Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the
fourth leading cause of cancer-related mortality worldwide,
although its incidence is lower compared with that of other types
(1). In addition to its aggressive
nature, the late onset of symptoms, failure to respond to
systematic therapy, and its radio and chemoresistance, contribute
to a 5-year survival rate of only 5% among pancreatic cancer (PC)
patients (2). In addition to
surgery, radiotherapy is a standard treatment used for PDAC.
However, PDAC cells inevitably develop resistance to radiotherapy.
Although extensive investigations have been performed on the
mechanisms underlying the radioresistance of cancers and the
multiple signaling pathways involved, such as the suppressed
P53-mediated apoptosis pathway, the pro-survival cytokine and
growth factor-mediated activation of the signal transducer and
activator of transcription (Stat)3 and nuclear factor (NF)-κB
pathways, and the impaired DNA damage repair pathway (3,4), the
molecular mechanisms that antagonize these pathways have not been
fully elucidated in PC cells. Therefore, it is important to explore
the mechanisms underlying cancer cell radiosensitivity in order to
overcome the radioresistance of PDAC and develop safe, effective
regimens.
In the clinical setting, radiotherapy is one of the
main adjuvant treatments for cancer and it is frequently applied to
patients with PC. Ionizing radiation (IR)-induced apoptosis is
considered to be one of the major cell death responses following
exposure to irradiation, including X-rays or γ-rays. After
irradiation, the DNA damage response cascade is activated, a number
of transcription factors, such as P53, are activated, and the DNA
repair process is impaired, with subsequent cell cycle arrest,
senescence and/or apoptosis (5).
While the P53-mediated apoptotic pathway is initiated,
radiation-induced cell cycle arrest, failure to repair damaged DNA
and inactivation of pro-survival pathways promote cell death
(6).
B-cell lymphoma (Bcl)-2 and related family members
are key regulators of mitochondrial-related apoptosis (7). This family of proteins is divided
into two subfamilies, the pro-apoptotic proteins and anti-apoptotic
proteins. The pro-apoptotic Bcl-2 family proteins, such as
Bcl-2-associated X (BAX), Bcl-2 homologous antagonist/killer (BAK),
Bcl-2-like protein 11 (Bim) and p53-upregulated modulator of
apoptosis (PUMA), serve critical roles in the P53-mediated
apoptotic pathway, which can be inhibited by inhibitor of apoptosis
proteins (IAPs), such as survivin (8). By contrast, the anti-apoptotic Bcl-2
family proteins, such as Bcl-2, Bcl-extra large (Bcl-xL) and
myeloid cell leukemia sequence-1 (Mcl-1), are associated with the
outer mitochondrial membrane, antagonize pro-apoptotic proteins and
protect cells from programmed cell death (PCD) (9). P53 can mediate transcriptional
repression of anti-apoptotic genes, including the Bcl-2 gene and
the IAP family member survivin (10,11).
Eventually, the caspase family becomes involved in the apoptotic
process, during which caspase-3 is activated by caspase-9, and then
executes the apoptosis (12).
Caspases exist in the cell as zymogens and are activated when the
cell encounters external or internal stimuli. The IR-induced
caspase cascade may also inactivate the poly(ADP-ribose) polymerase
(PARP), an enzyme critical for repairing damaged DNA, in order to
block DNA repair, thereby promoting IR-induced apoptotic cell
death. Indeed, both PARP-1 and PARP-2 knockout mice exhibit severe
deficiencies in DNA repair, showing increased sensitivity to
alkylating agents or IR (13).
The Stat3, a member of the Stat family, is a
transcription factor that transmits pro-survival signals from the
surface of the cell to the nucleus, playing a key role in the
development of human cancers (14). Cytokines, such as interleukin
(IL)-6, may activate Stat3 through tyrosine phosphorylation to
transactivate anti-apoptotic regulators, such as Bcl-2, Bcl-xL, and
other apoptosis-related genes (15). The NF-κB is a ubiquitous
transcription factor that is associated with inflammatory and
innate immune responses (16).
NF-ĸB is constitutively activated by various stimuli, including
inflammatory cytokines, such as tumor necrosis factor (TNF)-α,
IL-1β, epidermal growth factor (EGF), insulin-like growth factor
(IGF)-1, T and B-cell mitogens, bacteria and lipopolysaccharides,
viruses, viral proteins, double-stranded RNA, and physical and
chemical stress (17). Under
normal physiological conditions, by binding to the inhibitory
protein IĸBα in the cytoplasm, NF-κB assumes an inactive form
(18). Upon induction by various
stimuli, IκBα is ubiquitinated and degraded, thereby releasing
NF-ĸB to translocate from the cytoplasm to the nucleus. The
activated NF-ĸB in the nucleus regulates the transcription of a
wide range of target genes associated with cell proliferation,
survival and angiogenesis (19).
Although the Stat3 and NF-ĸB signaling pathways
described above contribute to the radioresistance of cancers, it is
not clear how they are overridden by the P53-mediated pathways in
radiosensitive cancer cells. In the present study, spontaneous
radiosensitive and radioresistant PC cell lines were screened and
their potential signaling pathways mediating radiosensitivity were
investigated following irradiation. The current study aimed to
provide a novel insight into the mechanisms underlying the
radiosensitivity of PC cells, particularly the unique functions of
adaptively expressed EGF and cyclin D1.
Materials and methods
Cell culture and reagents
The human PC cells SW1990, Capan-2, PANC-1, AsPC-1,
BxPC-3 and CFPAC-1 were obtained from the Type Culture Collection
of the Chinese Academy of Sciences (Shanghai, China). The cells
were grown in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (FBS) and maintained in a
humidified 5% CO2 atmosphere at 37°C. DMEM and FBS were
obtained from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). The Annexin V-fluorescein isothiocyanate (FITC) Apoptosis
Detection kit was purchased from BD Biosciences (San Jose, CA,
USA). Recombinant human EGF was purchased from PeproTech, Inc.
(Rocky Hill, NJ, USA). TRIzol reagent was purchased from Invitrogen
(Thermo Fisher Scientific, Inc.). Cell Counting Kit-8 (CCK-8) was
purchased from Dojindo Molecular Technologies, Inc. (Kumamoto,
Japan).
Clonogenic assay
PC cells at various concentrations were plated into
6-well plates (Corning Inc., Corning, NY, USA), according to the
dose of irradiation, and cultured for 24 h. After irradiation by
X-rays at 0, 2, 4, 6, 8 and 10 Gy, the cells were cultured for 2
weeks at 37°C. The cells were washed three times with PBS, fixed
with ice-cold methanol for 15 min, stained with 1% crystal violet
solution (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 15
min, and rinsed in distilled water to remove the excess dye. The
plates were allowed to dry prior to scanning. Only colonies of ≥50
cells were counted. Triplicate experiments were performed
independently. The surviving fractions were determined as ratios of
the plating efficiencies (PE = counted colonies/seeded cells ×100)
of the irradiated cells to the non-irradiated cells. The cell
survival curves were fitted with the linear-quadratic equation of
SF=exp [−(αD+βD2)] by optimizing variable parameters α and β.
X-ray irradiation
Human PC cell lines were irradiated by a linear
accelerator (Elekta Medical Systems, Stockholm, Sweden) with 8-MV
X-rays at a dose rate of 500 cGy/min, the cells were further
incubated for different time periods, and then harvested for the
subsequent experiments.
Cell proliferation assay
Cells were seeded into 96-well culture plates at a
density of 5,000 cells/well and allowed to adhere for 24 h. After
X-ray irradiation, the cells were incubated for different times in
a humidified chamber at 37°C. Each day for 3 consecutive days,
viable cells were evaluated with the CCK-8 assay, according to the
manufacturer’s instructions. CCK-8 solution was added to the cells
in 96-well plates incubated at 37°C for an additional 1 h, and the
absorbance at 450 nm was determined using a microplate reader
(ELX800; Bio-Tek Instruments, Inc., Winooski, VT, USA).
Flow cytometric detection of the cell
cycle
PC cells were harvested by trypsin. After
centrifugation, cells were washed twice with PBS and fixed with
ethanol for 1 h at −20°C. After washing twice with PBS, the cells
were stained with a solution containing 5 mg/ml propidium iodide
(PI) and 1 mg/ml RNase A (Sigma Aldrich; Merck KGaA) at 4°C for 30
min. The cell-cycle distribution was examined by flow cytometry (BD
Biosciences) and the proportion of cells in the G0-G1, S and G2-M
phases was determined. Cell cycle analysis was performed using
FlowJo 7.6.1 software (FlowJo LLC, Ashland, OR, USA).
Flow cytometric detection of
apoptosis
Cells were cultured in growth medium for 12 h at a
density of 2×105 cells per well in 6-well plates, and
irradiated at the indicated doses. Apoptotic cells were quantified
using an Annexin V-FITC/PI Apoptosis Detection kit and FACSCalibur
flow cytometry (BD Biosciences). The cells were harvested by
centrifugation after irradiation and washed twice with PBS. The
cells were then resuspended in 100 µl of Annexin V binding
buffer, incubated with 5 µl of Annexin V-FITC for 15 min at
room temperature, and counterstained with PI (final concentration 1
µg/ml). After the incubation period, the cells were diluted
with 190 µl of Annexin V binding buffer. A total of 10,000
counts were acquired per sample, and examined by flow cytometry (BD
Biosciences). Cells in the early stages of apoptosis were Annexin
V-positive, whereas cells that were Annexin V-positive and
PI-positive were in the late stages of apoptosis.
Nuclear protein extraction
Nuclear protein extracts were obtained using the
nuclear and cytoplasmic extraction kit (Thermo Fisher Scientific,
Inc.). Cells were harvested with trypsin and then centrifuged at
500 × g for 5 min at 4°C. Cells were resuspended with PBS and
centrifuged at 500 × g for 5 min at 4°C. The supernatant was
discarded, followed by addition of ice-cold CER I solution. The
tube was vortexed vigorously for 15 sec and incubated on ice for 10
min. Then, CER II solution was added, followed by vortexing for 5
sec and incubation for 1 min. After centrifugation at 16,000 × g
for 5 min at 4°C, the supernatant (cytoplasmic extract) was
collected and the insoluble fraction was washed with PBS. Finally,
NER solution was added to the tube and vortexed for 15 sec four
times, followed by centrifugation at 16,000 × g for 10 min at 4°C
and collection of the supernatant (nuclear extract).
Western blotting
The treated cells were collected and washed twice
with cold PBS. The cells were lysed in 200 µl RIPA buffer
(cat. no. 89900; Thermo Fisher Scientific, Inc.) and the lysates
were incubated on ice for 30 min, vortexed and centrifuged at
14,000 × g for 15 min at 4°C. The supernatant was collected and
protein concentration was determined using the Bradford assay.
After addition of sample loading buffer, protein samples (20
µg) were electrophoresed on a 10% SDS-polyacrylamide gel and
then transferred to PVDF membranes (Millipore Corporation,
Billerica, MA, USA). After blocking for 4 h in a solution of 5%
non-fat dry milk in Tris-buffered saline containing 0.1% Tween-20
(TBST) at room temperature for 1.5 h, the membranes were incubated
overnight at 4°C with primary antibodies (1:1,000 dilution). The
antibodies were: p53 (cat. no. sc-126; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), PUMA (cat. no. 4976), BAX (cat. no. 5023),
BAK (cat. no. 6947), Bcl-xL (cat. no. 2764), Bcl-2 (cat. no. 2870),
PARP (cat. no. 9532), phosphorylated (p-) Histone H2AX (cat. no.
9718S), Survivin (cat. no. 2808), cyclin D1 (cat. no. 2978), p-EGF
receptor (EGFR; cat. no. 3777), NF-ĸB p65 (cat. no. 8242), p-NF-ĸB
p65 (cat. no. 3033), cleaved caspase-3 (cat. no. 9664), Stat3 (4
cat. no. 904), p-Stat3 (cat. no. 9145), β-actin (cat. no. 4970)
(all from Cell Signaling Technology, Inc., Danvers, MA, USA), EGFR
(cat. no. 18986-1-AP), TATA-binding protein (TBP; cat. no.
66166-1-Ig), and GAPDH (cat. no. 60004-1-Ig) (all from ProteinTech
Group, Inc., Chicago, IL, USA). After washing four times, the
membranes were incubated with secondary anti-rabbit and anti-mouse
antibodies (cat. nos. 7074 and 7076; Cell Signaling Technology,
Inc.; 1:3,000 dilution) at room temperature for 1 h. The blots were
developed using an Immobilon Western Chemiluminescent detection
reagent (Millipore Corporation) and the results were recorded using
the ChemiDox XRS+ system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). Quantitative analysis was performed using Image Lab 6.0.1
software (National Institutes of Health, Bethesda, MD, USA).
RNA interference
Small interfering (si)RNA targeting BAX, cyclin D1
and control siRNA were purchased from GenePharma Co., Ltd.
(Shanghai, China). A total of 2×105 cells per well were
seeded in 6-well plates and incubated overnight, followed by
transfection with BAX siRNA-1 (5′-ACUUUGCCAGCAAACUGGUGCUCAA-3′ and
5′-UUGAGCACCAGUUUGCUGGCAAAGU-3′), BAX siRNA-2
(5′-ATCCAGGATCGAGCAGGGCG-3′ and 5′-GGTTCTGATCAGTTCCGGCA-3′), cyclin
D1 siRNA-1 (5′-CCCGCACGAUUUCAUUGAATT-3′ and
5′-UUCAAUGAAAUCGUGCGGGTT-3′), cyclin D1 siRNA-2
(5′-GUCUGCGAGGAACAGAAGUTT-3′ and 5′-ACUUCUGUUCCUCGCAGACTT-3′),
cyclin D1 siRNA-3 (5′-CCACAGAUGUGAAGUUCAUTT-3′ and
5′-AUGAACUUCACAUCUGUGGTT-3′), or negative control
(5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′),
using jetPRIME transfection reagent (Polyplus-transfection SA,
Illkirch, France) according to the manufacturer’s protocol. First,
110 pmole siRNA was diluted in 200 µl of jetPRIME buffer and
mixed by pipetting. Second, 4 µl jetPRIME reagent was added,
vortexed for 10 sec and spun down briefly, followed by incubation
for 10 min at room temperature. Third, the transfection mixture was
added dropwise to the cells in serum-containing medium, the plate
was gently rocked, and then returned to the incubator. After 24 h,
the transfection mixture was removed and fresh medium was added,
and the cells were further cultured for 24 h.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Cells were seeded in 6-well plates and allowed to
grow until semi-confluent prior to being irradiated in DMEM
supplemented with 10% FBS. After treatment, total RNA was extracted
using TRIzol reagent (Thermo Fisher Scientific, Inc.) according to
the manufacturers’ recommendations. cDNA was generated using equal
amounts of sample RNA with RevertAid First strand cDNA Synthesis
kit (Thermo Fisher Scientific, Inc.). Subsequently, 2 µl of
cDNA and Taq PCR Master Mix (Takara Biotechnology Co., Ltd.,
Dalian, China) was used for PCR. The primers sequences used for
qPCR were as follows: BAX, forward, AAGCTGAGCGAG TGTCTCAAG and
reverse, CAAAGTAGAAAAGGGCGAC AAC; cyclin D1, forward,
GTGTATCGAGAGGCCAAAGG and reverse, GCAACCAGAAATGCACAGAC; IGF-1,
forward, TTCAACAAGCCCACAGGGTA and reverse, GCAATACAT CTCCAGCCTCCT;
EGF, forward, GCTTCAGGACCACA ACCATT and reverse,
GGCATAAACCATTCCCATCTG; IL-6, forward, CAATGAGGAGACTTGCCTGG and
reverse, GGCATTTGTGGTTGGGTCAG; IGF-1R, forward, CCTGAA
AGGAAGCGGAGAG and reverse, GGGTCGGTGATGTTG TAGGT; EGFR, forward,
ATGCAGAAGGAGGCAAAGTG and reverse, AGGTCATCAACTCCCAAACG; IL-6R,
forward, GGTGAGAAGCAGAGGAAGGA and reverse, TGGGAG GTGGAGAAGAGAGA;
and GAPDH, forward ATGACAT CAAGAAGGTGGTG and reverse,
CATACCAGGAAATG AGCTTG. PCR amplifications were performed as
follows: 5 min at 94°C, followed by 25 cycles of 30 sec at 94°C, 30
sec at 55°C, 30 sec at 72°C, and a final extension step at 72°C for
5 min. qPCR was performed using the StepOnePlus Real-Time PCR
System (ABI, Applied Biosystems; Thermo Fisher Scientific, Inc.).
Relative fold changes in mRNA expression were calculated using the
formula 2−ΔΔCq.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA,
USA). Each experiment was performed three times. All data are
expressed as mean ± standard error of the mean, unless otherwise
specified. Comparison of data between two groups was performed
using a two-tailed Student’s t-test. Multiple comparisons were
assessed by one-way analysis of variance with Bonferroni’s post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Identification of radiosensitive and
radioresistant PC cells
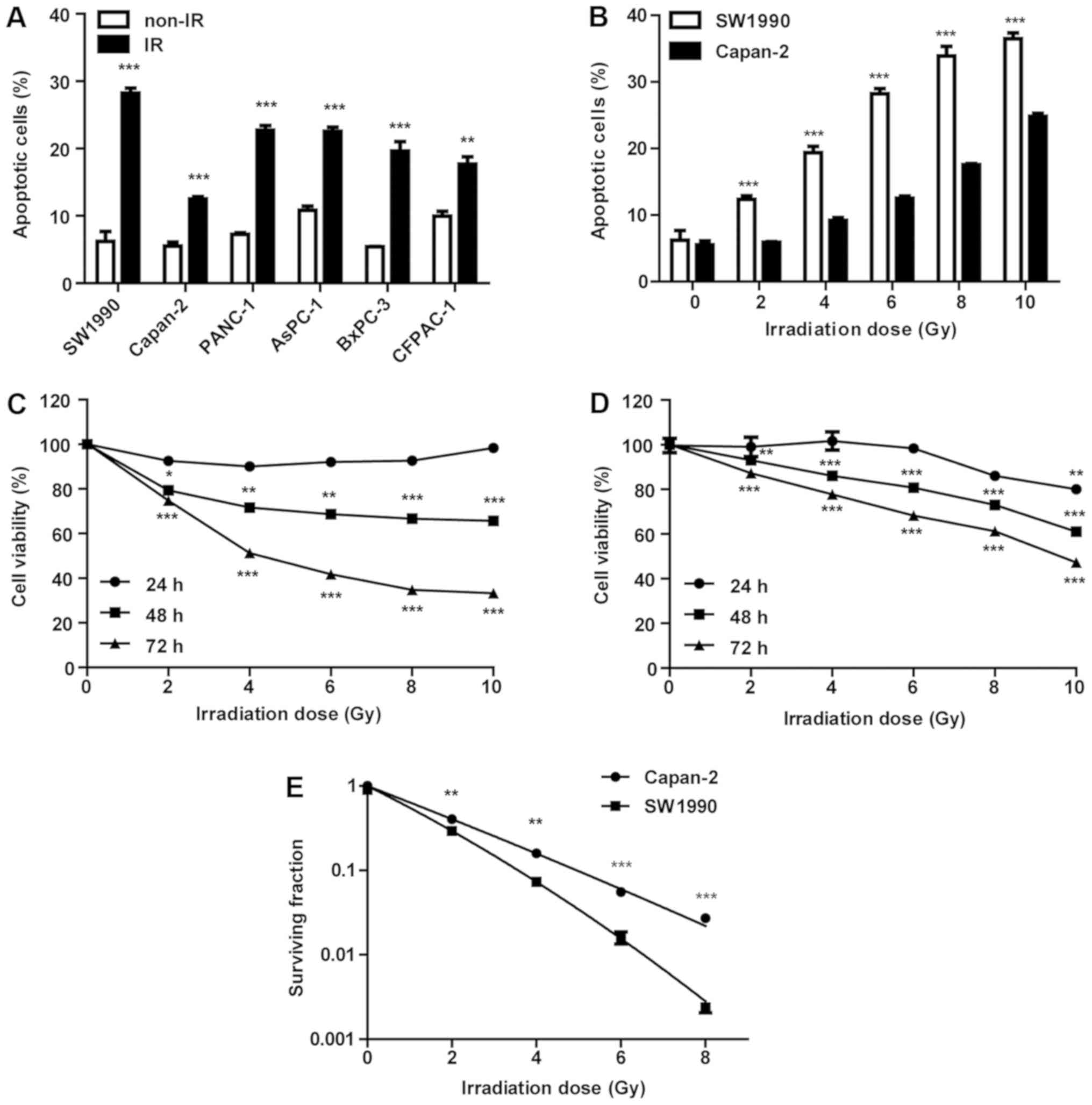
To evaluate the radiosensitivity of PC cells, six
human PC cell lines (SW1990, Capan-2, PANC-1, AsPC-1, BxPC-3 and
CFPAC-1) were irradiated with 6 Gy, and apoptotic cell rates were
measured by flow cytometry 72 h post-IR. As illustrated in Fig. 1A, the SW1990 cell line was most
sensitive to IR, whereas the Capan-2 cell line was the most
resistant. The PANC-1, AsPC-1, BxPC-3 and CFPAC-1 cell lines
exhibited comparable but relatively mild sensitivity to IR. Thus,
SW1990 and Capan-2 cells were irradiated with various doses (2, 4,
6, 8 and 10 Gy) and their viability was measured at 24, 48 and 72 h
post-IR (Fig. 1C and D). Within 24
h post-IR, the viability of SW1990 and Capan-2 cells was not
significantly affected. However, cell viability was significantly
decreased in a dose-dependent manner at 24 h post-IR (P<0.001).
At 72 h post-IR, the viability of SW1990 cells was decreased more
obviously compared with Capan-2 cells (Fig. 1C and D), confirming that SW1990 was
the most sensitive cell line and Capan-2 was the most resistant
cell line to IR. While SW1990 cells had a rate of spontaneous
apoptosis comparable with Capan-2 cells in the culture prior to IR,
their apoptosis rate increased ~2-fold that of Capan-2 cells
post-IR (Fig. 1B), suggesting a
differential adaptive response of these cells to IR. Clonogenic
formation assay was performed to examine its association with
radiosensitivity. The survival fraction results revealed that
SW1990 cells were more radiosensitive compared with Capan-2 cells
post-IR (P<0.001; Fig. 1E).
Therefore, SW1990 cells were identified as radiosensitive and
Capan-2 cells as radioresistant, and were used as a cell model to
investigate the molecular mechanisms underlying the adaptive
radiosensitivity of PC cells in the present study.
Activation of the P53-mediated apoptotic
pathway is more pronounced in radiosensitive compared with
radioresistant PC cells post-IR
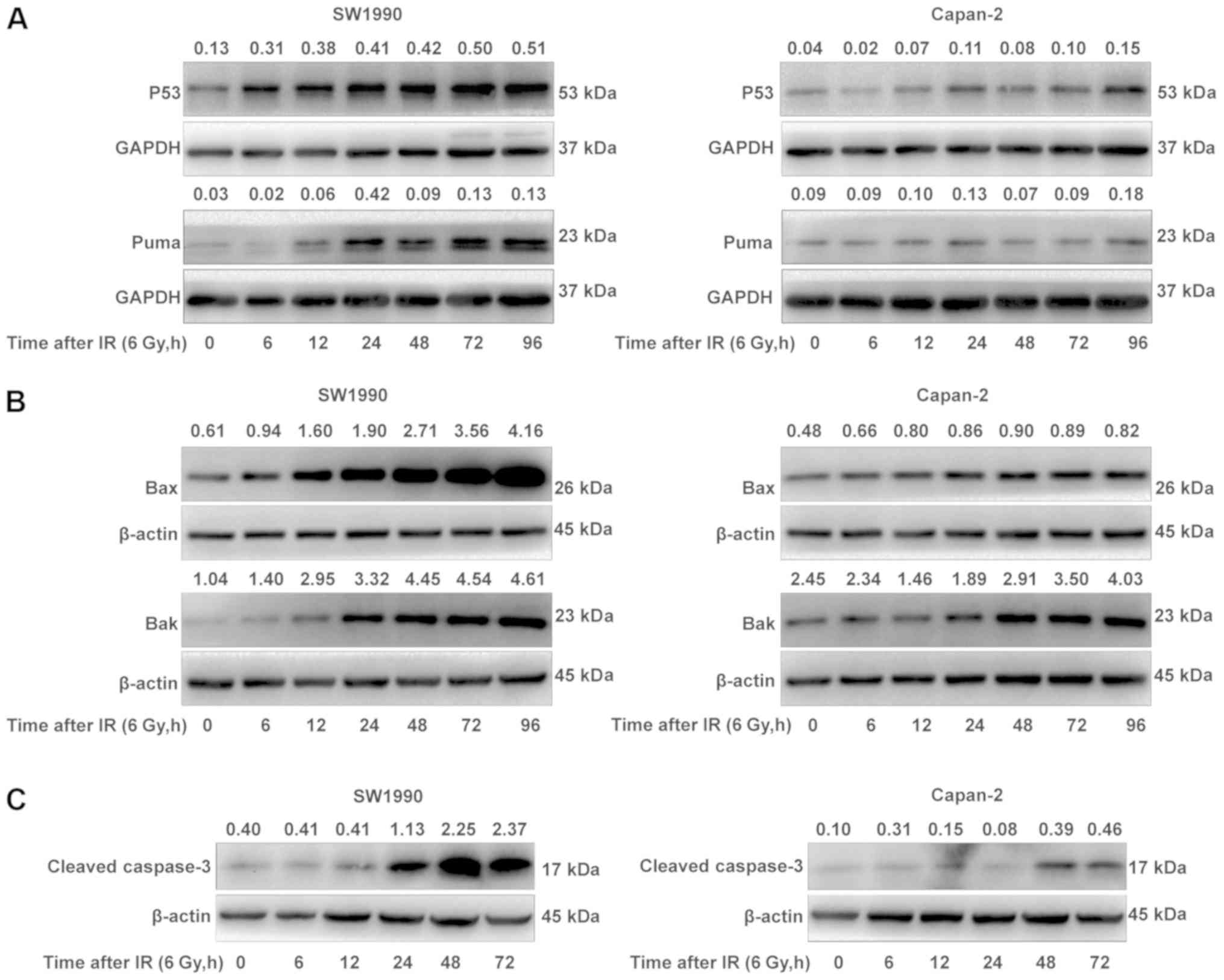
To elucidate the reason as to why SW1990 cells were
more sensitive to IR compared with Capan-2 cells, the adaptive
expression of the P53 protein in SW1990 cells and Capan-2 cells was
examined following IR treatment. P53 is a major sensor of DNA
damage mediating IR-induced cell death. While the basal levels of
P53 protein were comparable between SW1990 and Capan-2 cells prior
to IR, the P53 protein levels in the SW1990 cells were upregulated
to a markedly higher extent compared with Capan-2 cells between 6
and 96 h post-IR (Fig. 2A).
Consequently, PUMA, a protein downstream of P53 activation
(20), was also upregulated in a
similar manner in SW1990 cells post-IR (Fig. 2A). Additionally, the expression of
BAX and BAK, two proapoptotic proteins that interact with PUMA to
mediate the mitochondrial apoptosis pathway (21), was also upregulated, with kinetics
similar to that of P53 and PUMA (Fig.
2B). By contrast, P53, PUMA, BAX and BAK were less or not
upregulated in Capan-2 cells post-IR (Fig. 2A and B). As a consequence,
caspase-3, an executor of cell apoptosis, was also more markedly
activated/cleaved in the radiosensitive SW1990 cells compared with
the radioresistant Capan-2 cells, starting from 24 h post-IR
(Fig. 2C). These results indicated
that the P53-mediated mitochondrial apoptosis pathway was more
extensively upregulated in the radiosensitive SW1990 cells compared
with the radioresistant Capan-2 cells.
P53-mediated apoptotic pathway activation
in radiosensitive PC cells is associated with inactivation of
PARP
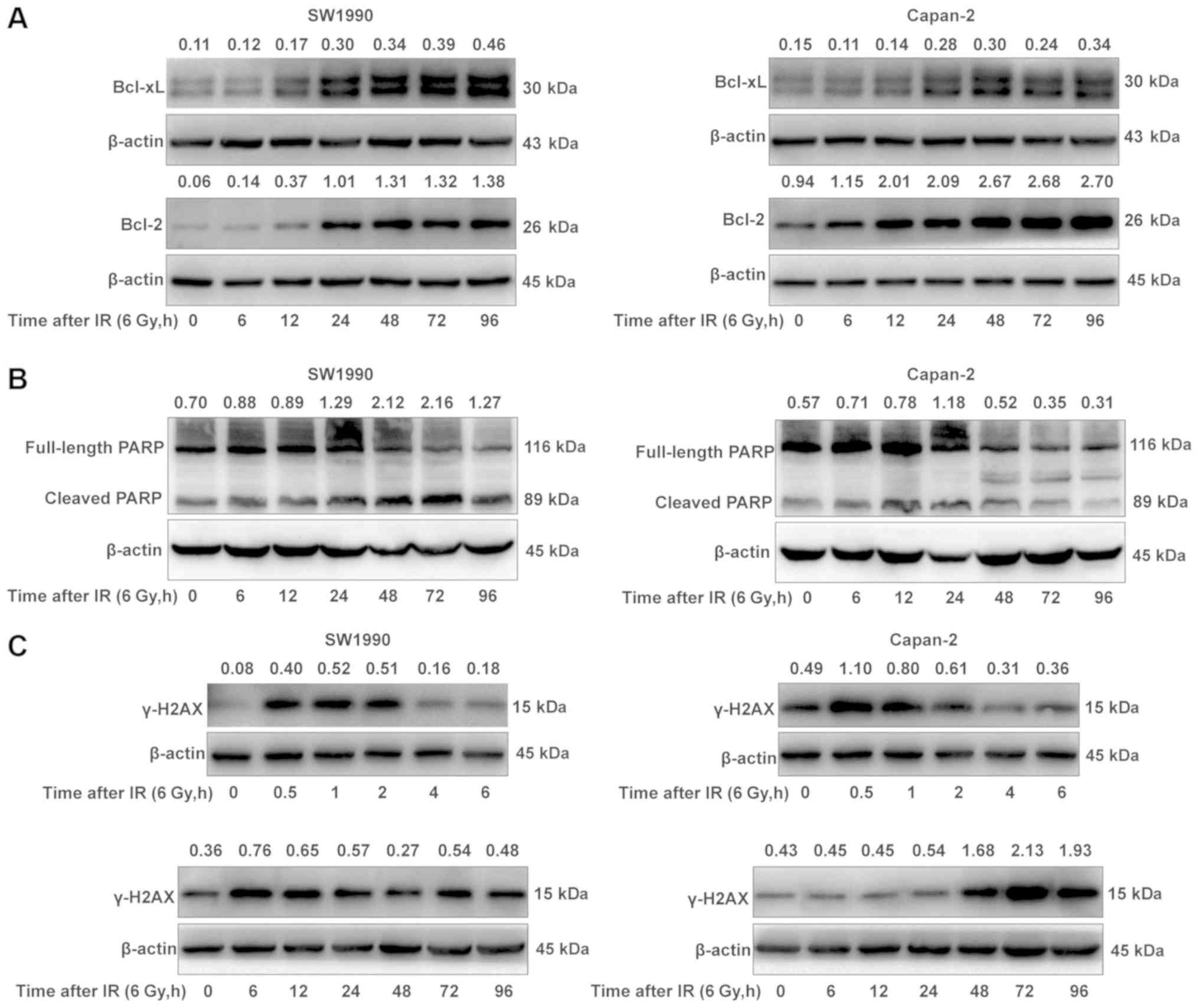
Enhanced activation of the
P53/PUMA/BAX/BAK/caspase-3 pathway is considered to be associated
with the suppressed expressions of Bcl-2 and Bcl-xL in the
radiosensitive PC cells following IR treatment. To test this
hypothesis, the adaptive expression of Bcl-2 and Bcl-xL was further
examined in SW1990 and Capan-2 cells post-IR, as Bcl-2 and Bcl-xL
can bind BAX or BAK to suppress their functions. Unexpectedly, the
expressions of Bcl-2 and Bcl-xL were not significantly decreased in
the radiosensitive SW1990 cells compared with the radioresistant
Capan-2 cells post-IR, although the basal level of Bcl-2 was higher
in the radioresistant Capan-2 cells (Fig. 3A). The results indicated that the
enhanced activation of the P53/PUMA/BAX/BAK pathway was likely not
associated with the expression of Bcl-2 and Bcl-xL in
radiosensitive PC cells, and other mechanisms may be
responsible.
It is known that IR-induced DNA damage activates
PARP to repair the damaged DNA, however activated caspase-3 may
cleave PARP to block the DNA repair, and the failure to repair the
damaged DNA may lead to cell death (22). Thus, the activation state of PARP
was examined in SW1990 cells and Capan-2 cells post-IR. As
illustrated in Fig. 3B, the
cleaved PARP levels were higher in the radiosensitive SW1990 cells
compared with the radioresistant Capan-2 cells at 72 h post-IR,
suggesting that PARP was cleaved by activated caspase-3 in SW1990
cells but not in Capan-2 cells. As a consequence, the DNA damage
repair was blocked by the cleaved PARP in SW1990 cells, resulting
in increased apoptosis or increased sensitivity to IR (Fig. 1). This hypothesis was further
confirmed by the decreased expression of γ-H2A histone family
member X (γ-H2AX), a marker of the efficiency of DNA repair
(23), in SW1990 cells at 72 h
post-IR, as compared to that in Capan-2 cells (Fig. 3C). At this time, SW1990 cells were
more susceptible to radiation-induced cell death (Fig. 1B-D). Notably, there were two peaks
of γ-H2AX expression in both radiosensitive and radioresistant PC
cells within 96 h post-IR. The first peak appeared between 0.5 and
2 h post-IR, and the second peak between 6 and 12 h or between 48
and 96 h post-IR in the radiosensitive SW1990 cells (Fig. 3C). The expression kinetics of
γ-H2AX was essentially consistent with that of cleaved PARP
(Fig. 3B). These results indicated
that the enhanced activation of the P53/PUMA/BAX/BAK/caspase-3
pathway following IR may sensitize PC cells through cleaving PARP
to block DNA damage repair.
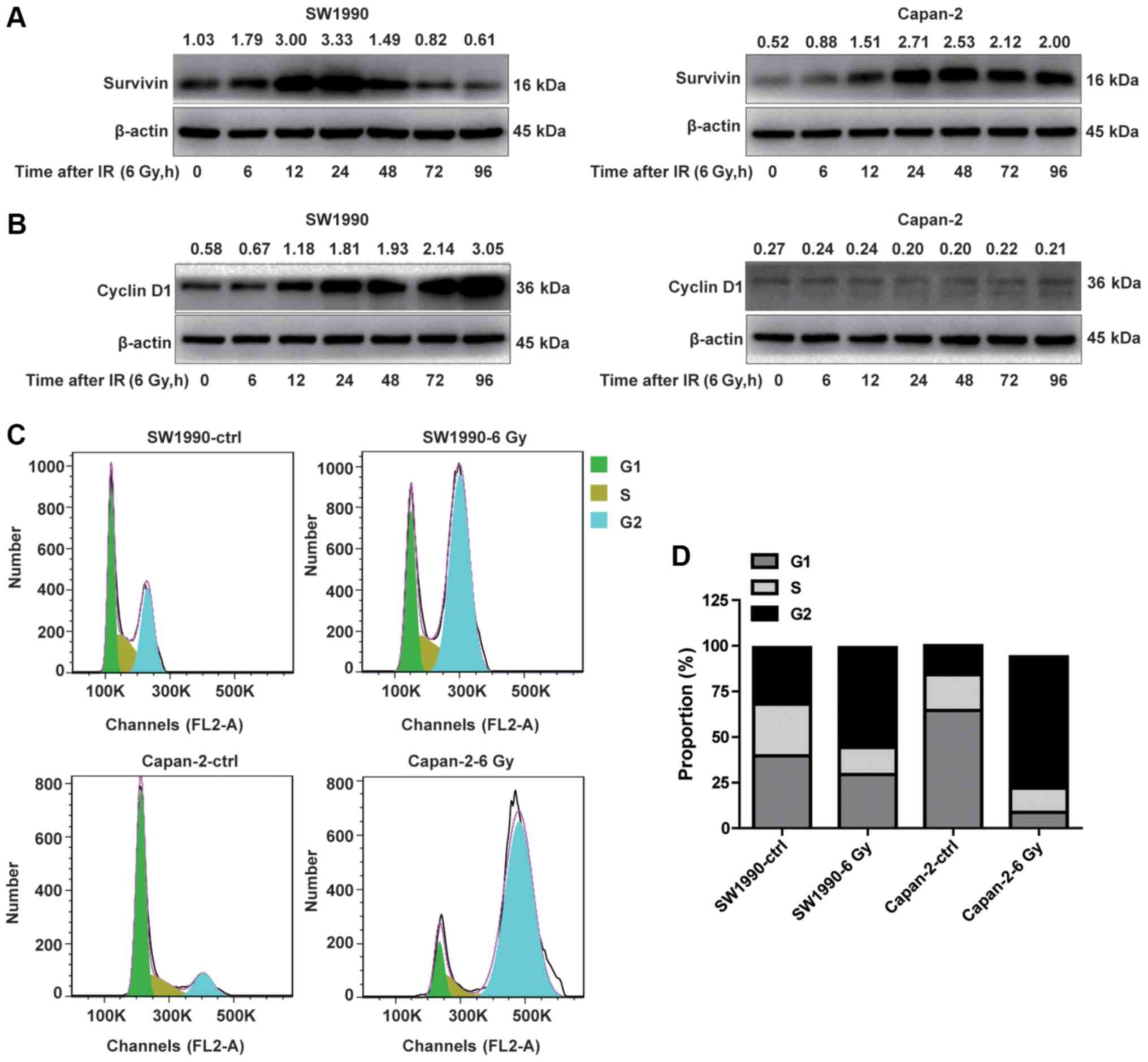
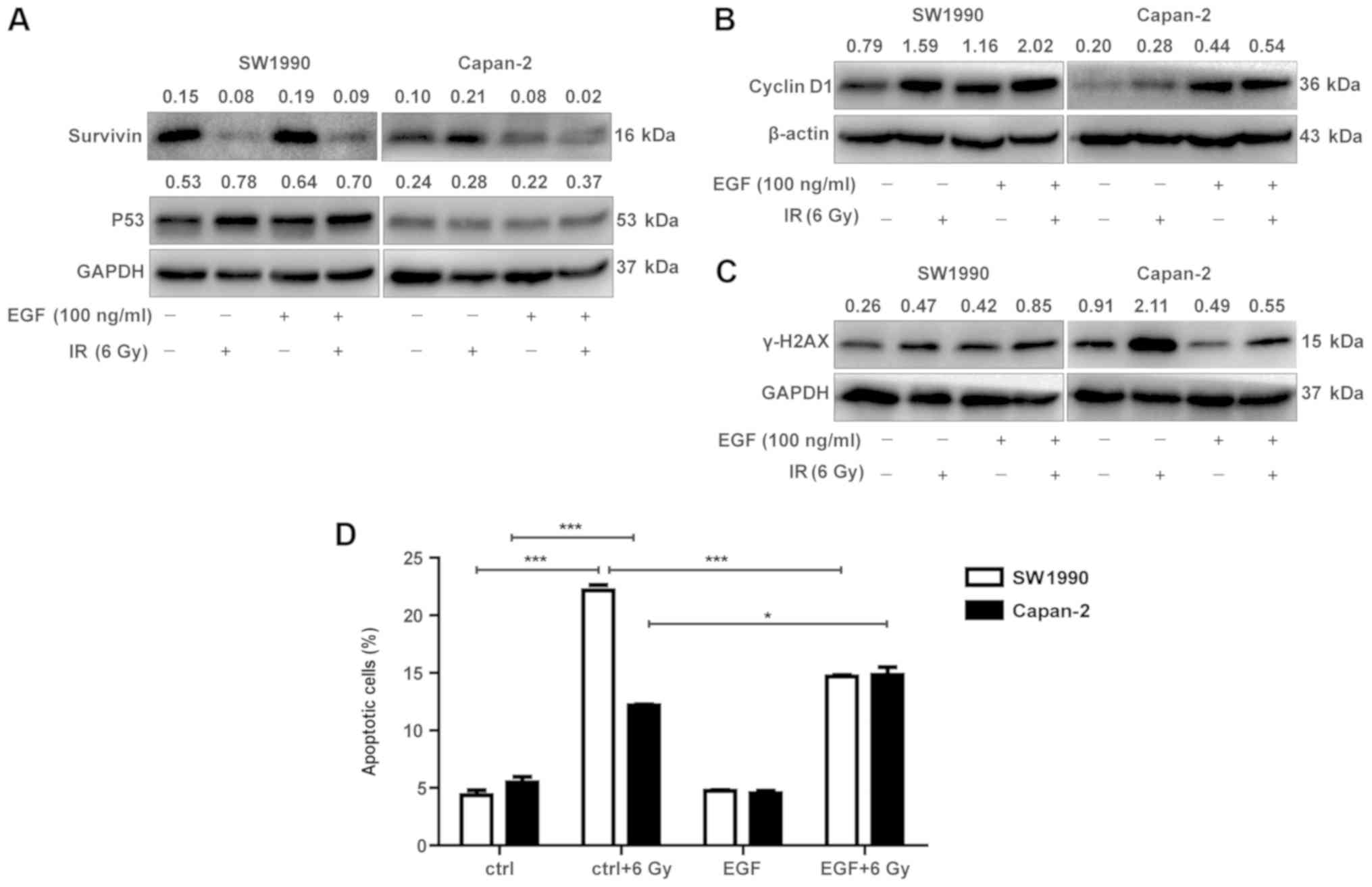
Cyclin D1 and survivin are distinctly
expressed in radiosensitive and radioresistant PC cells
To determine whether the activation of the
P53/PUMA/BAX/BAK/caspase-3 pathway in radiosensitive PC cells was
associated with the downregulation of IAPs, the adaptive expression
of survivin, a member of the IAP family, was investigated in
radioresistant and radiosensitive cancer cells. As illustrated in
Fig. 4A, survivin appeared to be
distinctively expressed in SW1990 and Capan-2 cells post-IR. While
survivin was constitutively expressed in Capan-2 cells at a higher
level compared with SW1990 cells (Fig.
4A), it was transiently upregulated in SW1990 cells between 12
and 24 h post-IR, followed by a marked downregulation starting from
48 h, resulting in levels lower than Capan-2 cells. The
downregulation of survivin was associated with the upregulation of
P53, PUMA, BAX and BAK, and cleaved caspase-3 (Fig. 2) in SW1990 cells. By contrast,
survivin was increasingly upregulated in Capan-2 cells post-IR,
without a subsequent decrease after 48 h, suggesting that the
P53-mediated apoptotic pathway was inhibited by survivin in the
radioresistant PC cells (Fig. 4A).
These results suggest that adaptive expression of survivin is
transient in radiosensitive cancer cells but persistent in
radioresistant cells, serving a critical role in modulating the
radiosensitivity of PC cells.
It is well-known that survivin is specifically
expressed in dividing cancer cells at the G2/M cell cycle phase,
during which time cyclin D1 is downregulated (24). Thus, we investigated whether the
adaptive expression of survivin in radioresistant PC cells was
associated with decreased expression of cyclin D1, a marker of G1/S
phase (25), and vice versa. As
illustrated in Fig. 4B, the
expression pattern of cyclin D1 was opposite to that of survivin in
the radiosensitive SW1990 cells within 96 h post-IR, whereas cyclin
D1 expression was suppressed in radioresistant PC cells (Fig. 4B). The kinetics of the adaptive
expression of cyclin D1 was similar to that of P53, PUMA, BAX and
BAK in SW1990 cells (Fig. 2A and
B). These results suggested that cyclin D1 and survivin were
oppositely expressed in radiosensitive and radioresistant PC
cells.
Since cyclin D1 and survivin are specifically
expressed during the G1/S and G2/M phases, respectively, the cell
cycle status of the SW1990 and Capan-2 cells was examined at 24 h
post-IR. The % of G1/S phase cells (68.06%) was decreased in SW1990
cells compared with Capan-2 cells (84.15%) prior to IR. By
contrast, the % of G2/M phase cells was markedly higher (30.94%) in
SW1990 cells compared with Capan-2 cells (15.92%) prior to IR
(Fig. 4C and D). This cell cycle
pattern was reversed following IR treatment, with more SW1990 cells
arrested at the G1/S phase, and more Capan-2 cells arrested at the
G2/M phase (Fig. 4C and D). These
results suggested that the survivin-associated G2/M phase cells
were less susceptible to IR-induced cell death compared with the
cyclin-D1-associated G1/S phase cells. Therefore, the adaptive
expression levels of cyclin D1 and survivin in PC cells after IR
may determine the susceptibility of cancer cells to
radiation-induced cell death.
EGF and IL-6/IGF-1 are distinctly
expressed in radiosensitive and radioresistant PC cells
Since the expression of cyclin D1 and survivin is
regulated by growth factors such as EGF, IL-6 and IGF-1 (26-31),
their expression was examined in the radiosensitive and
radioresistant PC cells following exposure to IR. As illustrated in
Fig. 5A, EGF transcripts were
constitutively expressed and adaptively upregulated in the
radiosensitive SW1990 cells, but less in the radioresistant Capan-2
cells post-IR. By contrast, IGF-1 and IL-6 were constitutively
expressed and adaptively upregulated in the radioresistant Capan-2
cells, but less so or not at all in the radiosensitive SW1990 cells
(Fig. 5D and F). Notably, EGFR
transcripts were constitutively expressed at a higher level in
Capan-2 cells compared with SW1990 cells (Fig. 5B). At the protein level, EGFR also
appeared to be less expressed in SW1990 cells. However, the levels
of p-EGFR were significantly upregulated in Capan-2 cells post-IR,
while they were less or not upregulated in SW1990 cells (Fig. 5C). Therefore, the adaptively
increased expression of EGF and activation of EGFR appeared to be
associated with cell proliferation and survival. These results
suggested that the adaptively upregulated EGF may be associated
with the upregulation of cyclin D1 in radiosensitive PC cells
post-IR (Fig. 4B).
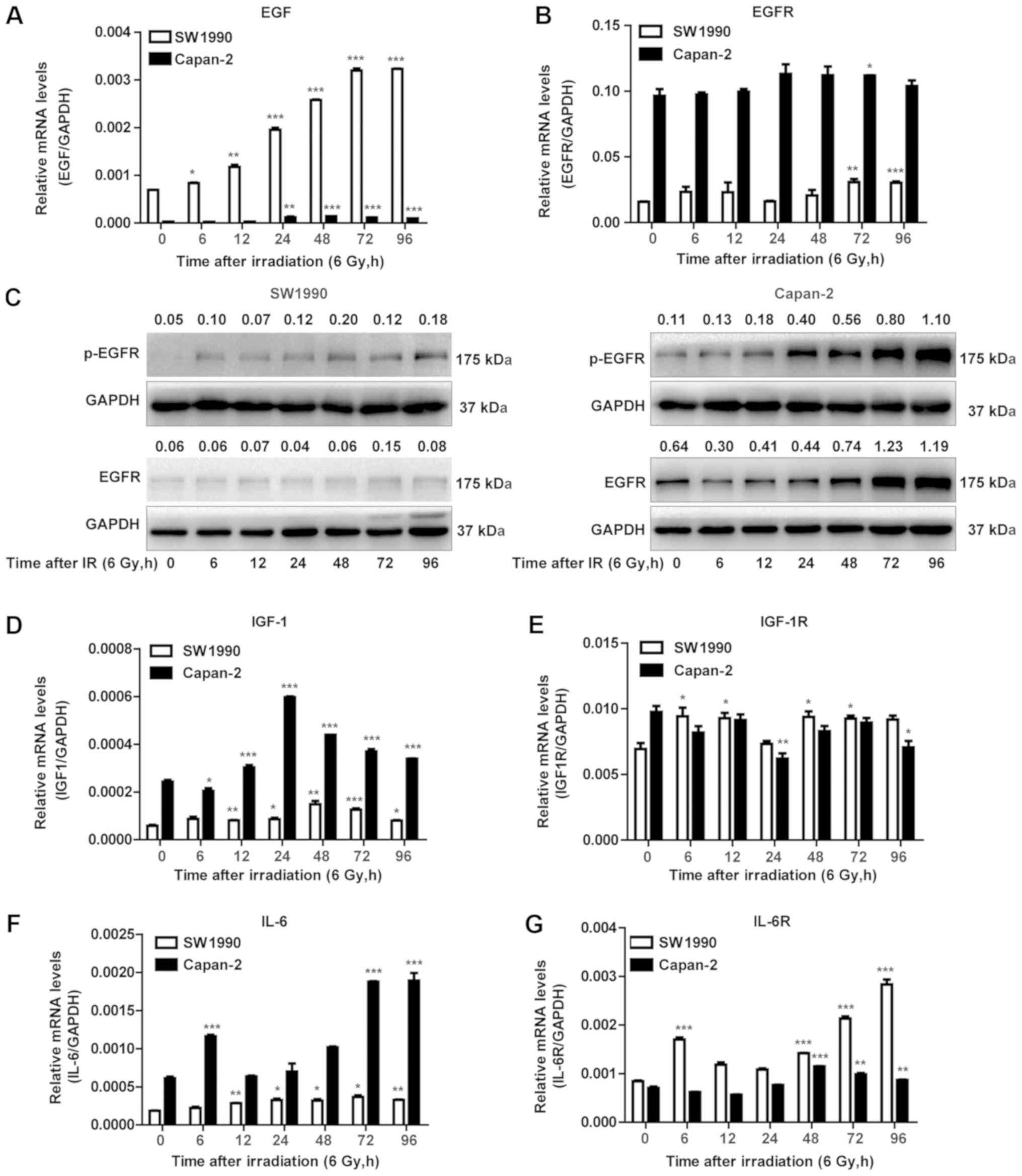 | Figure 5EGF and IL-6/IGF-1 are distinctly
expressed in radiosensitive and radioresistant pancreatic cancer
cells. (A) RT-qPCR was performed to measure the mRNA expression of
EGF. *P<0.05, **P<0.01 and
***P<0.001 vs. non-IR. (B) RT-qPCR was performed to
measure the mRNA expression of EGFR. *P<0.05,
**P<0.01 and ***P<0.001 vs. non-IR. (C)
Western blot analysis was performed using antibodies against the
p-EGFR and total EGFR. GAPDH was used as a loading control. (D)
RT-qPCR of IGF-1 levels. *P<0.05,
**P<0.01 and ***P<0.001 vs. non-IR. (E)
RT-qPCR of IGF-1R levels. *P<0.05 and
**P<0.01 vs. non-IR. (F) RT-qPCR of IL-6 levels and
(G) RT-qPCR of IL-6R levels. **P<0.01 and
***P<0.001 vs. non-IR. (H) Western blot analysis was
performed using antibodies against NF-κB p65 and NF-κB p-p65.
β-actin was used as a loading control for cytoplasmic protein. (I)
Western blot analysis was performed using antibodies against the
NF-κB p65 and NF-κB p-p65. TBP was used as a loading control for
nuclear protein. (J) Western blot analysis was performed using
antibodies against p-Stat3 and total Stat3. β-actin was used as a
loading control. EGF, epidermal growth factor; IL, interleukin;
IGF, insulin-like growth factor; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; EGFR, EGF
receptor; p-, phosphorylated; IGF-1R; IGF-1 receptor; IL-6R, IL-6
receptor; NF, nuclear factor; TBP, TATA-binding protein; Stat,
signal transducer and activator of transcription. Numbers above the
bands indicate quantified protein levels normalized to loading
control. |
Of note, similar to EGF expression, the adaptive
upregulation of IGF-1 and IL-6 in the radioresistant Capan-2 cells
was not necessarily correlated with the IGF-1R and IL-6R. The IL-6R
mRNA was upregulated in SW1990 cells, while the IGF-1R mRNA levels
were comparable between SW1990 and Capan-2 cells (Fig. 5E and G). The NF-κB protein, a
downstream signaling protein of IGF-1, was differentially activated
in SW1990 and Capan-2 cells. The activated NF-κB (p65) protein
(p-p65) was detected in the cytoplasm (Fig. 5H) and nuclei (Fig. 5I) of Capan-2 cells more prominently
compared with SW1990 cells. In addition, Stat3, which is induced
and activated by IL-6, was more highly expressed in Capan-2 cells
compared with SW1990 cells before and after IR (Fig. 5J). These results indicated that
IGF-1 and IL-6, rather than EGF, may contribute to the
radioresistance of PC cells through activation of the NF-κB and
Stat3 pathways, respectively.
Adaptive expression of EGF is required
for sensitization of PC cells through activation of the cyclin
D1/P53 pathway
Since cyclin D1 and EGF were adaptively upregulated
in the radiosensitive SW1990 cells with similar kinetics, we
investigated whether adaptive expression of EGF could induce cyclin
D1 expression in radioresistant cancer cells. To test this
hypothesis, exogenous EGF was added into the cultures of the
radioresistant Capan-2 cells and its effects on cyclin D1
expression in the radioresistant cells were examined at 72 h
post-IR. As illustrated in Fig. 6,
exogenous EGF promoted cyclin D1 expression in Capan-2 cells at 72
h post-IR (Fig. 6B), suggesting
that continuous supply (autocrine) of EGF may be required for the
radioresistant PC cells to continuously express cyclin D1. As a
control, addition of exogenous EGF to SW1990 cells exerted no
additive effect on the expression of cyclin D1 at 72 h post-IR
(Fig. 6B). Consistently with
cyclin D1 expression, exogenous EGF also promoted P53 expression in
Capan-2 cells at 72 h post-IR, although to a relatively lower
extent compared with the SW1990 cells (Fig. 6A). The enhanced P53 expression was
associated with decreased γ-H2AX expression (Fig. 6C). These results were supported by
the fact that exogenous EGF promoted IR-induced cell death in
Capan-2 cells, although moderately (Fig. 6D). Therefore, the adaptive
expression of EGF may sensitize cancer cells to IR through
upregulation of cyclin D1 and P53.
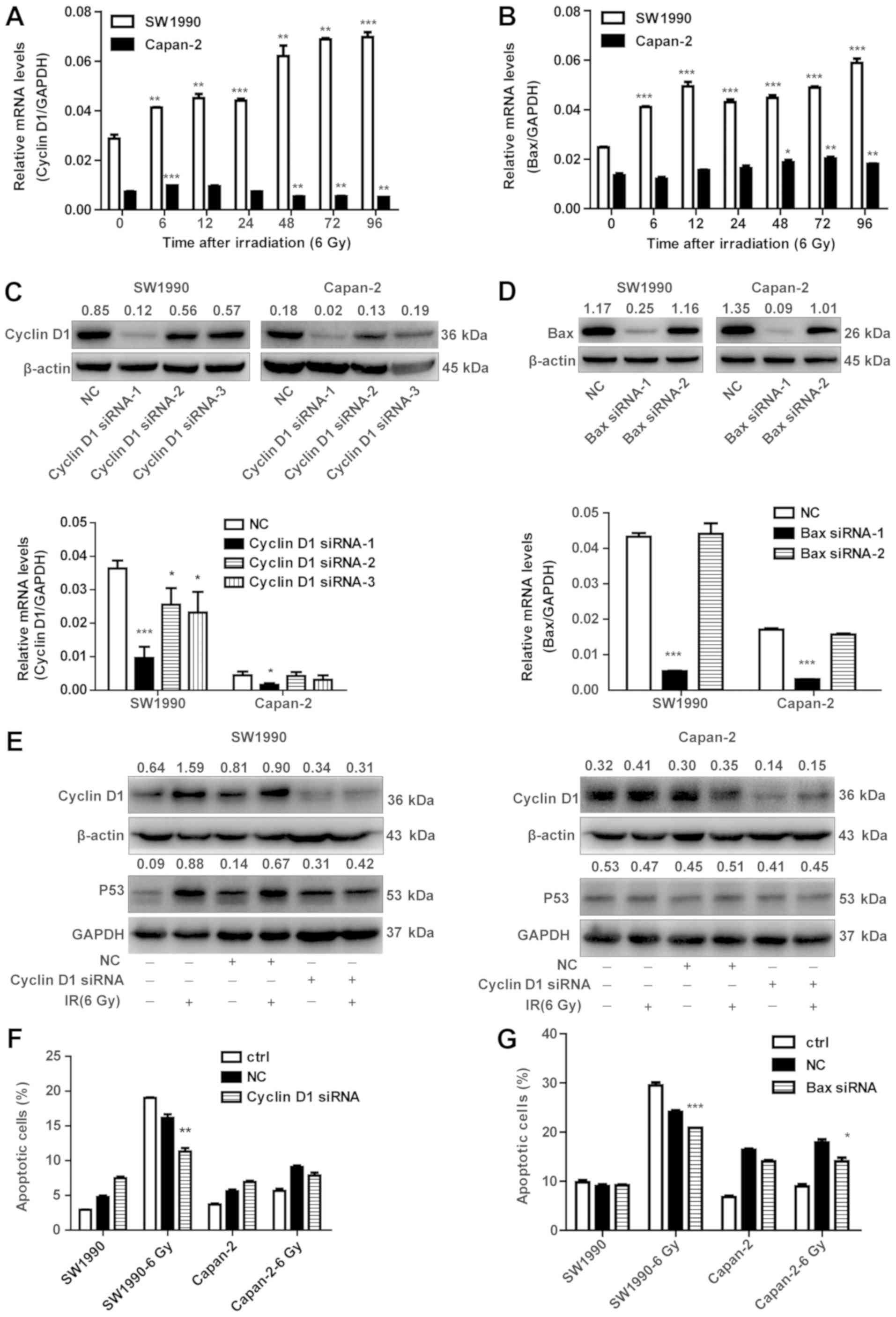
Knockdown of cyclin D1 and BAX prevents
IR-induced apoptotic cell death
To further confirm that the EGF-dependent expression
of cyclin D1 is critically involved in the radiosensitization of
cancer cells, cyclin D1 or BAX were silenced by siRNA in SW1990 and
Capan-2 cells, in order to examine their effects on apoptotic cell
death of PC cells upon IR exposure. First, it was confirmed that
cyclin D1 and BAX were transcriptionally upregulated in SW1990
cells but less so in Capan-2 cells upon IR exposure (Fig. 7A and B). These data were in
accordance with the results observed for the protein levels in
Fig. 4. siRNAs specific for cyclin
D1 and BAX were screened and used for subsequent experiments
(Fig. 7C and D). Cyclin D1
knockdown resulted in decreased P53 in SW1990 cells after
irradiation (Fig. 7E). However,
the expression of P53 was not significantly downregulated in
Capan-2 cells (Fig. 7E).
Consistently, the % of apoptotic cells was decreased in the
radiosensitive SW1990 cells, but not in the radioresistant Capan-2
cells post-IR (Fig. 7F). However,
BAX knockdown resulted in decreased apoptosis in both SW1990 and
Capan-2 cells (Fig. 7G). These
results confirm that cyclin D1 has an important role in the
radiosensitization of PC cells.
Discussion
Radiotherapy is one of the common approaches to the
treatment of cancer, including PC. IR induces cancer cell
apoptosis, however, irradiation can also induce various responses,
such as radioresistance, which may contribute to tumor recurrence
and metastasis (32). It has been
reported that cancer cells can adaptively respond to irradiation by
regulating several signaling pathways, including those mediated by
P53, Stat3 and NF-ĸB (33-35). It is well-known that the
P53-mediated pathway is enhanced, but the Stat3 and NF-ĸB-mediated
pathways are suppressed, in radiosensitive cancer cells. However,
it remains unclear how these pathways are differentially regulated
and utilized in radiosensitive vs. radioresistant cancer cells. To
the best of our knowledge, the present study is the first to
demonstrate that the radiosensitivity of PC cells may be determined
and modulated at least partially by the status of adaptive
expression of EGF or IL-6 and IGF-1 in irradiated cancer cells
(Fig. 8). In radiosensitive cancer
cells, the adaptively expressed endogenous EGF may promote cyclin
D1 expression, which in turn enhances the activity of the
P53/PUMA/BAX/caspase-3 pathway, reducing survivin expression and
eventually preventing DNA repair through inactivation of PARP. By
contrast, the radioresistance of cancer cells may be mediated by
the IGF-1 and/or IL-6 pathways, which is characteristic of
increased activation of Stat3 and NF-ĸB, respectively. Furthermore,
the P53-mediated apoptotic pathway is suppressed by survivin in
radioresistant cancer cells (Fig.
8). These findings are of great significance in understanding
the mechanisms underlying the radiosensitivity of cancer cells and
providing valuable prognostic and therapeutic markers for cancer
radiotherapy.
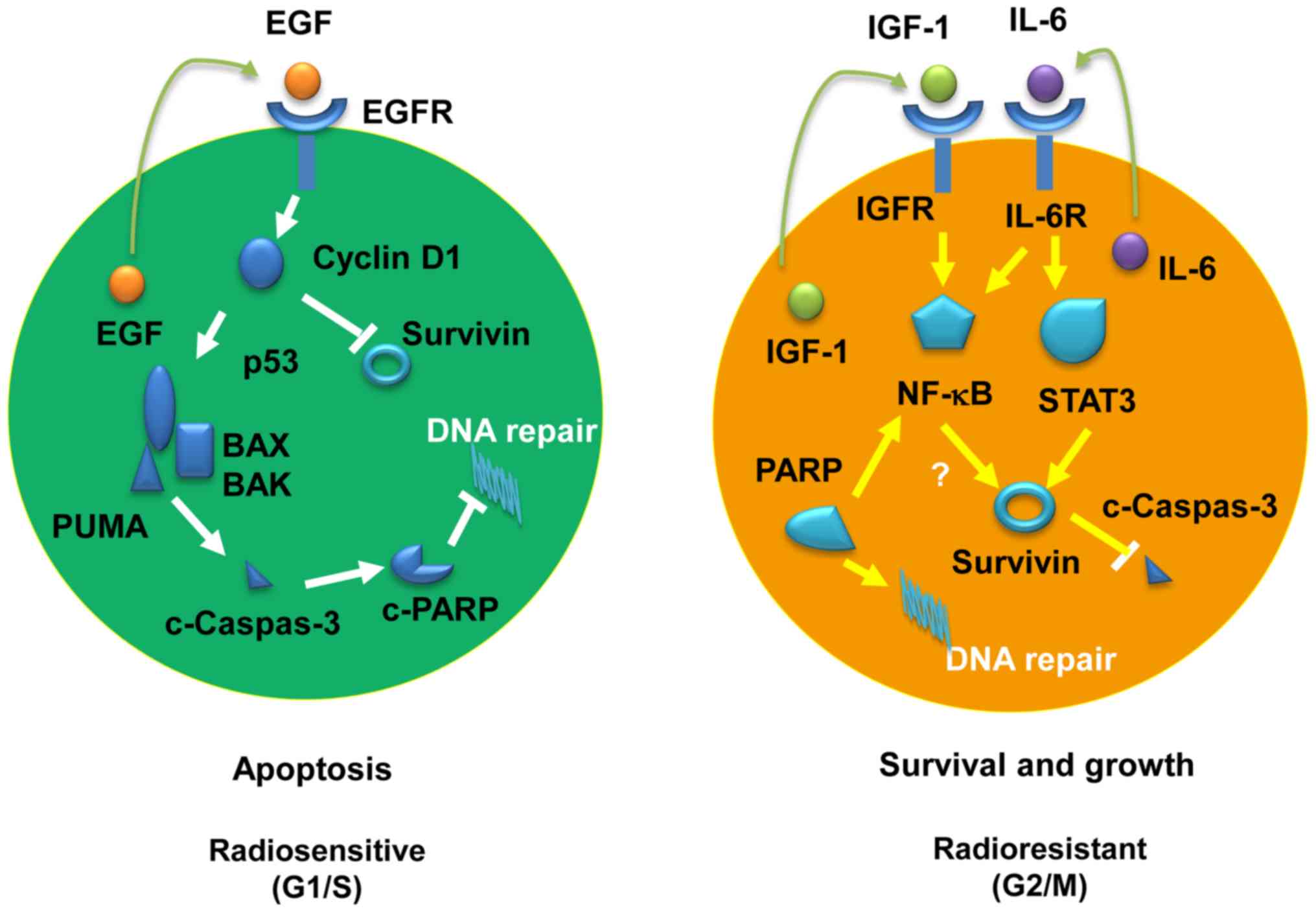 | Figure 8Schematic diagrams of the adaptive
molecular pathways for radiosensitive and radioresistant pancreatic
cancer cells. Left panel, radiosensitive pathway; right panel,
radioresistant pathway. EGF, epidermal growth factor; BAX,
Bcl-2-associated X; BAK, Bcl-2 homologous antagonist/killer; PUMA,
p53-upregulated modulator of apoptosis; PARP, poly(ADP- ribose)
polymerase; IGF, insulin-like growth factor; IL, interleukin; NF,
nuclear factor; STAT, signal transducer and activator of
transcription. |
A number of proapoptotic and antiapoptotic factors
have been reported to be involved in the radiosensitivity of cancer
cells. P53 can initiate apoptosis and programmed cell death, if DNA
damage proves to be irreparable. The present study provided
evidence that P53-mediated apoptosis of cancer cells post-IR is
associated with inactivation of PARP, and thus irreparable DNA
damage. P53 and its downstream signaling proteins PUMA, BAX, BAK
and caspase-3 were more active in the radiosensitive SW1990 cells
compared with the radioresistant Capan-2 cells. In particular, the
kinetics of caspase-3 activation was similar to that of PARP
inactivation, suggesting that caspase-3 inactivated PARP, leading
to failure of DNA repair and cell death. The DNA damage-dependent
PARP activation is an immediate cellular response to metabolic,
chemical, or radiation-induced DNA damage. Both PARP-1 and PARP-2
knockout mice exhibit severe deficiencies in DNA repair mechanisms,
with ensuing increased sensitivity to alkylating agents or IR
(36). PARP inactivation by
cleaved caspase-3 may result in defective DNA repair in
radiosensitive PC cells. However, the expressions of Bcl-2 and
Bcl-xL was not defective in radiosensitive PC cells, suggesting
that the Bcl-2-mediated antiapoptotic functions in these cells were
unaffected. Therefore, the P53/PARP pathway may override the
Bcl-2/Bcl-xL pathway in radiosensitive cancer cells upon exposure
to IR.
There remains the question of how the P53/PARP
pathway was modulated in radiosensitive vs radioresistant PC cells.
Analysis of autocrine factors revealed that EGF and IGF-1/IL-6 were
differentially expressed in radiosensitive and radioresistant PC
cells after IR. This finding suggests that adaptive expression of
EGF may single out the P53/PARP pathway and thus increase the
radiosensitivity of cancer cells. Indeed, the kinetics of
transcriptional expression of EGF is similar to that of P53
expression. In addition, the kinetics of cyclin D1 expression is
also similar to P53, suggesting a functional association between
EGF, P53 and cyclin D1. This hypothesis was confirmed by the fact
that radioresistant Capan-2 cells became more apoptotic post-IR
when exogenous EGF was added to the culture, which was accompanied
by increased expression of cyclin D1 and P53. Of note, exogenous
EGF exerted no further effects on the expression of P53 and cyclin
D1 in the radiosensitive SW1990 cells post-IR, suggesting that
endogenous EGF was sufficient to initiate the cyclin D1/P53/PARP
pathway. Furthermore, knockdown of cyclin D1 decreased P53
expression in the radiosensitive PC cells only following IR
treatment. These results indicate that in radiosensitive cancer
cells, EGF may be upregulated and in turn stimulate cyclin D1
expression followed by upregulation of P53 (EGF/cyclin D1/P53
pathway). Therefore, a mechanism was identified (namely the
EGF/cyclin D1/P53/PUMA/BAX/caspase-3/PARP pathway), through which
radiosensitive cancer cells may undergo apoptosis post-IR. However,
this pathway is less or not activated in radioresistant cancer
cells post-IR.
EGFR is usually overexpressed in various types of
tumor cells, and mediates tumor cell proliferation, invasion,
metastasis and resistance to radiotherapy and chemotherapy
(37,38). It has been reported that EGFR can
perform its function by translocating from the plasma membrane to
the nucleus through two pathways (39-41).
In addition to directing phosphorylation of proliferating cell
nuclear antigen (PCNA) (42),
nuclear EGFR can also interact with Stat3 and E2F1, regulating
transcription of cyclin D1, inducible nitric oxide synthase (iNOS),
MYB proto-oncogene like 2 (B-Myb) and Aurora kinase A (43-45).
The present study revealed a distinct but opposite expression
pattern of EGF/EGFR between radiosensitive and radioresistant PC
cells. Adaptive expression of EGF transcripts was observed in
radiosensitive but not in radioresistant PC cells. Furthermore,
EGFR was expressed at a higher level in radioresistant compared
with radiosensitive cells. However, it is likely that a high level
of endogenous EGF may promote p-EGFR translocation to the nucleus
to induce cyclin D1 transcription in radiosensitive PC cells,
although the level of p-EGFR is low. Consistent with this
hypothesis, the present study observed that cyclin D1 was
upregulated in the radiosensitive SW1990 cells compared with the
radioresistant Capan-2 cells. However, the low levels of endogenous
EGF may not be sufficient to stimulate EGFR translocation, despite
the fact that EGFR is highly expressed in radioresistant Capan-2
cells. These results suggest that the level of autocrine expression
of EGF is crucial for the cancer cells to acquire radiosensitivity
upon irradiation.
Cyclin D1 is known to be involved in cell-cycle
arrest in DNA-damage responses and it serves a key role in
maintaining the integrity of the G1/S checkpoint via the activation
of apoptotic pathways following exposure to IR in vitro
(46). Cyclin D1 prevents cell
apoptosis when it is sequestered in the cytoplasm, but may induce
apoptosis when it is localized in the nucleus (47). The localization of cyclin D1 is
associated with the radiation dose. It has been reported that
high-dose of irradiation (5 Gy of γ-rays) may enhance cyclin D1
translocation from the cytoplasm to the nucleus. By contrast,
low-dose of irradiation (10 cGy X-rays) may induce cyclin D1
accumulation in the cytoplasm by dissociating the complex of cyclin
D1 and chaperone 14-3-3, as demonstrated in human keratinocytes
(48). Furthermore, cyclin D1 can
directly interact with the pro-apoptotic BAX protein to prevent
apoptosis via improving the mitochondrial membrane potential (Dwm)
after irradiation. Blocking cyclin D1/BAX complexes using cyclin
D1-specific siRNA reversed the mitochondrial membrane potential and
suppressed the apoptotic response after irradiation. In the present
study, it was demonstrated that knockdown of cyclin D1 using
specific siRNAs promoted apoptosis in both radiosensitive and
radioresistant PC cells, but decreased apoptosis in PC cells
post-IR, especially more so in radiosensitive PC cells. These
results suggest that radiation-induced cyclin D1 is more likely
translocated to the nucleus, mediating cell death. However, it is
unlikely that cyclin D1 forms a complex with BAX to prevent
apoptosis in radiosensitive cancer cells post-IR, although the
levels of BAX were found to be high in the radiosensitive SW1990
cells. Further investigation is required to verify this
hypothesis.
The present study demonstrated that cyclin D1
expression is mutually exclusive to survivin expression in a
time‐dependent manner, consistent with the kinetics of cell
apoptosis post‐IR. Cyclin D1 is expressed in the G1/S phase,
whereas survivin is expressed in the G2/M phase (49). Increased cyclin D1 expression in
the radiosensitive SW1990 cells was found to be associated with
decreased expression of survivin at 48‐72 h post‐IR when cell
apoptosis reaches a peak, suggesting that the cells arrested in the
G1/S phase are more susceptible to apoptosis. By contrast, higher
levels of survivin expression were observed in the radioresistant
Capan‐2 cells, suggesting that the radioresistant cells that have
progressed to the G2/M phase are resistant to apoptotic cell death
induced by irradiation. Therefore, cyclin D1 and survivin appear to
be potential markers for evaluating the radiosensitivity of cancer
cells.
While EGF is adaptively expressed in radiosensitive
PC cells to mediate apoptosis post-IR, IL-6 and IGF-1 are
adaptively expressed in radioresistant PC cells. Both IL-6 and
IGF-1 are reportedly associated with radioresistance of cancer
(50,51). IL-6, as an inflammatory cytokine,
can activate the Janus kinase (Jak)/Stat3 signaling pathway in both
a pro-inflammatory and an anti-inflammatory manner. In human
esophageal carcinoma cells, IL-6 exerts its anti-apoptotic function
through activation of both Stat3 and mitogen-activated protein
kinase pathways (51).
Constitutive activation of Stat3 leads to an increase in the
oncogenes that drive proliferation and inhibit apoptosis (52). In addition, IL-6 can also activate
the NF-ĸB pathway, further driving IL-6 production (53). IGF-1-mediated phosphoinositide
3-kinase (PI3K)/Akt signaling can enhance NF-ĸB signaling (54), which exerts strong anti-apoptotic
effects on cancer cells. In the present study, we also observed
that Stat3 and NF-ĸB were not only constitutively but also
adaptively expressed and highly activated in radioresistant PC
cells compared with radiosensitive PC cells, suggesting that both
autocrine IL-6 and IGF-1 may coordinately have an important role in
mediating radioresistance of PC cells upon IR exposure. The precise
mechanisms through which IL-6 and IGF-1 interact to promote
radioresistance of cancer cells require further investigation. In
addition, these findings are based on cell models in vitro,
and therefore further verification will be required in patients
undergoing radiotherapy.
In summary, the present study comprehensively
analyzed the pathways mediating radiosensitivity of PC cells and
revealed the mechanisms through which radiosensitivity is
determined. The adaptively expressed EGF may sensitize PC cells to
radiation therapy through induction of the cyclin D1/P53/PARP
signaling pathway, and IL-6/IGF-1 may contribute to the
radioresistance of PC cells through coordinately activating the
Stat3 and NF-ĸB pathways. These findings are novel, and comparable
to the reported pathways involved in the radioresistance in cancer
stem cells, including the PI3K/Akt/mammalian target of rapamycin,
extracellular signal-regulated kinase, glycolysis, vascular
endothelial growth factor, autophagy, non-homologous end joining
and homologous recombination DNA repair pathways (55). Furthermore, the present study has
for the first time integrated these pathways to delineate the
distinct signaling between the radiosensitive and radioresistant
cancer cells.
Funding
The present study was supported by grants from the
Science and Technology Committee of Shanghai Municipality (grant
no. 114119a7400 to YB); the National Natural Science Foundation of
China (grant nos. 81372188 and 81672713 to JG, and 81402287 to LL);
the State Key Laboratory of Oncogenes and Related Genes in China
(grant no. 901406 to JG); the Special Fund for Innovation and
Development of Science and Technology and Cultivation Fund for
Major Projects and Innovative Team (grant no. 13X190030003 to JG);
Shanghai Cancer Institute, China; the University Doctorate Research
Fund for Freshly Recruited Teachers (grant no. 20130073120010 to
LL); Ministry of National Education, China; the SJTU
Interdisciplinary Research Grant (grant no. YG2015MS56 to LL).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors’ contributions
XL performed experiments and drafted the manuscript.
YH and HC performed the statistical analysis. XM, MY and RH
contributed reagents/methods/analysis tools. HC and LX participated
in irradiation of cells. BH, ML and LL helped prepare the figures.
JG and YB designed the experiments. All the authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neoptolemos JP, Kleeff J, Michl P,
Costello E, Greenhalf W and Palmer DH: Therapeutic developments in
pancreatic cancer: Current and future perspectives. Nat Rev
Gastroenterol Hepatol. 15:333–348. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yogev O, Barker K, Sikka A, Almeida GS,
Hallsworth A, Smith LM, Jamin Y, Ruddle R, Koers A, Webber HT, et
al: p53 loss in MYC-driven neuroblastoma leads to metabolic
adaptations supporting radioresistance. Cancer Res. 76:3025–3035.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maier P, Hartmann L, Wenz F and Herskind
C: Cellular pathways in response to ionizing radiation and their
targetability for tumor radiosensitization. Int J Mol Sci.
17:1022016. View Article : Google Scholar :
|
|
5
|
Ziegler V, Henninger C, Simiantonakis I,
Buchholzer M, Ahmadian MR, Budach W and Fritz G: Rho inhibition by
lovastatin affects apoptosis and DSB repair of primary human lung
cells in vitro and lung tissue in vivo following fractionated
irradiation. Cell Death Dis. 8:e29782017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kashiwagi H, Shiraishi K, Sakaguchi K,
Nakahama T and Kodama S: Repair kinetics of DNA double-strand
breaks and incidence of apoptosis in mouse neural stem/progenitor
cells and their differentiated neurons exposed to ionizing
radiation. J Radiat Res (Tokyo). 59:261–271. 2018. View Article : Google Scholar
|
|
7
|
Adams JM and Cory S: The BCL-2 arbiters of
apoptosis and their growing role as cancer targets. Cell Death
Differ. 25:27–36. 2018. View Article : Google Scholar
|
|
8
|
Tamm I, Wang Y, Sausville E, Scudiero DA,
Vigna N, Oltersdorf T and Reed JC: IAP-family protein survivin
inhibits caspase activity and apoptosis induced by Fas (CD95), Bax,
caspases, and anticancer drugs. Cancer Res. 58:5315–5320.
1998.PubMed/NCBI
|
|
9
|
Sochalska M, Tuzlak S, Egle A and
Villunger A: Lessons from gain- and loss-of-function models of
pro-survival Bcl2 family proteins: Implications for targeted
therapy. FEBS J. 282:834–849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mirakhor Samani S, Ezazi Bojnordi T,
Zarghampour M, Merat S and Fouladi DF: Expression of p53, Bcl-2 and
Bax in endometrial carcinoma, endometrial hyperplasia and normal
endometrium: a histopathological study. J Obstet Gynaecol. 1–6.
2018.
|
|
11
|
Murley JS, Miller RC, Weichselbaum RR and
Grdina DJ: TP53 mutational status and ROS effect the expression of
the survivin-associated radio-adaptive response. Radiat Res.
188:579–590. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Parida PK, Mahata B, Santra A, Chakraborty
S, Ghosh Z, Raha S, Misra AK, Biswas K and Jana K: Inhibition of
cancer progression by a novel trans-stilbene derivative through
disruption of microtubule dynamics, driving G2/M arrest, and
p53-dependent apoptosis. Cell Death Dis. 9:4482018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Balart J, Pueyo G, de Llobet LI, Baro M,
Sole X, Marin S, Casanovas O, Mesia R and Capella G: The use of
caspase inhibitors in pulsed-field gel electrophoresis may improve
the estimation of radiation-induced DNA repair and apoptosis.
Radiat Oncol. 6:62011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Venturutti L, Romero LV, Urtreger AJ,
Chervo MF, Cordo Russo RI, Mercogliano MF, Inurrigarro G, Pereyra
MG, Proietti CJ, Izzo F, et al: Stat3 regulates ErbB-2 expression
and co-opts ErbB-2 nuclear function to induce miR-21 expression,
PDCD4 downregulation and breast cancer metastasis. Oncogene.
2015.PubMed/NCBI
|
|
15
|
Braun DA, Fribourg M and Sealfon SC:
Cytokine response is determined by duration of receptor and signal
transducers and activators of transcription 3 (STAT3) activation. J
Biol Chem. 288:2986–2993. 2013. View Article : Google Scholar :
|
|
16
|
Arora R, Yates C, Gary BD, McClellan S,
Tan M, Xi Y, Reed E, Piazza GA, Owen LB and Dean-Colomb W:
Panepoxydone targets NF-κB and FOXM1 to inhibit proliferation,
induce apoptosis and reverse epithelial to mesenchymal transition
in breast cancer. PLoS One. 9:e983702014. View Article : Google Scholar
|
|
17
|
Zeng Z, Sun Z, Huang M, Zhang W, Liu J,
Chen L, Chen F, Zhou Y, Lin J, Huang F, et al: Nitrostyrene
derivatives act as RXRα ligands to inhibit TNFα activation of
NF-κB. Cancer Res. 75:2049–2060. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang X, Lv B, Zhang S, Dai Q, Chen BB and
Meng LN: Effects of radix curcumae-derived diterpenoid C on
Helicobacter pylori-induced inflammation and nuclear factor kappa B
signal pathways. World J Gastroenterol. 19:5085–5093. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baens M, Bonsignore L, Somers R,
Vanderheydt C, Weeks SD, Gunnarsson J, Nilsson E, Roth RG, Thome M
and Marynen P: MALT1 auto-proteolysis is essential for
NF-κB-dependent gene transcription in activated lymphocytes. PLoS
One. 9:e1037742014. View Article : Google Scholar
|
|
20
|
Nakano K and Vousden KH: PUMA, a novel
proapoptotic gene, is induced by p53. Mol Cell. 7:683–694. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ren D, Tu HC, Kim H, Wang GX, Bean GR,
Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ and Cheng EH: BID,
BIM, and PUMA are essential for activation of the BAX- and
BAK-dependent cell death program. Science. 330:1390–1393. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cook PJ, Ju BG, Telese F, Wang X, Glass CK
and Rosenfeld MG: Tyrosine dephosphorylation of H2AX modulates
apoptosis and survival decisions. Nature. 458:591–596. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chandele A, Prasad V, Jagtap JC, Shukla R
and Shastry PR: Upregulation of survivin in G2/M cells and
inhibition of caspase 9 activity enhances resistance in
staurosporine-induced apoptosis. Neoplasia. 6:29–40. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pardo FS, Su M and Borek C: Cyclin D1
induced apoptosis maintains the integrity of the G1/S checkpoint
following ionizing radiation irradiation. Somat Cell Mol Genet.
22:135–144. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Borowiec A-S, Hague F, Gouilleux-Gruart V,
Lassoued K and Ouadid-Ahidouch H: Regulation of IGF-1-dependent
cyclin D1 and E expression by hEag1 channels in MCF-7 cells: The
critical role of hEag1 channels in G1 phase progression. Biochim
Biophys Acta. 1813:723–730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Botto S, Streblow DN, DeFilippis V, White
L, Kreklywich CN, Smith PP and Caposio P: IL-6 in human
cytomegalovirus secretome promotes angiogenesis and survival of
endothelial cells through the stimulation of survivin. Blood.
117:352–361. 2011. View Article : Google Scholar :
|
|
28
|
Hov H, Våtsveen TK, Waage A, Sundan A and
Borset M: Induction of Cyclin D1 by HGF, IGF-1 and IL-6 in a human
myeloma cell line with a t(11:14). Translocation Blood.
108:50552006.
|
|
29
|
Ravitz MJ, Yan S, Dolce C, Kinniburgh AJ
and Wenner CE: Differential regulation of p27 and cyclin D1 by
TGF-beta and EGF in C3H 10T1/2 mouse fibroblasts. J Cell Physiol.
168:510–520. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vaira V, Lee CW, Goel HL, Bosari S,
Languino LR and Altieri DC: Regulation of survivin expression by
IGF-1/mTOR signaling. Oncogene. 26:2678–2684. 2007. View Article : Google Scholar
|
|
31
|
Wang H, Gambosova K, Cooper ZA, Holloway
MP, Kassai A, Izquierdo D, Cleveland K, Boney CM and Altura RA: EGF
regulates survivin stability through the Raf-1/ERK pathway in
insulin-secreting pancreatic β-cells. BMC Mol Biol. 11:662010.
View Article : Google Scholar
|
|
32
|
Chi HC, Tsai CY, Tsai MM, Yeh CT and Lin
KH: Roles of long noncoding RNAs in recurrence and metastasis of
radiotherapy-resistant cancer stem cells. Int J Mol Sci. 18:182017.
View Article : Google Scholar
|
|
33
|
Hage-Sleiman R, Bahmad H, Kobeissy H,
Dakdouk Z, Kobeissy F and Dbaibo G: Genomic alterations during
p53-dependent apoptosis induced by γ-irradiation of Molt-4 leukemia
cells. PLoS One. 12:e01902212017. View Article : Google Scholar
|
|
34
|
Deng WW, Hu Q, Liu ZR, Chen QH, Wang WX,
Zhang HG, Zhang Q, Huang YL and Zhang XK: KDM4B promotes DNA damage
response via STAT3 signaling and is a target of CREB in colorectal
cancer cells. Mol Cell Biochem. 449:81–90. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chishti AA, Baumstark-Khan C, Koch K,
Kolanus W, Feles S, Konda B, Azhar A, Spitta LF, Henschenmacher B,
Diegeler S, et al: Linear energy transfer modulates
radiation-induced NF-kappa B activation and expression of its
downstream target genes. Radiat Res. 189:354–370. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ménissier de Murcia J, Ricoul M, Tartier
L, Niedergang C, Huber A, Dantzer F, Schreiber V, Amé JC, Dierich
A, LeMeur M, et al: Functional interaction between PARP-1 and
PARP-2 in chromosome stability and embryonic development in mouse.
EMBO J. 22:2255–2263. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee SL, Ryu H, Son AR, Seo B, Kim J, Jung
SY, Song JY, Hwang SG and Ahn J: TGF-β and hypoxia/reoxygenation
promote radioresistance of A549 lung cancer cells through
activation of Nrf2 and EGFR. Oxid Med Cell Longev.
2016:68234712016. View Article : Google Scholar
|
|
38
|
Tang J, Guo F, Du Y, Liu X, Qin Q, Liu X,
Yin T, Jiang L and Wang Y: Continuous exposure of non-small cell
lung cancer cells with wild-type EGFR to an inhibitor of EGFR
tyrosine kinase induces chemoresistance by activating STAT3. Int J
Oncol. 46:2083–2095. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Diluvio G, Del Gaudio F, Giuli MV,
Franciosa G, Giuliani E, Palermo R, Besharat ZM, Pignataro MG,
Vacca A, d’Amati G, et al: NOTCH3 inactivation increases triple
negative breast cancer sensitivity to gefitinib by promoting EGFR
tyrosine dephosphorylation and its intracellular arrest.
Oncogenesis. 7:422018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li C, Iida M, Dunn EF, Ghia AJ and Wheeler
DL: Nuclear EGFR contributes to acquired resistance to cetuximab.
Oncogene. 28:3801–3813. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang YN, Lee HH, Lee HJ, Du Y, Yamaguchi H
and Hung MC: Membrane-bound trafficking regulates nuclear transport
of integral epidermal growth factor receptor (EGFR) and ErbB-2. J
Biol Chem. 287:16869–16879. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tong D, Ortega J, Kim C, Huang J, Gu L and
Li GM: Arsenic inhibits DNA mismatch repair by promoting EGFR
expression and PCNA phosphorylation. J Biol Chem. 290:14536–14541.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y
and Hung MC: Co-regulation of B-Myb expression by E2F1 and EGF
receptor. Mol Carcinog. 45:10–17. 2006. View Article : Google Scholar
|
|
44
|
Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia
W, Wei Y, Bartholomeusz G, Shih JY and Hung MC: Nuclear interaction
of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer
Cell. 7:575–589. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lo HW and Hung MC: Nuclear EGFR signalling
network in cancers: Linking EGFR pathway to cell cycle progression,
nitric oxide pathway and patient survival. Br J Cancer. 94:184–188.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li SJ, Liang XY, Li HJ, Li W, Zhou L, Chen
HQ, Ye SG, Yu DH and Cui JW: Low-dose irradiation promotes
proliferation of the human breast cancer MDA-MB-231 cells through
accumulation of mutant P53. Int J Oncol. 50:290–296. 2017.
View Article : Google Scholar
|
|
47
|
Sumrejkanchanakij P, Tamamori-Adachi M,
Matsunaga Y, Eto K and Ikeda MA: Role of cyclin D1 cytoplasmic
sequestration in the survival of postmitotic neurons. Oncogene.
22:8723–8730. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ahmed KM, Fan M, Nantajit D, Cao N and Li
JJ: Cyclin D1 in low-dose radiation-induced adaptive resistance.
Oncogene. 27:6738–6748. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sheng L, Wan B, Feng P, Sun J, Rigo F,
Bennett CF, Akerman M, Krainer AR and Hua Y: Downregulation of
Survivin contributes to cell-cycle arrest during postnatal cardiac
development in a severe spinal muscular atrophy mouse model. Hum
Mol Genet. 27:486–498. 2018. View Article : Google Scholar :
|
|
50
|
Tamari Y, Kashino G and Mori H:
Acquisition of radioresistance by IL-6 treatment is caused by
suppression of oxidative stress derived from mitochondria after
γ-irradiation. J Radiat Res (Tokyo). 58:412–420. 2017. View Article : Google Scholar
|
|
51
|
Yang HY, Qu RM, Lin XS, Liu TX, Sun QQ,
Yang C, Li XH, Lu W, Hu XF, Dai JX, et al: IGF-1 from
adipose-derived mesenchymal stem cells promotes radioresistance of
breast cancer cells. Asian Pac J Cancer Prev. 15:10115–10119. 2014.
View Article : Google Scholar
|
|
52
|
Yang N, Han F, Cui H, Huang J, Wang T,
Zhou Y and Zhou J: Matrine suppresses proliferation and induces
apoptosis in human cholangiocarcinoma cells through suppression of
JAK2/STAT3 signaling. Pharmacol Rep. 67:388–393. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Iliopoulos D, Hirsch HA and Struhl K: An
epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and
IL6 links inflammation to cell transformation. Cell. 139:693–706.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ma J, Sawai H, Matsuo Y, Ochi N, Yasuda A,
Takahashi H, Wakasugi T, Funahashi H, Sato M and Takeyama H: IGF-1
mediates PTEN suppression and enhances cell invasion and
proliferation via activation of the IGF-1/PI3K/Akt signaling
pathway in pancreatic cancer cells. J Surg Res. 160:90–101. 2010.
View Article : Google Scholar
|
|
55
|
Chang L, Graham P, Hao J, Ni J, Deng J,
Bucci J, Malouf D, Gillatt D and Li Y: Cancer stem cells and
signaling pathways in radioresistance. Oncotarget. 7:11002–11017.
2016.
|