Introduction
Synovial sarcoma (SS) is an aggressive malignant
tumor of mesenchymal origin, with high risk of local recurrence and
distant metastasis, and is also characterized by poor prognosis,
high mortality and a poor curative rate (1,2). SS
frequently originates at the extremities of young adults and
constitutes 10–20% of all soft tissue sarcomas (3). Current therapy for SS comprises
surgery with or without adjuvant chemotherapy and/or radiotherapy,
which provides a good opportunity for healing of localized disease.
However, early and late recurrences often occur (4,5), and
the 5-year survival rate is estimated to be only 27–55% (6). Mounting evidence indicates that
certain proteins are dysregulated in primary tumors and are
associated with the development and progression of SS (7–9).
Understanding the specific functions and disclosing the underlying
mechanisms of these proteins involved in the occurrence and
development of SS may facilitate identification of effective
targeting strategies for SS.
Feline leukemia virus subgroup C cellular receptor 1
(FLVCR1), as a member of the major facilitator superfamily
transporters, has important roles in controlling the size of the
cytoplasmic free-heme pool (10).
There are two different isoforms of FLVCR1: FLVCR1a and FLVCR1b,
the former is a plasma membrane heme exporter mediating heme export
out of the cell (11,12), and the latter resides in the
mitochondria and is involved in heme trafficking between the
mitochondria and cytosol (13).
FLVCR1a is required for preventing hemorrhages and edema, whereas
FLVCR1b regulates erythropoiesis by controlling mitochondrial heme
efflux (10). Mercurio et
al (14) reported that FLVCR1a
is crucial for the proliferation of committed erythroid
progenitors, and FLVCR1b is crucial for terminal differentiation.
Additionally, Fiorito et al (13) demonstrated that the expression of
FLVCR1 was regulated by hypoxia in human colorectal adenocarcinoma
Caco2 cells through hypoxia inducible factors 2α (HIF2α) and v-ets
avian erythroblastosis virus E26 oncogene homolog 1 (ETS1), thus
demonstrating the role of FLVCR1 in hypoxia-stimulated processes,
including erythropoiesis, vasculogenesis, and potentially heme
absorption. FLVCR1 is reported to be involved in sensory neuron
maintenance and posterior column ataxia, and retinitis pigmentosa
(15,16). Chiabrando et al (15) reported that FLVCR1 is important for
the survival of neuroblastoma cells, and they further demonstrated
that FLVCR1 is crucial in mediating oxidative stress and regulating
the sensitivity to programmed cell death in several cell types,
including erythroid progenitors, hepatocytes and intestinal cells
(17–19). Together, these findings suggest
that FLVCR1 may have vital roles in a variety of biological
processes, including cell proliferation, cell death, apoptosis,
oxidative stress response, cellular differentiation and
metabolism.
In the current study, the expression of FLVCR1 was
markedly elevated in SS cells and clinical SS samples. However, the
biological roles of the overexpressed FLVCR1 in SS remain unknown.
According to aforementioned roles of FLVCR1, we hypothesized that
FLVCR1 may contribute to the occurrence and development of SS. By
silencing FLVCR1 using specific short hairpin RNA (RNA),
proliferation and tumorigenicity of SS cells were inhibited in
vitro and in vivo. FLVCR1 promoted the proliferation and
tumorigenicity of SS by regulating cytotoxic autophagy. To the best
of our knowledge, these findings were the first to demonstrate that
FLVCR1 functions as an oncoprotein during SS progression, which
indicates that FLVCR1 may be a novel therapeutic target for SS.
Materials and methods
Tissue samples
Samples of human SS tissues and paired adjacent
non-tumor tissues that were 5 cm away from the tumors were obtained
from 8 patients histopathologically diagnosed with SS that
underwent surgical resections between May 2014 and September 2017
at the Second Hospital of Shandong University (Jinan, China). None
of the patients had received anticancer therapy prior to the
sampling. Of 8 patients, 5 were male and 3 female. The age of the
patients at the time of presentation ranged between 18 and 76 years
(mean, 38.5 years). Fibroblast-like synovialcytes were isolated
from 2 patients (1 male aged 65 years, 1 female aged 57 years)
diagnosed with osteoarthritis who underwent surgical resections in
July 2017. The samples were obtained with patients' informed
consent. All research involving human participants have been
approved by the Second Hospital of Shandong University ethics
committees and in accordance with the Declaration of Helsinki.
Cell lines
The human HS-SY-II SS cell line was provided by Dr
Yi Guo (University of California, Irvine, CA, USA), and cultured in
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Human SW982 (no. HTB-93) SS
cell line was obtained from the American Type Culture Collection
(Manassas, VA, USA), and propagated in L-15 medium (Gibco; Thermo
Fisher Scientific, Inc.). Human fibroblast-like synovialcytes
derived from patients with osteoarthritis were cultured in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.). All the
media were supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin. Cells were cultured at 37°C under a 5% CO2
atmosphere.
Immunohistochemistry
The immunohistochemical procedures were performed as
previously reported (20,21). Antibody against FLVCR1 (cat. no.
PA5-61270) was purchased from Invitrogen (Thermo Fisher Scientific,
Inc.). Briefly, 10% formalin-fixed, paraffin-embedded human SS or
paired adjacent non-tumor samples were cut into 5-µm
sections, placed on glass slides, heated at 70°C for 30 min, and
then deparaffinized with xylene and ethanol. For antigen retrieval,
the deparaffinized and rehydrated slides were heated in 10 mM
citrate buffer for 20 min in a microwave oven and allowed to cool
for 20 min at room temperature. Slides were incubated with 3%
H2O2 in methanol for 15 min at room
temperature to eliminate endogenous peroxidase activity. Then
slides were incubated at 4°C overnight with antibody against the
rabbit polyclonal anti-FLVCR1 antibody (1:50). Sections incubated
with nonimmune rabbit serum (1:100; cat. no. FK-MB594J; Shanghai
Fanke Biotech Co., Ltd., Shanghai, China) in PBS instead of primary
antibody served as negative controls. After incubation in a 1:2,000
dilution of secondary antibody (cat. no. ab6720; Abcam, Cambridge,
MA, USA) for 4 h at room temperature, the slides were incubated
with a streptavidin-peroxidase complex at room temperature for 20
min. Immunohistochemistry staining was performed using
3,3′-diaminobenzidine Enhanced Liquid Substrate System
tetrahydrochloride (cat. no. D3939; Sigma-Aldrich, Merck KGaA,
Darmstadt, Germany) and counterstained with haematoxylin. Cells
exhibiting positive staining on cell membranes and in the cytoplasm
and nucleus were counted in at least 10 representative fields (in
total 80 sections were used; magnification, ×200) and the mean
percentage of positive cells was calculated. Immunostaining was
evaluated by two independent pathologists blinded to clinical
characteristics and outcomes.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), and RT was performed
to cDNA using the M-MLV reverse transcriptase kit (Promega
Corporation, Madison, WI, USA) in accordance to the manufacturer's
instructions. The PCR primers for FLVCR1 and GAPDH were as follows:
FLVCR1 forward, 5′-CCCACAGACCAAGAACC-3′ and reverse,
5′-CCACCACATACAAACCC-3′; GAPDH forward, 5′-TGACTTCAACAGCGACACCCA-3
and reverse, 5′-CACCCTGTTGCTGTAGCCAAA-3′. qPCR was performed with
the SYBR-Green Real-Time PCR assay kit (Takara Bio, Inc., Otsu,
Japan) on an ABI PRISM 7300 Sequence Detection System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermal cycling
conditions were as follows: initial denaturation at 95°C for 10
min, followed by 35 amplification cycles at 95°C for 15 sec, 60°C
for 1 min. The level of GAPDH was used as an internal control. Fold
changes in expression were calculated using the 2−ΔΔCq
method (22).
Vectors and lentivirus infection
For depletion of FLVCR1, a human FLVCR1-targeting
shRNA sequence was cloned into pGCSIL-green fluorescent protein
(GFP) vector to generate pGCSIL-GFP-FLVCR1-RNAi, with target
sequence 5′-CCCTGAAGAGTACTCCTATAA-3′ (synthesized by Shanghai
GeneChem Co., Ltd., Shanghai, China), or control scrambled shRNA
(Lv-shCon sequence, 5′-TTCTCCGAACGTGTCACGT-3′). Lentiviruses
production and infection were performed as previously reported
(7). Stable cell lines (SW982 and
HS-SY-II) expressing FLVCR1 shRNAs were selected for 10 days with
0.5 µg/ml puromycin. At 3 weeks later, the cells were
essentially 100% GFP positive, and processed for RT-qPCR and
western blot.
Western blotting
Standard western blot assays were performed as
described previously (7,23). Briefly, cells were harvested in the
NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (cat. no.
78833; Thermo Fisher Scientific, Inc.), and protein was quantified
with Pierce™ BCA Protein Assay kit (cat. no. 23227; Pierce; Thermo
Fisher Scientific, Inc.). Then, equal amounts of protein (30
µg) were electrophoresed by 10% SDS-PAGE and transferred
onto polyvinylidene fluoride membranes. Then membranes were blocked
overnight at 4°C in 5% skim milk and incubated for 2 h at room
temperature with the primary antibodies. The membranes were then
washed with 1X TBS-Tween-20 three times and incubated with
horseradish peroxidase-conjugated antibodies (1:10,000; cat. no.
7074; Cell Signaling Technology, Inc., Danvers, MA, USA) at room
temperature for 1 h. The specific proteins were measured using an
enhanced chemiluminescence Western blotting kit (cat. no. RPN2108;
GE Healthcare, Chicago, IL, USA) according to the instructions from
the manufacturer. Antibodies against FLVCR1 (cat. no. PA5-28463;
1:1,000) was purchased from Invitrogen (Thermo Fisher Scientific,
Inc.). Microtubule associated protein 1 light chain 3β (LC3; cat.
no. L7543; 1:1,000) and nucleoporin p62 (cat. no. N1163; 1:1,000)
antibodies were purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). An internal control GAPDH (cat. no. sc-47724;
1:1,000) or β-actin (cat. no. sc-8432; 1:1,000) antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
The quantitative densitometry analysis of immunoreactive band was
performed using ImageJ bundled with 64-bit Java 1.6.0_24 program
for Windows from the National Institutes of Health (Bethesda, MD,
USA).
MTT assay
Cell viability was determined via MTT assay as
previously reported (24).
Briefly, cells were plated in 96-well plates at initial density of
2×103 cells/well. At each time point, cells were stained
with 100 ml sterile MTT dye (0.5 mg/ml; Sigma-Aldrich; Merck KGaA)
for 4 h at 37°C, followed by removal of the culture medium and
addition of 100 ml of dimethyl sulfoxide (Sigma-Aldrich; Merck
KGaA). The optical density (OD) was measured at 490 nm using an
EL-311SX ELISA reader (BioTek Instruments, Inc., Winooski, Vermont,
USA). All experiments were performed in triplicate.
Colony formation assay
Colony formation assay was performed as described
previously (7). Briefly, cells
were seeded in 6-well plates (5×102 cells) and
maintained for 10 days. The colonies were stained with 1% crystal
violet for 30 sec after fixation with 4% formaldehyde (both from
Sigma-Aldrich; Merck KGaA) for 5 min. Colonies were counted and the
results were presented as the fold change compared to vector
control cells.
Flow cytometry analysis of the cell cycle
and apoptosis
Cell cycle and apoptosis analysis by using flow
cytometry were performed as previously reported (7,25).
Cells were seeded in 6-well plates. Following trypsinization, cells
were centrifuged at 400 × g for 5 min at 4°C, rinsed twice with ice
cold PBS, and fixed at 4°C in 70% alcohol for >1 h.
Subsequently, fixed cells were resuspended in PBS containing 0.5%
Triton X-100 and 100 µg/ml RNase on ice for 30 min, and
stained with 50 µg/ml propidium iodide (PI; Sigma-Aldrich;
Merck KGaA) in the dark for 30 min. Cells were analyzed for cell
cycle distribution using a FACScan flow cytometer and BD ModFit
software (both from BD Biosciences, San Jose, CA, USA) was used to
analyze the data in accordance with the manufacturer's
guidelines.
For apoptosis analysis, the same procedures were
conducted as those for cell cycle assay described above and cell
apoptosis was detected using Annexin V-FITC/PI Apoptosis Detection
kit (cat. no. BMS500FI-100; eBioscience; Thermo Fisher Scientific,
Inc.) and analyzed with to a FACScan flow cytometer (BD
Biosciences), according to the manufacturer's protocol.
Xenografted tumor model
The xenografted tumor model was performed as
previously described (7). For the
SS xenograft model, 4-week-old NOD/SCID nude mice (14–20 g; Vital
River Laboratory Animal Technology Co., Ltd., Beijing, China) were
housed in a temperature-controlled, pathogen-free environment and
used for experimentation. All experimental procedures were approved
by the Institutional Animal Care and Use Committee of Shandong
University (Jinan, China). The mice were randomly divided into two
groups (n=4 per group), and HS-SY-II cells (2×106)
transduced with lentivirus expressing shFLVCR1 or negative control
vector were subcutaneously injected into the right flank of nude
mice. Tumor length (L), width (W) and diameter were measured every
week from week 1 to week 6; tumor volume (mm3) was
calculated using the following formula: L × W2/2
(7,26). After 6 weeks, mice were sacrificed
and tumors were harvested.
Statistical analysis
Data are presented as the mean ± standard deviation
from at least three separate experiments. Statistical analysis was
performed with one-way analysis of variance with post hoc
Bonferroni testing for multiple comparison or Student's t-test
using GraphPad Prism 6 software (GraphPad Software Inc., La Jolla,
CA, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
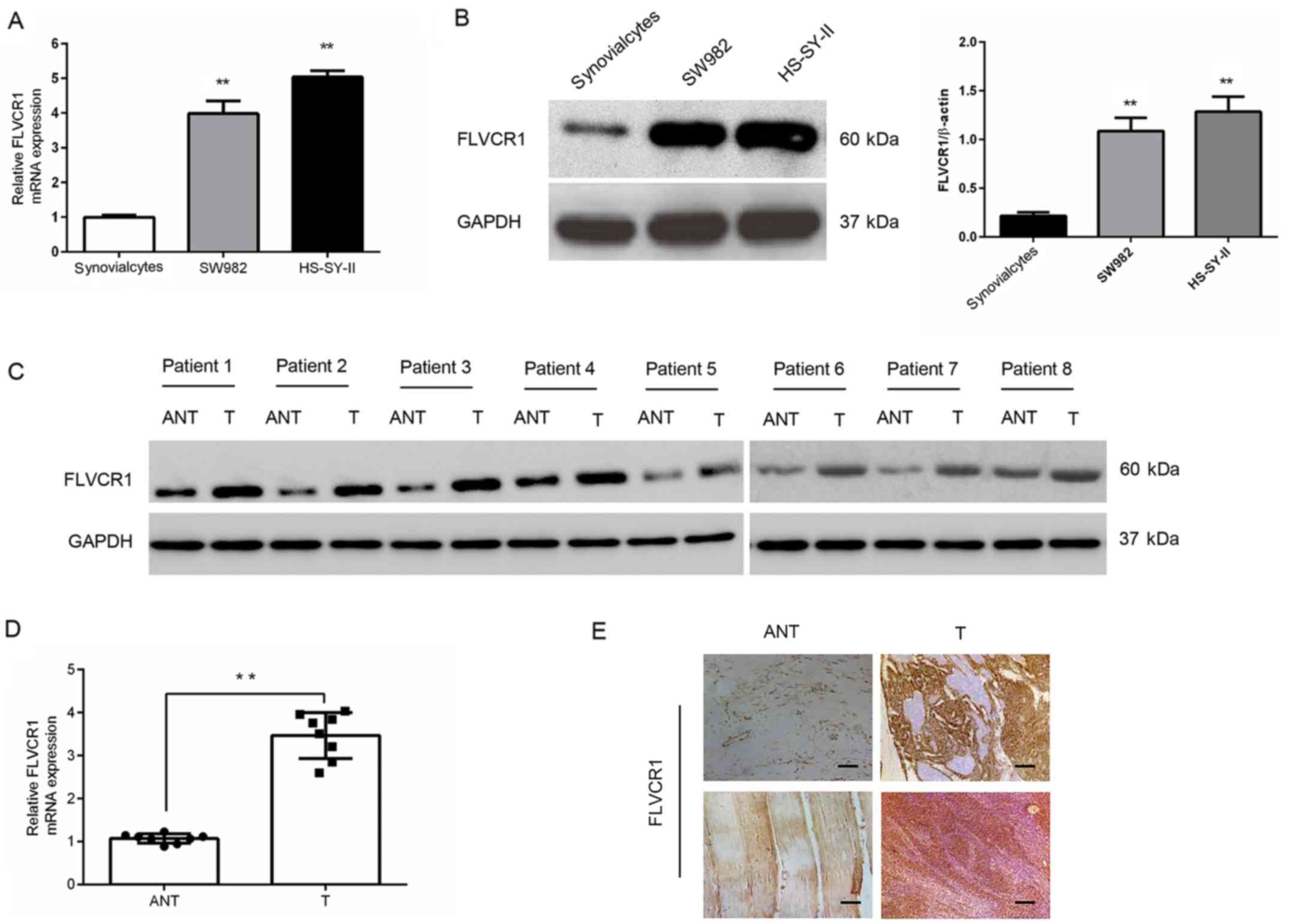
FLVCR1 is upregulated in human SS cells
and tissues
Gene and protein expression analysis in SW982 and
HS-SY-II SS cell lines demonstrated that FLVCR1 was markedly
upregulated in SS cell lines compared with synovialcytes derived
from patients with osteoarthritis (Fig. 1A and B). Additionally, in eight
pairs of human SS tissues, the mRNA and protein levels of FLVCR1
were higher than those in matched adjacent nontumor tissues
(Fig. 1C and D). In accordance
with the western blot results, the protein levels of FLVCR1 in
eight specimens measured by immunohistochemistry further
demonstrated higher expression of FLVCR1 in SS tissues than those
in matched adjacent nontumor tissues (Fig. 1E). These data collectively
suggested that FLVCR1 expression is upregulated in SS.
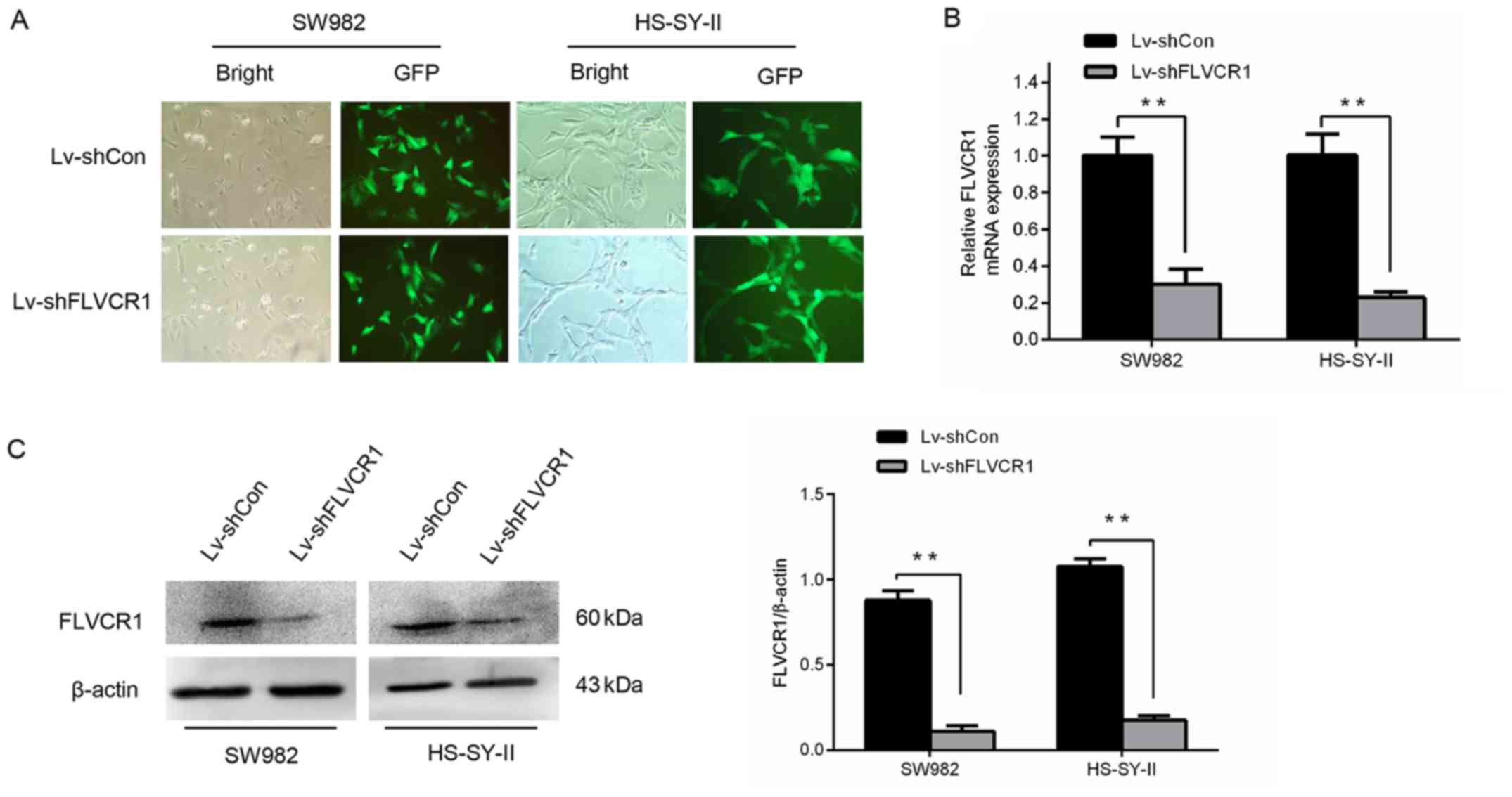
Silencing of FLVCR1 expression in SS
cells by lentivirus mediated RNA interference
To determine the potential roles of FLVCR1 in SS,
FLVCR1 was silenced in the SS cell lines, SW982 and HS-SY-II,
through lentivirus-mediated gene infection. As demonstrated in
Fig. 2A, the majority of SW982 and
HS-SY-II cells were GFP-positive following infection with
lentivirus expressing shRNA targeting FLVCR1 (Lv-shFLVCR1) or
Lv-shCon, indicating that the SS cells were effectively infected by
the recombinant lentivirus. As demonstrated in Fig. 2B and C, the gene and protein levels
of FLVCR1 in Lv-shFLVCR1 infected SS cells were significantly
suppressed, further corroborating the knockdown efficiency. These
findings demonstrated that recombinant lentivirus carrying shFLVCR1
effectively inhibited the expression of endogenous FLVCR1 in SS
cells.
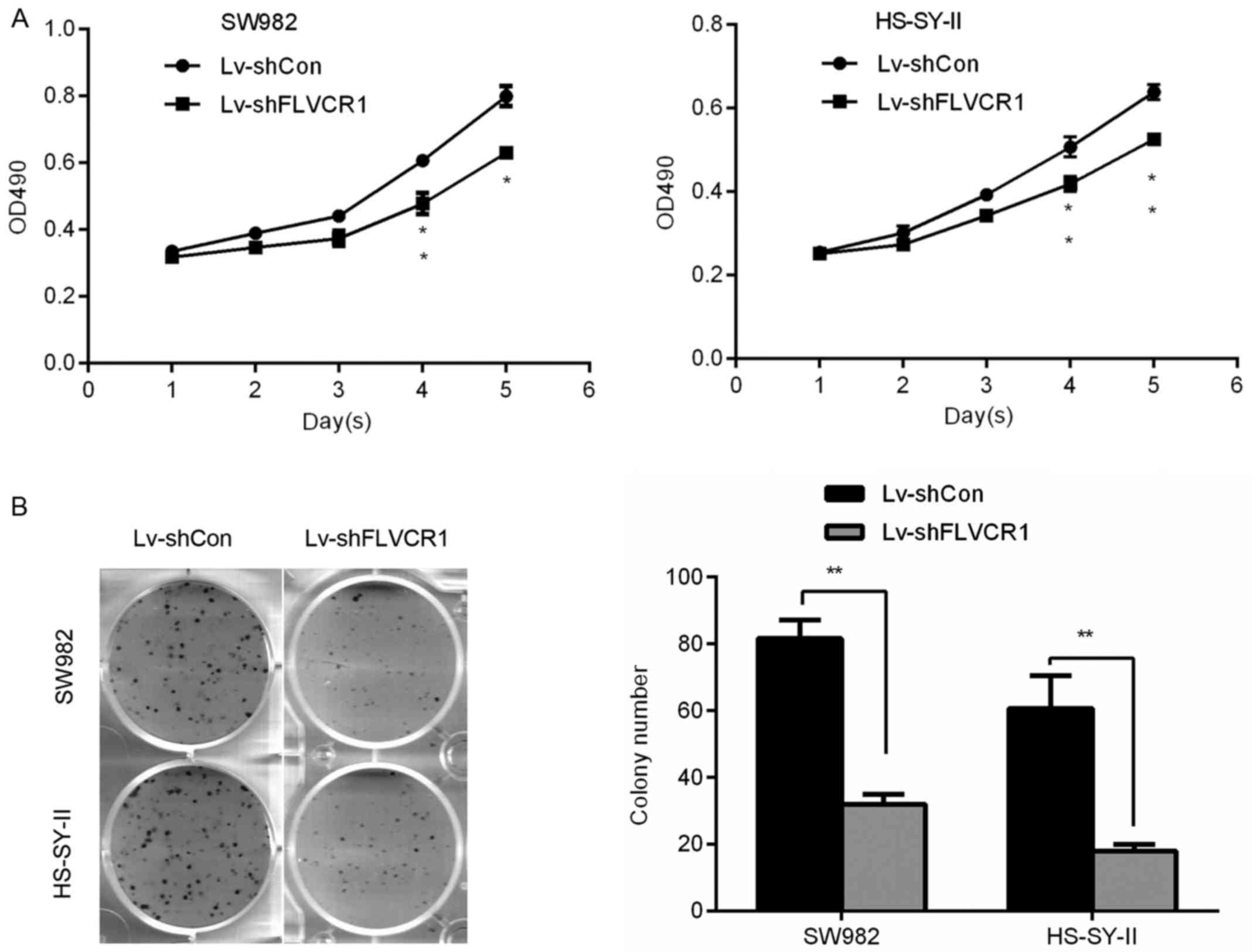
Silencing of FLVCR1 reduces the
proliferation and colony formation ability of SS cells
To investigate the specific role of FLVCR1 in the
proliferation and colony formation ability of SS cells, a 5-day MTT
and colony formation assay were performed, respectively. Fig. 3A demonstrates that Lv-shFLVCR1
infected SS cells exhibited a slower growth rate compared with
Lv-shCon infected cells. On day 5, the OD490 of Lv-shFLVCR1
infected SW982 and HS-SY-II cells were 0.63±0.02 and 0.52±0.01,
respectively, whereas they were 0.79±0.03 and 0.64±0.01 in the
Lv-shCon infected cells. Consistently, colony formation assay
further confirmed these results, Lv-shFLVCR1 infected SW982 and
HS-SY-II cells produced less (32.0±3.0 and 15.67±3.21) and smaller
colonies compared with Lv-shCon infected cells (81.67±5.51 and
60.67±9.86) (Fig. 3B). Thus, these
data suggested that FLVCR1 promoted proliferation and colony
formation of SS cells.
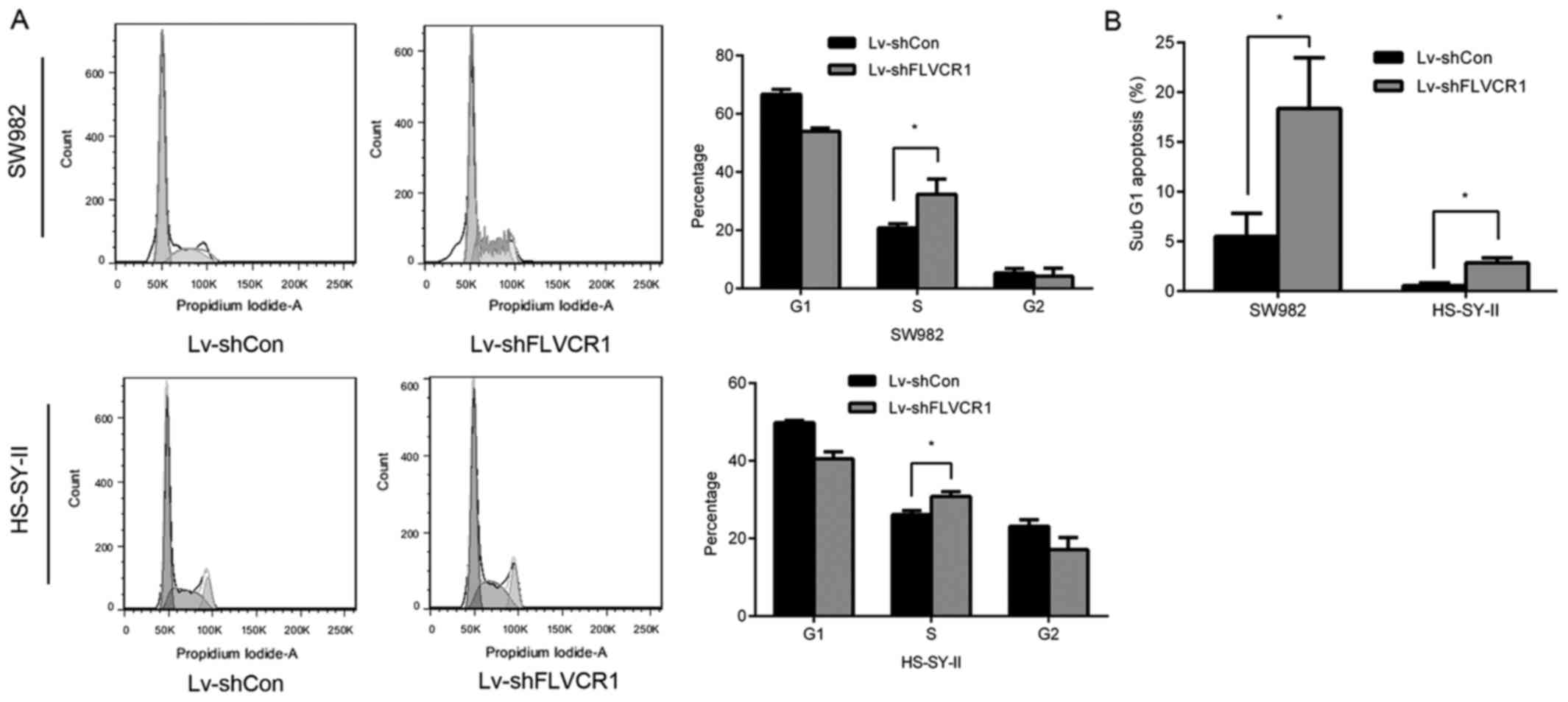
Inhibition of FLVCR1 impairs cell cycle
progression of SS cells
To further investigate the underlying mechanisms of
inhibition of cell proliferation and colony formation, cell cycle
phases of SS cells were detected using PI staining and flow
cytometry following FLVCR1 knockdown. As presented in Fig. 4A, 53.96±1.14 and 40.54±1.78% of
cells were at the G0/G1 phase in Lv-shFLVCR1-infected SW982 and
HS-SY-II cells, which was lower than Lv-shCon-infected SW982
(66.70±1.68%) and HS-SY-II cells (49.85±0.51%). Additionally, there
were significantly more cells at S phase following Lv-shFLVCR1
infection in SW982 and HS-SY-II cells (32.69±4.77 and 30.81±1.22%),
compared with Lv-shCon infected SW982 and HS-SY-II cells
(20.78±1.45 and 26.14±1.02%) (Fig.
4A). These results suggested that cell cycle progression was
impaired in FLVCR1-silenced SS cells. Furthermore, 18.36±2.34 and
2.84±0.04% cells were in the sub-G1 phase in Lv-shFLVCR1-infected
SW982 and HS-SY-II cells, higher in Lv-shCon-infected SW982 and
HS-SY-II cells (5.52±0.05 and 0.55±0.02%) (Fig. 4B), suggesting that there were more
apoptotic cells in the group with FLVCR1 suppression.
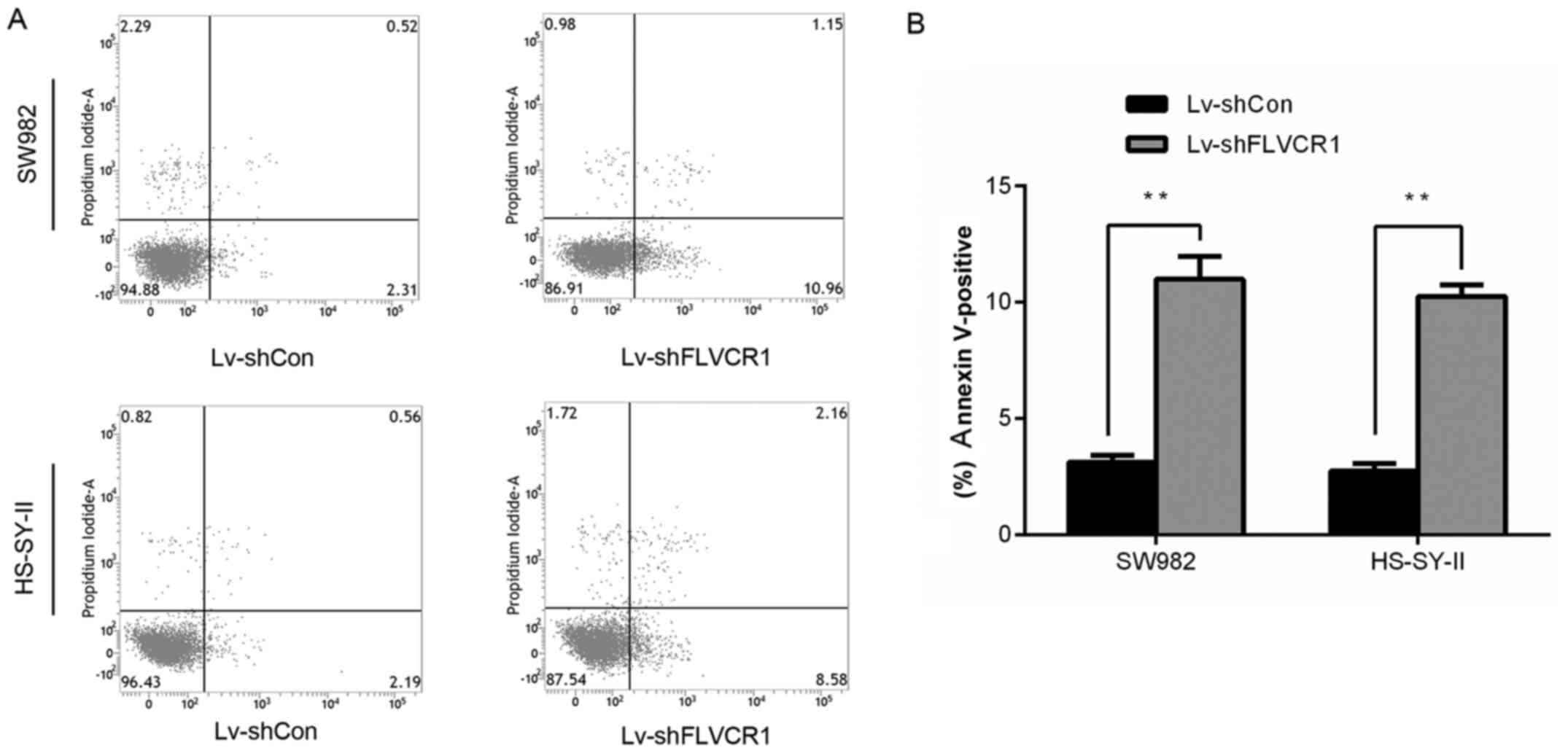
Silencing of FLVCR1 induced apoptosis of
SS cells
As presented in Fig.
5, the percentage of early and late apoptotic SW982 and
HS-SY-II cells increased to 10.99±0.97 and 10.26±0.48%,
respectively, in Lv-shFLVCR1 cells, which was significantly higher
than that of the control (3.12±0.29 and 2.74±0.33%), indicating
FLVCR1 silencing promoted SS cell apoptosis.
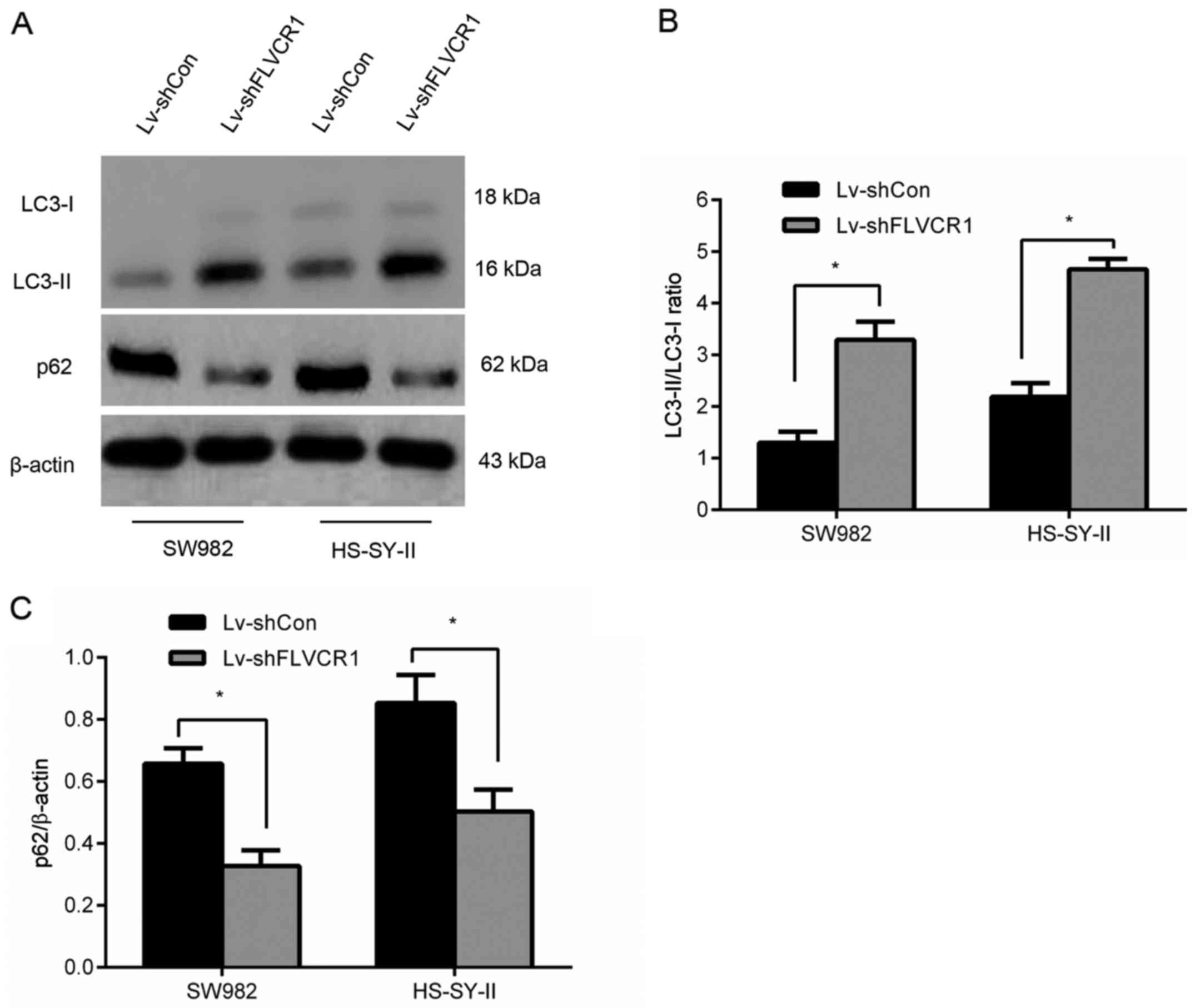
Silencing of FLVCR1 induced cytotoxic
autophagy of SS cells
To assess whether FLVCR1 affects autophagy, the
expression of LC3-II, LC3-I and nucleoporin p62 (a known autophagy
substrate) in SS cells was assayed by western blot, and the
LC3-II/LC3-I ratio (a specific indicator of autophagy level) was
calculated. As presented in Fig.
6, an increased accumulation of LC3-II and reduced expression
of p62 were observed in Lv-shFLVCR1-infected cells compared to
those in Lv-shCon-infected cells. Collectively, the results suggest
that silencing of FLVCR1 induced cytotoxic autophagy, thus,
promoting apoptosis under certain conditions of metabolic
stress.
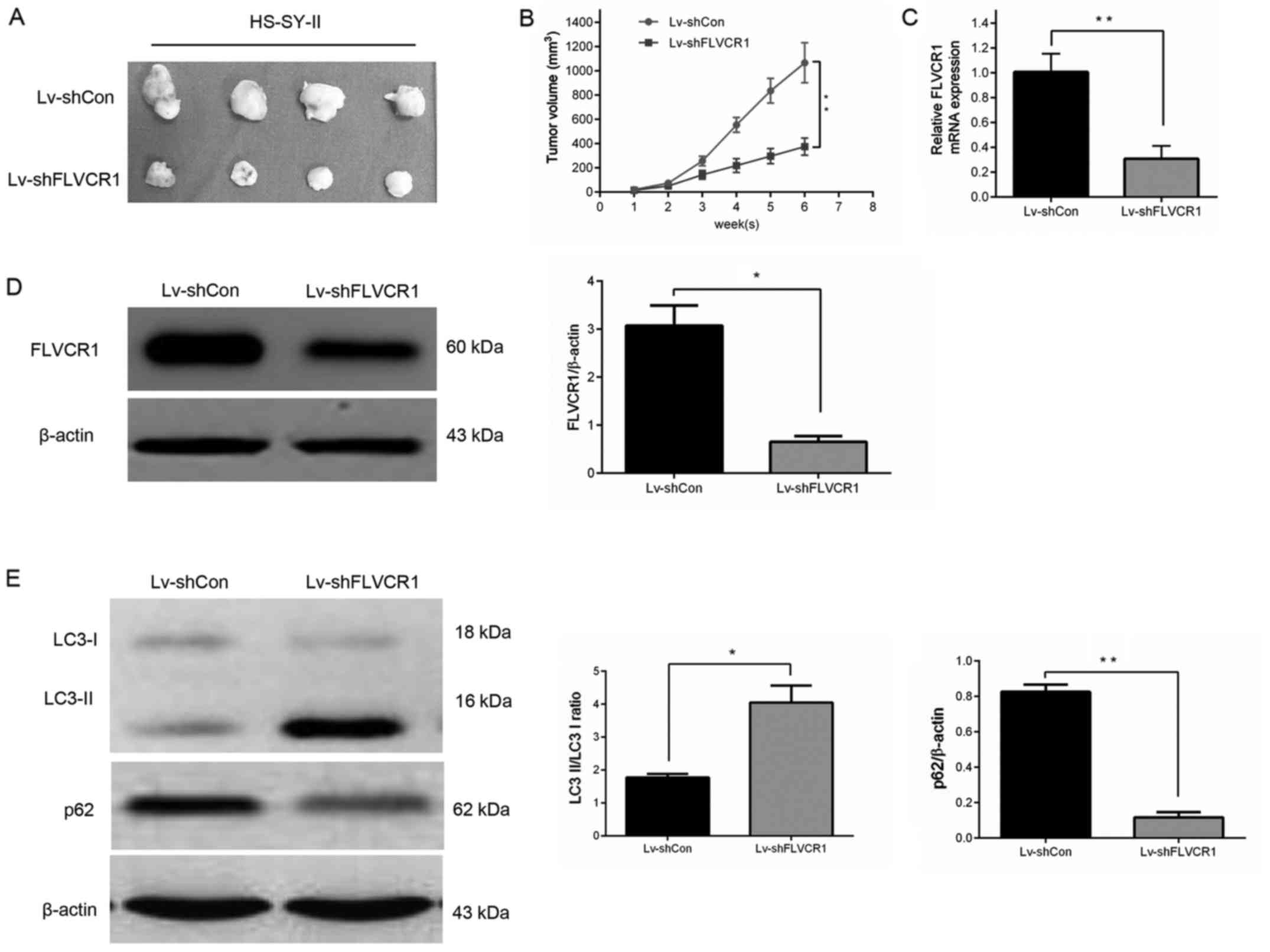
Silencing of FLVCR1 suppressed SS cell
growth in vivo
A subcutaneous SS nude mouse xenograft model was
established to confirm whether silencing of FLVCR1 inhibited the
growth of SS cells in vivo. As displayed in Fig. 7A and B, the mouse group infected
with Lv-shFLVCR1 lentivirus had a lower proliferation rate, and
formed markedly smaller tumors than the Lv-shCon lentivirus group.
The tumor size at the time of sacrifice in the Lv-shFLVCR1
lentivirus group was 375±40.7 mm3, which was
significantly smaller than in the Lv-shCon lentivirus group
(1,067±95.2 mm3). Additionally, in agreement with the
above in vitro results, reduced expression of FLVCR1 mRNA
(Fig. 7C) and protein (Fig. 7D) were observed in tumor tissues
following the inhibition of FLVCR1 compared with control tumor
tissues.
It was also investigated whether Lv-shFLVCR1 induces
cytotoxic autophagy of SS cells in vivo. Western blot
analysis revealed increased accumulation of LC3-II and degradation
of p62 in Lv-shFLVCR1 tumors, as compared with Lv-shCon tumors
(Fig. 7E). These results, taken
together, suggest that FLVCR1 silencing suppressed SS
tumorigenicity in vivo through, partially inducing cytotoxic
autophagy.
Discussion
The current study investigated a novel role for
FLVCR1 in promoting the proliferation and tumorigenicity of SS,
which has not been reported previously, to the best of our
knowledge. The data revealed that FLVCR1 enhances the proliferation
and tumorigenicity of SS cells in vitro and in vivo.
FLVCR1 silencing reduced the proliferation and tumorigenicity of SS
cells via effects on the cell cycle and autophagy. The results from
the current study provided novel insights and evidence that FLVCR1
play a crucial role in promoting the tumorigenicity and progression
of human SS, indicating that targeting FLVCR1 may be useful as a
novel therapeutic strategy in the treatment of patients with
SS.
Overexpression of FLVCR1 in human SS cells suggested
that FLVCR1 may have a key role in the development and progression
of SS. To examine its roles, lentivirus-mediated knockdown was used
to specifically suppress FLVCR1 gene expression. MTT and colony
formation assays demonstrated that silencing of FLVCR1
significantly inhibited cell proliferation and colony formation
compared with control cells. Through flow cytometry analysis, it
was demonstrated that downregulation of FLVCR1 resulted in impaired
cell cycle progression and increased apoptosis in SW982 and
HS-SY-II cells. The results suggested that the observed
growth-promoting effect of FLVCR1 on SS cells may be mediated
through modulation of cell cycle progression. This pro-growth role
of FLVCR1 is similar to a previous study, which revealed that
FLVCR1 is important for the survival of neuroblastoma cells
(15). Notably, in vivo
studies demonstrated that FLVCR1 knockdown significantly suppressed
the growth of SS in a xenograft mouse model. Clearly, findings from
the present study have highlighted the important role of FLVCR1 in
tumor growth, indicating its potential as a novel therapeutic
target.
Previous studies demonstrated that depletion of
FLVCR1 did not affect the cell cycle of K562 leukemia cells and
Caco2 colon carcinoma cells (18,19);
however, in the current study, FLVCR1 silencing impaired cell cycle
progression of SS cell lines. A number of factor may account for
the differences in the effects on cell cycle arrest in the current
study compared with previous studies: i) The requirement for FLVCR1
in cell cycle progression may vary in different cancer cell lines
due to differences in mechanisms to bypass cell cycle arrest; ii)
the G1 arrest observed in K562 and Caco2 cells with FLVCR1
knockdown, occurred when cells were concurrently treated with
sodium butyrate (18,19), raising the possibility that the
arrest may be an indirect effect; and iii) variations in the degree
of FLVCR1 knockdown may result in elaboration of different cell
cycle phenotypes. For example, such differences have been observed
with DNA methyltransferase 1 (DNMT1) depletion, where different
levels of DNMT1 loss result in varying cell cycle effects (27).
Apoptosis and autophagy are two key mechanisms
controlling cell survival and cell death (21,28).
The important role of apoptosis (the first known programmed cell
death mechanism) in the pathogenesis of various diseases has been
well demonstrated. As an important homeostatic mechanism, autophagy
removes unnecessary or damaged proteins and dysfunctional cellular
organelles, and recycles cytoplasmic contents in all living cells
(29). Autophagy initially was
described as a cell survival mechanism in response to stress
(30), and in certain
circumstances, autophagy may contribute to cell survival by
averting apoptosis (31).
Autophagy is closely associated with the apoptosis induced by
hypoxia (32,33). It has previously been reported that
hypoxia modulates FLVCR1 gene expression via key transcription
regulatory proteins, including HIF2α and ETS1 (13). Additionally, FLVCR1 has a vital
role in the survival of committed erythroid progenitors and
neuroblastoma cells (10,15). Based on these findings and the
results of the current study in SS cells, we hypothesized that
FLVCR1 may affect survival of SS cells via regulation of autophagy.
Indeed, to the best of out knowledge, the present study was the
first to demonstrate that knocking down FLVCR1 expression induces
autophagy of SS cells in vitro and in vivo. Thus, the
results demonstrated that the inhibition of growth in SS cells by
FLVCR1 depletion may be, at least in part, due to the induction of
autophagy.
Notably, the finding of the current study regarding
autophagy are in contrast to what was reported by Petrillo et
al (34), where western blot
analysis exhibited no elevated LC3-II levels in CD31+
FLVCR1-knockout endothelial cells compared with FLVCR1-null
endothelial cells. SS is a heterogeneous tumor, which exhibits
different morphological and phenotypic profiles, including
variation in cellular morphology, gene expression, metabolism,
motility, proliferation and metastatic potential. Thus, the
observed difference between the data of the current study and the
previous study is potentially attributable to differences in the
cell lines used. In addition, the effect of autophagy varies in
different diseases, even in different time periods of one specific
disease model (35).
Cancer development and progression is a complex
process that involves a host of functional and genetic
abnormalities, with various signaling pathways, including Wnt, Akt,
SRC, MAPK, IGF-1R and TGF-β signaling, implicated in the
development of SS (2,7,23,36).
Whether FLVCR1 expression can affect these signaling pathways is
largely unknown. Thus, the association between FLVCR1 and these
signaling pathways in SS cell proliferation and metastasis required
further investigation.
Use of the Basic Local Alignment Search Tool
(blast.ncbi.nlm.nih.gov/Blast.cgi) demonstrated that the portion of
FLVCR1 mRNA targeted by shRNA in the current study is the common
area of FLVCR1b and FLVCR1a. Thus, it is unclear which isoform of
FLVCR1 was involved in promoting the proliferation and
tumorigenicity of SS. Additionally, only the silencing effects of
FLVCR1 were investigated in the present study; the effects of
FLVCR1 overexpression on the tumorigenicity of SS will be addressed
in future experiments.
In conclusion, the findings of the current study
demonstrated that silencing of FLVCR1 expression by shRNA knockdown
significantly inhibited the proliferation and tumorigenicity of SS
cells in vitro and in vivo. In addition, FLVCR1 may
affect survival of SS cells via regulation of autophagy, although
the underlying molecular mechanism requires further investigation.
The novel role of FLVCR1 in promoting tumorigenicity of SS cells
suggests that it may be a potential target gene for treatment of
patients with SS.
Acknowledgments
Not applicable.
Notes
[1]
Funding
This study was funded by the National Natural
Science Foundation of China (grant no. 81301727) and the Youth
Foundation of the Second Hospital of Shandong University (grant no.
2018YT28).
[2] Availability
of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
CLP and YS conceived and designed the study. XLL,
FW, DJW, SZM, and XWW performed the experiments. WC, XYW, and CZG
analyzed and interpreted the data. CLP and YS wrote the manuscript.
All authors read and approved the final manuscript.
[4] Ethics
approval and consent to participate
Samples were obtained with patients' informed
consent. All research involving human participants was been
approved by the Second Hospital of Shandong University ethics
committees (Jinan, China) and in accordance with the Declaration of
Helsinki. All experimental procedures using animals were approved
by the Institutional Animal Care and Use Committee of Shandong
University (Jinan, China).
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Haldar M, Hancock JD, Coffin CM, Lessnick
SL and Capecchi MR: A conditional mouse model of synovial sarcoma:
Insights into a myogenic origin. Cancer Cell. 11:375–388. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Trautmann M, Sievers E, Aretz S, Kindler
D, Michels S, Friedrichs N, Renner M, Kirfel J, Steiner S, Huss S,
et al: SS18-SSX fusion protein-induced Wnt/β-catenin signaling is a
therapeutic target in synovial sarcoma. Oncogene. 33:5006–5016.
2014. View Article : Google Scholar
|
|
3
|
Herzog CE: Overview of sarcomas in the
adolescent and young adult population. J Pediatr Hematol Oncol.
27:215–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lewis JJ, Antonescu CR, Leung DH, Blumberg
D, Healey JH, Woodruff JM and Brennan MF: Synovial sarcoma: A
multivariate analysis of prognostic factors in 112 patients with
primary localized tumors of the extremity. J Clin Oncol.
18:2087–2094. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ladanyi M, Antonescu CR, Leung DH,
Woodruff JM, Kawai A, Healey JH, Brennan MF, Bridge JA, Neff JR,
Barr FG, et al: Impact of SYT-SSX fusion type on the clinical
behavior of synovial sarcoma: A multi-institutional retrospective
study of 243 patients. Cancer Res. 62:135–140. 2002.PubMed/NCBI
|
|
6
|
Palmerini E, Staals EL, Alberghini M,
Zanella L, Ferrari C, Benassi MS, Picci P, Mercuri M, Bacci G and
Ferrari S: Synovial sarcoma: Retrospective analysis of 250 patients
treated at a single institution. Cancer. 115:2988–2998. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peng C, Zhao H, Chen W, Song Y, Wang X, Li
J, Qiao Y, Wu D, Ma S, Wang X, et al: Identification of SHCBP1 as a
novel downstream target gene of SS18-SSX1 and its functional
analysis in progression of synovial sarcoma. Oncotarget.
7:66822–66834. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qi Y, Wang N, He Y, Zhang J, Zou H, Zhang
W, Gu W, Huang Y, Lian X, Hu J, et al: Transforming growth
factor-β1 signaling promotes epithelial-mesenchymal transition-like
phenomena, cell motility, and cell invasion in synovial sarcoma
cells. PLoS One. 12:e01826802017. View Article : Google Scholar
|
|
9
|
Przybyl J, Jurkowska M, Rutkowski P,
Debiec-Rychter M and Siedlecki JA: Downstream and intermediate
interactions of synovial sarcoma-associated fusion oncoproteins and
their implication for targeted therapy. Sarcoma. 2012:2492192012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chiabrando D, Marro S, Mercurio S, Giorgi
C, Petrillo S, Vinchi F, Fiorito V, Fagoonee S, Camporeale A, Turco
E, et al: The mitochondrial heme exporter FLVCR1b mediates
erythroid differentiation. J Clin Invest. 122:4569–4579. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Keel SB, Doty RT, Yang Z, Quigley JG, Chen
J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, et
al: A heme export protein is required for red blood cell
differentiation and iron homeostasis. Science. 319:825–828. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Quigley JG, Yang Z, Worthington MT,
Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL and
Abkowitz JL: Identification of a human heme exporter that is
essential for erythropoiesis. Cell. 118:757–766. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fiorito V, Neri F, Pala V, Silengo L,
Oliviero S, Altruda F and Tolosano E: Hypoxia controls Flvcr1 gene
expression in Caco2 cells through HIF2α and ETS1. Biochim Biophys
Acta. 1839:259–264. 2014. View Article : Google Scholar
|
|
14
|
Mercurio S, Petrillo S, Chiabrando D,
Bassi ZI, Gays D, Camporeale A, Vacaru A, Miniscalco B, Valperga G,
Silengo L, et al: The heme exporter Flvcr1 regulates expansion and
differentiation of committed erythroid progenitors by controlling
intracellular heme accumulation. Haematologica. 100:720–729. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chiabrando D, Castori M, di Rocco M,
Ungelenk M, Giesselmann S, Di Capua M, Madeo A, Grammatico P,
Bartsch S, Hübner CA, et al: Mutations in the heme exporter FLVCR1
cause sensory neurodegeneration with loss of pain perception. PLoS
Genet. 12:e10064612016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rajadhyaksha AM, Elemento O, Puffenberger
EG, Schierberl KC, Xiang JZ, Putorti ML, Berciano J, Poulin C,
Brais B, Michaelides M, et al: Mutations in FLVCR1 cause posterior
column ataxia and retinitis pigmentosa. Am J Hum Genet. 87:643–654.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vinchi F, Ingoglia G, Chiabrando D,
Mercurio S, Turco E, Silengo L, Altruda F and Tolosano E: Heme
exporter FLVCR1a regulates heme synthesis and degradation and
controls activity of cytochromes P450. Gastroenterology.
146:1325–1338. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fiorito V, Forni M, Silengo L, Altruda F
and Tolosano E: Crucial role of FLVCR1a in the maintenance of
intestinal heme homeostasis. Antioxid Redox Signal. 23:1410–1423.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mercurio S, Aspesi A, Silengo L, Altruda
F, Dianzani I and Chiabrando D: Alteration of heme metabolism in a
cellular model of Diamond-Blackfan anemia. Eur J Haematol.
96:367–374. 2016. View Article : Google Scholar
|
|
20
|
Zeng Z, Lin H, Zhao X, Liu G, Wang X, Xu
R, Chen K, Li J and Song L: Overexpression of GOLPH3 promotes
proliferation and tumorigenicity in breast cancer via suppression
of the FOXO1 transcription factor. Clin Cancer Res. 18:4059–4069.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jiao G, Guo W, Ren T, Lu Q, Sun Y, Liang
W, Ren C, Yang K and Sun K: BMPR2 inhibition induced apoptosis and
autophagy via destabilization of XIAP in human chondrosarcoma
cells. Cell Death Dis. 5:e15712014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Peng C, Zhao H, Song Y, Chen W, Wang X,
Liu X, Zhang C, Zhao J, Li J, Cheng G, et al: SHCBP1 promotes
synovial sarcoma cell metastasis via targeting TGF-β1/Smad
signaling pathway and is associated with poor prognosis. J Exp Clin
Cancer Res. 36:1412017. View Article : Google Scholar
|
|
24
|
Han RL, Wang FP, Zhang PA, Zhou XY and Li
Y: miR-383 inhibits ovarian cancer cell proliferation, invasion and
aerobic glycolysis by targeting LDHA. Neoplasma. 64:244–252. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Peng C, Guo W, Yang Y and Zhao H:
Downregulation of SS18-SSX1 expression by small interfering RNA
inhibits growth and induces apoptosis in human synovial sarcoma
cell line HS-SY-II in vitro. Eur J Cancer Prev. 17:392–398. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang S, Liu H, Ren L, Pan Y and Zhang Y:
Inhibiting colorectal carcinoma growth and metastasis by blocking
the expression of VEGF using RNA interference. Neoplasia.
10:399–407. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brown KD and Robertson KD: DNMT1 knockout
delivers a strong blow to genome stability and cell viability. Nat
Genet. 39:289–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun Y, Guo W, Ren T, Liang W, Zhou W, Lu
Q, Jiao G and Yan T: Gli1 inhibition suppressed cell growth and
cell cycle progression and induced apoptosis as well as autophagy
depending on ERK1/2 activity in human chondrosarcoma cells. Cell
Death Dis. 5:e9792014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vangamudi B, Paul TA, Shah PK,
Kost-Alimova M, Nottebaum L, Shi X, Zhan Y, Leo E, Mahadeshwar HS,
Protopopov A, et al: The SMARCA2/4 ATPase domain surpasses the
bromodomain as a drug target in SWI/SNF-mutant cancers: Insights
from cDNA rescue and PFI-3 inhibitor studies. Cancer Res.
75:3865–3878. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fang Y, Tan J and Zhang Q: Signaling
pathways and mechanisms of hypoxia-induced autophagy in the animal
cells. Cell Biol Int. 39:891–898. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Petrillo S, Chiabrando D, Genova T,
Fiorito V, Ingoglia G, Vinchi F, Mussano F, Carossa S, Silengo L,
Altruda F, et al: Heme accumulation in endothelial cells impairs
angiogenesis by triggering paraptosis. Cell Death Differ. Dec
11–2017.Epub ahead of print. PubMed/NCBI
|
|
35
|
Wang L, Jin Z, Wang J, Chen S, Dai L, Lin
D, Wu L and Gao W: Detrimental effect of hypoxia-inducible
factor-1α-induced autophagy on multiterritory perforator flap
survival in rats. Sci Rep. 7:117912017. View Article : Google Scholar
|
|
36
|
Michels S, Trautmann M, Sievers E, Kindler
D, Huss S, Renner M, Friedrichs N, Kirfel J, Steiner S, Endl E, et
al: SRC signaling is crucial in the growth of synovial sarcoma
cells. Cancer Res. 73:2518–2528. 2013. View Article : Google Scholar : PubMed/NCBI
|