through the
genome is highly coincident with estrogen receptor α binding,
resulting in widespread crosstalk of RA and estrogen signaling to
antagonistically regulate breast-cancer associated genes (29). Our gene expression studies
determined 443 genes which defined a signature of ATRA
re-programming effect in early transformed breast epithelial cells;
these genes were dysregulated in the early transformed cells and
they reached normal levels after the cells were treated with
10−6 M ATRA. Genes from the aryl hydrocarbon receptor
(AhR), retinoic acid receptor (RAR) and the xenobiotic pathways
were dysregulated in the early transformed breast epithelial cells
and their expression reached normal levels after ATRA treatment. It
has been shown that there is an interaction between AhR and RAR
activation and that AhR not only binds to polycyclic aromatic
hydrocarbon family of environmental contaminants but also to some
synthetic retinoids (30,31).
N-(4-hydoxyphenyl) retinamide (fenretinide or 4HPR)
is a synthetic retinoid that is currently one of the most promising
clinically tested retinoids. The modification of the carboxyl end
of all-trans RA with N-4-hydroxyphenyl group resulted in increased
efficacy as a chemoprevention agent as well as reduced toxicity
when compared with other retinoids (32). Animal models have demonstrated that
treatment with fenretinide prevents chemically induced cancers of
the breast, prostate, bladder and skin (33–36).
In conclusion, our results showed that 1 μM
ATRA was able to re-differentiate transformed cells at early stages
of the neoplastic process and antagonistically regulated breast
cancer associated genes. Our results support previous findings that
1 μM ATRA could be used as a chemo-preventive agent to
inhibit the progression of premalignant lesions of the breast.
Acknowledgements
We thank Karen Trush for helping in
the preparation of the final figures. This study was supported by
The Pennsylvania Breast Cancer Coalition and Friends for an Earlier
Breast Cancer Test.
References
|
1.
|
Theodosiou M, Laudet V and Schubert M:
From carrot to clinic: an overview of the retinoic acid signaling
pathway. Cell Mol Life Sci. 67:1423–1445. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Duester G: Retinoic acid synthesis and
signaling during early organogenesis. Cell. 134:921–931. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Mongan NP and Gudas LJ: Diverse actions of
retinoid receptors in cancer prevention and treatment.
Differentiation. 75:853–870. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Connolly R, Nguyen NK and Sukumar S:
Molecular pathways: current role and future directions of the
retinoic acid pathway in cancer prevention and treatment. Clin
Cancer Res. 19:1651–1659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Bushue N and Wan YJ: Retinoid pathway and
cancer therapeutics. Adv Drug Deliv Rev. 62:1285–1298. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Aagaard MM, Siersbaek R and Mandrup S:
Molecular basis for gene-specific transactivation by nuclear
receptors. Biochim Biophys Acta. 1812.824–835. 2011.PubMed/NCBI
|
|
7.
|
Tsai MJ and O’Malley BW: Molecular
mechanisms of action of steroid/thyroid receptor superfamily
members. Annu Rev Biochem. 63:451–486. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Rochette-Egly C and Germain P: Dynamic and
combinatorial control of gene expression by nuclear retinoic acid
receptors (RARs). Nucl Recept Signal. 7:e0052009.PubMed/NCBI
|
|
9.
|
Heyman RA, Mangelsdorf DJ, Dyck JA, et al:
9-cis retinoic acid is a high affinity ligand for the retinoid X
receptor. Cell. 68:397–406. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Garattini E, Gianni M and Terao M:
Cytodifferentiation by retinoids, a novel therapeutic option in
oncology: rational combinations with other therapeutic agents.
Vitam Horm. 75:301–354. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Zanardi S, Serrano D, Argusti A, Barile M,
Puntoni M and Decensi A: Clinical trials with retinoids for breast
cancer chemoprevention. Endocr Relat Cancer. 13:51–68. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Seewaldt VL, Johnson BS, Parker MB,
Collins SJ and Swisshelm K: Expression of retinoic acid receptor
beta mediates retinoic acid-induced growth arrest and apoptosis in
breast cancer cells. Cell Growth Differ. 6:1077–1088.
1995.PubMed/NCBI
|
|
13.
|
Swisshelm K, Ryan K, Lee X, Tsou HC,
Peacocke M and Sager R: Down-regulation of retinoic acid receptor
beta in mammary carcinoma cell lines and its up-regulation in
senescing normal mammary epithelial cells. Cell Growth Differ.
5:133–141. 1994.PubMed/NCBI
|
|
14.
|
Widschwendter M, Berger J, Muller HM,
Zeimet AG and Marth C: Epigenetic downregulation of the retinoic
acid receptor-beta2 gene in breast cancer. J Mammary Gland Biol
Neoplasia. 6:193–201. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Russo J, Fernandez SV, Russo PA, et al:
17-Beta-estradiol induces transformation and tumorigenesis in human
breast epithelial cells. FASEB J. 20:1622–1634. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Huang Y, Fernandez SV, Goodwin S, et al:
Epithelial to mesenchymal transition in human breast epithelial
cells transformed by 17beta-estradiol. Cancer Res. 67:11147–11157.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Fernandez SV and Russo J: Estrogen and
xenoestrogens in breast cancer. Toxicol Pathol. 38:110–122. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Hebner C, Weaver VM and Debnath J:
Modeling morphogenesis and oncogenesis in three-dimensional breast
epithelial cultures. Annu Rev Pathol. 3:313–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Snider KE, Ehya H, Russo J and Fernandez
SV: NRG1 and RARB hypermethylation in breast cancer progression.
In: Proceedings of the ACCR 102nd Anual Meeting; Orlando, FL..
Cancer Res. 71(Suppl 1): abs. 75. pp. 2011
|
|
20.
|
Glass CK and Rosenfeld MG: The coregulator
exchange in transcriptional functions of nuclear receptors. Genes
Dev. 14:121–141. 2000.PubMed/NCBI
|
|
21.
|
Hartman HB, Yu J, Alenghat T, Ishizuka T
and Lazar MA: The histone-binding code of nuclear receptor
co-repressors matches the substrate specificity of histone
deacetylase 3. EMBO Rep. 6:445–451. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Lefebvre B, Ozato K and Lefebvre P:
Phosphorylation of histone H3 is functionally linked to retinoic
acid receptor beta promoter activation. EMBO Rep. 3:335–340. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
McKenna NJ and O’Malley BW: Combinatorial
control of gene expression by nuclear receptors and coregulators.
Cell. 108:465–474. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Rochette-Egly C: Dynamic combinatorial
networks in nuclear receptor-mediated transcription. J Biol Chem.
280:32565–32568. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Chen H, Lin RJ, Xie W, Wilpitz D and Evans
RM: Regulation of hormone-induced histone hyperacetylation and gene
activation via acetylation of an acetylase. Cell. 98:675–686. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Bhat-Nakshatri P, Goswami CP, Badve S,
Sledge GW Jr and Nakshatri H: Identification of FDA-approved drugs
targeting breast cancer stem cells along with biomarkers of
sensitivity. Sci Rep. 3:25302013. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Gudas LJ: Retinoids induce stem cell
differentiation via epigenetic changes. Semin Cell Dev Biol.
24:701–705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Zhang XK, Liu Y and Lee MO: Retinoid
receptors in human lung cancer and breast cancer. Mutat Res.
350:267–277. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Hua S, Kittler R and White KP: Genomic
antagonism between retinoic acid and estrogen signaling in breast
cancer. Cell. 137:1259–1271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Soprano DR and Soprano KJ: Pharmacological
doses of some synthetic retinoids can modulate both the aryl
hydrocarbon receptor and retinoid receptor pathways. J Nutr.
133:277S–281S. 2003.PubMed/NCBI
|
|
31.
|
Murphy KA, Quadro L and White LA: The
intersection between the aryl hydrocarbon receptor (AhR)- and
retinoic acid-signaling pathways. Vitam Horm. 75:33–67. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Hail N Jr, Kim HJ and Lotan R: Mechanisms
of fenretinide-induced apoptosis. Apoptosis. 11:1677–1694. 2006.
View Article : Google Scholar
|
|
33.
|
McCormick DL, Becci PJ and Moon RC:
Inhibition of mammary and urinary bladder carcinogenesis by a
retinoid and a maleic anhydride-divinyl ether copolymer (MVE-2).
Carcinogenesis. 3:1473–1476. 1982. View Article : Google Scholar
|
|
34.
|
McCormick DL and Moon RC: Antipromotional
activity of dietary N-(4-hydroxyphenyl)retinamide in two-stage skin
tumorigenesis in CD-1 and SENCAR mice. Cancer Lett. 31:133–138.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Moon RC, Pritchard JF, Mehta RG, Nomides
CT, Thomas CF and Dinger NM: Suppression of rat mammary cancer
development by N-(4-hydroxyphenyl)retinamide (4-HPR) following
surgical removal of first palpable tumor. Carcinogenesis.
10:1645–1649. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Pollard M, Luckert PH and Sporn MB:
Prevention of primary prostate cancer in Lobund-Wistar rats by
N-(4-hydroxyphenyl) retinamide. Cancer Res. 51:3610–3611.
1991.PubMed/NCBI
|