Introduction
Under normal conditions, hematopoietic stem cells
(HSCs) can differentiate into mature blood cells of all lineages
(1). Genetic stability of HSCs
requires strict regulation, since genetic alterations can
compromise lineage commitment (2).
Myelodysplastic syndrome (MDS) is a malignant disorder that is
characterized by the aberrant differentiation of HSCs (3). During MDS, genomic instability in HSCs
leads to the accumulation of somatic mutations, defective
differentiation and subsequent progression to leukemia (3). Genomic instability can result in DNA
damage, which can be caused by multiple sources, including exposure
to environmental genotoxic substances, such as polycyclic aromatic
hydrocarbons or pesticides, or to endogenous generation of reactive
compounds of metabolic origin, such as reactive oxygen species
(4). However, the human genome
harbors a plethora of potential insertional mutagens within its own
architecture in the form of transposable elements (TEs). The
deleterious effects of TEs primarily lie in their inherent mobile
nature, where their expression can lead to insertion mutagenesis
and chromosomal rearrangements, thereby causing genomic instability
(5). This can in turn lead to the
development of different types of cancer, including leukemia
(5). During evolution, a key
mechanism that functions to protect genome integrity against the
potentially harmful mobilization of TEs has emerged in the form of
a family of small non-coding RNAs, known as P-element-induced WImpy
testis (Piwi)-interacting RNAs (piRNAs) (6).
Despite considerable advances in the understanding
of MDS pathogenesis, the influence of TEs and piRNAs on genome
instability in hematopoietic cells has not been defined
sufficiently. Therefore, the present review summarizes the current
knowledge of the transcriptional activity and function of TEs and
piRNAs in hemato-oncology, with particular emphasis on the
potential involvement of these non-coding molecules in the
pathogenesis of myelodysplasia.
Myelodysplastic syndrome (MDS)
MDS represents a spectrum of HSC disorders that is
characterized by impaired hematopoiesis, peripheral blood cytopenia
and a tendency toward leukemic transformation (3). Acute myeloid leukemia (AML) with
myelodysplasia-related changes (AML-MRC) gradually develops in
30-40% patients with MDS (7). In
recent years, several new therapeutic agents have been approved for
the treatment of MDS, where hypomethylating agents, such as
azacitide or decitabine, have been found to be effective for
treating MDS and AML-MRC (8,9). These
hypomethylating agents can improve the overall survival, clinical
outcomes and quality of life in a proportion of patients (overall
response rate, 40-50%) (10,11).
Although the precise mechanism of their action remains the subject
of scientific investigation, DNA hypomethylation is hypothesized to
reverse the inactivation of tumor-suppressor gene transcription
(9).
MDS is a dynamic disorder in which clonal evolution
triggers disease onset and progression (3). Clonal evolution is a multistep process
that involves the successive acquisition of abnormalities in the
genome of normal HSCs (12). These
aberrations in turn lead to the expansion of MDS clones and
subsequent transformation into AML (13). A vast body of evidence has been
accumulated on the spectrum of cytogenetic abnormalities,
epigenetic modifications, gene expression and signaling pathways,
including apoptosis, proliferation, immune response, RNA-splicing
machinery, microenvironment interactions, genome instability and
DNA damage response, associated with the disease (14,15).
Over the last decade, a number of breakthrough observations have
been reported describing the presence of multiple somatic mutations
in MDS (16–18). These mutations have been
characterized in a number of key components of the spliceosome,
regulator of DNA methylation, chromatin modification,
transcription, signal transduction and cell cycle control machinery
(19). However, the precise
mechanism of MDS development remain a subject of intense research
at present.
Transposable elements (TEs)
TEs are DNA sequences with the ability to move
(transpose) into new locations in the genome, which frequently
results in the creation of new copies of themselves (20). Since TEs were discovered in the
maize crop in 1950 (21), they have
become gradually recognized to be major components of eukaryotic
genome. It is now estimated that they constitute ~45% of the human
genome in a variety of forms and structures (5).
According to their mechanism of transposition, two
major classes of TEs have been designated: Class I retrotransposons
and Class II DNA transposons (22).
Furthermore, each class can be divided further into several
subclasses and families (22). DNA
transposons move by a non-replicative mechanism from one genomic
location to another (23). They
encode a transposase enzyme that cleaves the TE from the genomic
sequence and then mediate its reintegration into another loci in
the genome in a so-called ‘cut-and-paste’ mechanism (23). DNA transposons can be divided into
two subclasses based on the number of DNA strands that they can cut
during transposition (24). DNA
transposons also tend to be less common, where they are estimated
to constitute ≥3% of the human genome (24).
By contrast, retrotransposons replicate using an RNA
intermediate that is reverse transcribed into cDNA and reintegrated
as an additional copy elsewhere in the genome in a ‘copy-and-paste’
retro-transposition mechanism (25). Retrotransposons are divided based on
the presence or absence of long terminal repeats (LTRs) at their
ends, which flank a central coding region (25). Endogenous retroviruses (ERVs) are a
type of LTR retrotransposons that share similarities with exogenous
retroviral proviruses and have integrated into host DNA following
past infections (26). The majority
of ERVs encode two proteins closely related to Gag (the original
structural matrix and capsid) and Pol (original reverse
transcriptase and integrase) proteins of retroviruses, but do not
encode the Env (envelope) protein and therefore have lost their
ability to exit the cell (27).
However, ERV sequences generally only consist of solitary LTRs that
are most likely generated by homologous recombination between the
5′ and 3′ LTRs (27).
Non-LTR retrotransposons do not have LTR sequences
and resemble integrated RNA (28).
They can be classified as autonomous or nonautonomous elements
(29). Autonomous elements are also
known as long interspersed elements (LINEs) and encode two
proteins, Orf1 and Orf2 (30). Orf1
and Orf2 have endonuclease and reverse transcriptase activities,
respectively and are able to self-mobilize (31,32).
LINEs constitute the vast majority of transposable elements in the
human genome (33). The LINE-1
element is one example of LINE, of which there are ~500,000 copies
in the human genome, where ~99.9% of these copies are fixed and are
no longer mobile (34). However,
each individual is estimated to carry a set of ~100 potentially
mobile LINE-1 elements (34).
Non-autonomous elements, such as short interspersed nuclear
elements (SINEs), depend on LINE-encoded proteins for their own
cycle of retrotranscription (35).
Human Arthrobacter luteus (alu) elements are ~300 bp-long
SINEs that are among the most abundant SINEs observed (36). There are >1×106 copies
of Alu in the human genome (37).
In addition, other important members of the SINE family include
mammalian-wide interspersed repeats (MIRs), which are ~260 bp in
length and account for ~3% of the human genome (36).
Although they are generally assigned to their
specific sub-families, the TE taxonomy and nomenclature is in a
constant state of flux due to the discovery of new TEs and the
necessity to introduce novel classification criteria (38). Diverse families of TEs take up a
substantial proportion of the genome, where their propagation is
regulated not only by their intrinsic properties but also by
natural selection and genetic drift forces (39). Insertions that are potently
deleterious are rapidly removed from the population, whereas
insertions that exert little to no effects on the host become fixed
in the genome (39). Therefore, it
is not surprising that TEs are rarely randomly distributed in the
genome but rather exhibit various types of insertion preferences,
such that some TEs are more likely to be retained at certain
genomic locations compared with others (40). For example, although de novo
LINE-1 retrotransposon insertions readily occur within gene exons,
these elements have rarely been observed to be fixed within the
coding regions of humans (41).
The means by which TEs can lead to changes in DNA
sequences are heterogeneous (20).
Transposition is an imperfect mechanism that consists of repeated
cycles of TE insertion and removal from the genome (42). Therefore, this process can also
result in changes in the surrounding host sequences, including
rearrangement of target genes or regulatory elements (39,43).
Integration of TEs into exons can create frameshift mutations,
leading to premature stop codons and nonsense-mediated decay or by
inducing exon skipping (44,45).
Furthermore, TEs, even those that have lost their mobilization
capacity, can provide a large repertoire of homologous sequences
scattered throughout the genome (39). This may promote nonallelic
homologous recombination at distant genomic regions, resulting in
large-scale deletions, duplications and/or inversions (39).
A number of TEs have been domesticated in the host
genome (46). Following their
integration, the vast majority of TEs become inactivated as a
result of accumulating mutations that prevent further autonomous
mobilization (47). However, in
some cases these accumulating mutations can cause the
neo-functionalization of these inserted TEs, such that they can
even confer beneficial cellular effects due to the novel downstream
gene product (48). Although the
majority of domesticated TEs remain functionally uncharacterized,
those that have been studied have been previously found to regulate
a variety of cellular processes, including transcriptional
regulation, proliferation, cell cycle progression and apoptosis
(47).
TE silencing by P-element-induced WImpy
testis-interacting RNAs (piRNAs)
The activity of TEs poses a serious threat to genome
stability. Therefore, organisms have evolved a complex arsenal of
mechanisms to control these potentially harmful mobilizations of
TEs (49). One of the key
TE-silencing mechanisms involves a large family of
Krüppel-associated box (Krab) zinc-finger proteins, which bind to
TEs and recruits Krab-associated protein 1 (Kap1; also known as
Trim28) to form repressive chromatin complexes in association with
multiple interacting partners, including the histone H3K9me3
methyltransferase, Set Domain Bifurcated 1 (SETDB1), the histone
deacetylase containing NuRD complex and heterochromatin protein 1
(HP1) (50,51). Another important mechanism of TE
regulation is mediated by epigenetic regulation, particularly
through DNA methylation and histone modifications, which serve to
maintain the viability of repressive heterochromatin (52) and small RNA molecules, called
piRNAs.
piRNAs form one of the three main classes of
regulatory small non-coding RNAs (sncRNAs), together with small
interfering RNAs (siRNAs) and microRNAs (miRNAs) (53). These classes differ in terms of
their biogenesis and mode of target regulation, but share a number
of common features, including their ability to guide Argonaute
(Ago) proteins to target nucleic acids in a sequence-dependent
manner (54). piRNAs are
single-stranded sncRNAs that are 26-31 nucleotides in length with
2′-O-methyl modification sites at the 3′ terminus (55,56).
They comprise the largest class of sncRNAs expressed in animal
cells (57). According to piRBase,
the number of piRNA sequences reported has reached 173 million at
present, with ~8.5 million unique human piRNA sequences (58).
piRNAs are expressed from repetitive intergenic
elements in the genome called piRNA clusters using a
Dicer-independent mechanism (Fig.
1). These clusters span wide regions of the genome and are
mostly comprised of various TEs and their remnants (59). Primary piRNAs are processed from the
piRNA clusters through the primary processing pathway, which then
form functional complexes exclusively with Piwi proteins (60). In the germline of several organisms,
primary piRNAs are subjected to an amplification system called the
‘ping-pong’ cycle to reinforce their expression (61). Within this mechanism, primary piRNAs
bind to Ago3 or Aubergine proteins, recognize complementary targets
to induce their cleavage, producing secondary piRNAs (61). This ping-pong mechanism has been
identified in Drosophila melanogaster, zebrafish and
primitive animal-like sponges, such as those in the phylum
Porifera (62,63), but not in mice, suggesting that the
ping-pong mechanism only exists during the early stages of
evolution (64).
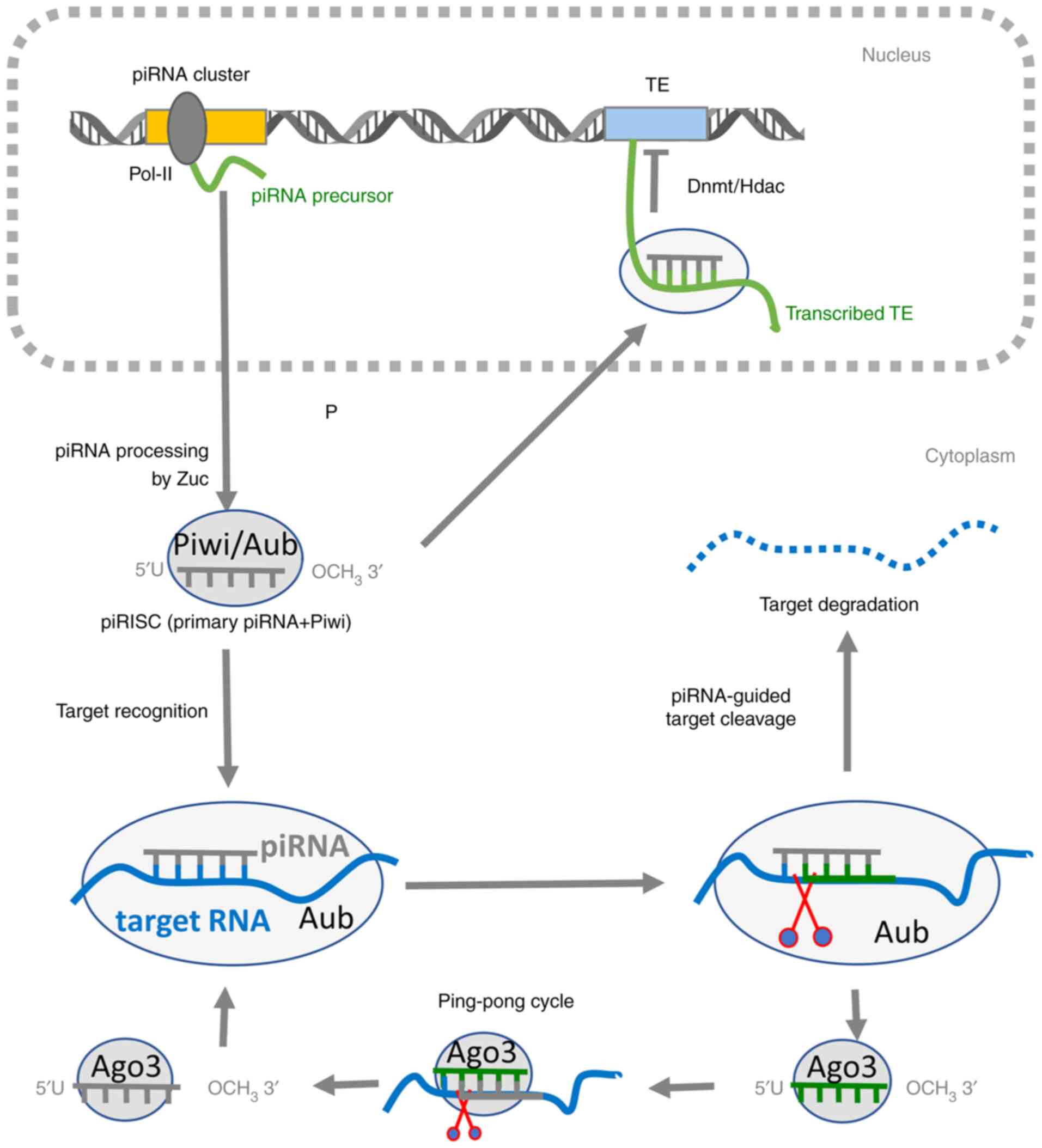 | Figure 1.Mechanism of the piRNA pathway. In
the nucleus, the piRNA precursor is transcribed from a piRNA
cluster by Pol-II. After its export to the cytoplasm, the precursor
is cleaved by the Zuc ribonuclease, generating a 5′-phosphate
residue. The 3′ fragment of the transcript is incorporated into
Piwi (gray ovals), forming the piRISC complex and trimmed to its
final length. The 2′-hydroxyl group at the 3′ end is then
methylated by the Hen1 2′-O-methyltransferase, rendering piRNA more
stable. This piRISC complex can migrate back into the nucleus,
where it can mediate co-transcriptional TE silencing. piRNA
recruits DNMT or HDAC to its complementary, nascent TE transcript
to repress its expression by making DNA/histone modifications. In
the cytoplasm, piRISC can mediate the post-transcriptional gene
regulation of complementary TEs or mRNAs. piRNAs typically target
the complementary RNA sequence in the 3′-untranslated region,
resulting in RNA degradation, translational inhibition or
intracellular localization changes. piRNA molecules can also enter
the ‘ping-pong’ cycle, where the piRNA-Aub complex recognizes and
cleaves the complementary TE transcript or a transcript derived
from the opposite strand of the same piRNA cluster. This cleavage
produces a new piRNA that is loaded into the Ago3 protein and in
turn induce the cleavage of its complementary RNA, generating a new
copy of piRNA that is identical in sequence to the piRNA that
initiated the cycle. PIWI, P-element-induced WImpy testis; piRNA,
PIWI-interacting RNA; RISC, RNA-induced silencing complex; Pol-II,
DNA polymerase II; TE, transposable element; DMNT, DNA
methyltransferase; HDAC, histone deacetylase; Zuc, Zucchini
ribonuclease; Aub, aubergine; Ago, argonaute. |
Piwi proteins are a class of Argonaute proteins that
are expressed in several stem cell populations across various
organisms and tissues (62).
However, they are most robustly expressed in the male germline of
adult mammals (62). These proteins
form specific RNA-induced silencing complexes (RISCs) with piRNAs
called piRISCs (65). There are
four known human Piwi proteins to date: Hiwi (also known as
Piwil1), Hili (Piwil2), Hiwi2 (Piwil4) and Hiwi3 (Piwil3) (66). Piwi proteins are predominantly
localized to the nucleus, where they colocalize with known
epigenetic modifiers, such as polycomb group proteins (67). They enhance DNA methylation, thereby
operating at the chromatin level and can modify the histone code to
repress the transcription of TEs (68).
After its formation, the piRISC enters the nucleus,
where it targets an actively transcribed nascent TE by recognizing
its sequence, which is complementary to the piRNA molecule
(69). The piRISC then prevents
further transcription of the TE by recruiting histone deacetylases
and DNA methyltransferases, leading to the enrichment of repressive
signals, such as H3K9me3, and formation of the
transcriptionally-silent heterochromatin (70). In addition to chromatin silencing,
piRNAs can control TE and mRNA expression in the cytoplasm by
mediating their degradation, translational inhibition and
regulation of intracellular localization (62). In addition, >25% human mRNAs
possess a retrotransposon in their 3′ untranslated region,
rendering them potential targets for piRISCs (71).
Methods for the exploration of TEs and
piRNAs
Recent progress in genomic sequencing, coupled with
the rapid generation of large sequencing datasets and continuously
improving bioinformatic technologies, has sparked interest in the
previously unexplored, non-coding parts of the human genome. This
has enabled the association between their structure, transcription
and functional relevance for human health and disease to be
accurately studied. Although standard next-generation sequencing
(NGS) methods are frequently applied for the identification of TEs
and piRNAs, their successful detection in genome-wide data requires
specific tools, for which experimental design brings a number of
novel challenges. Since the genome harbors multiple copies of
similar TEs and piRNA sequences, degree of uniqueness and/or
repetitiveness are important parameters that need to be considered,
especially when aligning NGS reads that could be originated from
multiple genomic locations (72).
In this context, novel technologies based on long-read sequencing
are providing new opportunities for reducing the complexity of TE
detection (73).
Goerner-Potvin et al (74) previously published a comprehensive
review of the available bioinformatics resources, databases and
classification tools developed to detect and analyze TEs.
Therefore, the present study only briefly describes the current
bioinformatics approaches applied to analyze TEs. Discovery and
annotation of TEs in NGS reads can be performed with or without a
genome assembly, where various tools exist to perform such tasks.
Currently, the ‘gold-standard’ strategy is by using a
repository-based annotation, whereby sequences are queried against
known TE consensus sequences or TE motifs (74). RepeatMasker (75) is the most widely accepted database
query tool that is used to identify, classify and mask repetitive
elements, including low-complexity sequences and interspersed
repeats. RepeatMasker searches for repetitive sequences by aligning
the input sequences against TE repositories (75). The repositories can be classified
into the following three specific types: TE-centric (consensus
sequences associated with each TE family); genome-centric (all
individual TE instances within a reference genome); and
polymorphism-centric (polymorphic insertions in individuals
diverging from the annotated reference genome) (Table I) (74). Furthermore, RepBase (76) is a popular source for probing
consensus sequences of eukaryotic TEs. Dfam (77) is another TE database where TE
families are defined by multiple sequence alignments through hidden
Markov models. Dfam is the only TE repository for individual TE
instances annotated in mammalian genomes, where the TE sequences
and their genomic locations are also available through the
RepeatMasker tracks in the genome browsers (77). An alternative method of TE discovery
and annotation that does not require a genome assembly is de
novo annotation, which offers the potential to identify novel
TE families (74). The most
commonly used de novo annotation tools include RepeatModeler
(78) or RepeatExplorer (79). Focusing on TE transcription,
TEtranscripts (80) is a
differential expression analysis software tool that has been shown
to be the most accurate for analyzing TE expression in
RNA-sequencing data.
 | Table I.Public resources specialized in human
TE and piRNA data. |
Table I.
Public resources specialized in human
TE and piRNA data.
| A, TE
databases |
|---|
|
|---|
| Database | Description | First author/s,
year | Web page | (Refs.) |
|---|
| RepBase | This largest
collection of eukaryotic transposons and repetitive sequences
includes >44,000 sequences (mostly family consensus). It
functions as a standard reference for annotating repetitive DNA in
genomic data. | Bao et al,
2015 | www.girinst.org/repbase | (76) |
| Dfam | Database of
repetitive DNA elements organized around multiple sequence
alignments of TE families. This seed alignment provides a
model-neutral representation of a sequence family from which both a
traditional consensus sequence and a hidden Markov model profile
can be built. | Wheeler et
al, 2013 | www.dfam.org | (77) |
| TranspoGene | Collection of TEs
that are located in the protein-coding genes of seven eukaryotic
organisms. | Levy et al,
2008 | www.transpogene.tau.ac.il | (153) |
| Line Fusion | Database of human
LINEs with information on LINE structure, expression pattern,
tissue distribution and chromosomal location of the host genes, in
addition to the domain structure altered by LINE integration. | Kim et al,
2006 | www.primate.or.kr/line | (154) |
| RCPedia | Database of
retrocopies (processed pseudogenes and retrogenes) from six primate
genomes. | Navarro and
Galante, 2013 | www.bioinfo.mochsl.org.br/rcpedia | (155) |
| dbVar | NCBI database of
human genomic structural variations, including mobile element
variations. | Lappalainen et
al, 2013 | www.ncbi.nlm.nih.gov/dbvar | (156) |
| dbRip | Database of human
retrotransposon insertion polymorphisms that contains all currently
known Alu, L1 and short interspersed nuclear elements-variable
number tandem repeat-Alu polymorphic insertion loci in the human
genome. | Wang et al,
2006 | www.dbrip.brocku.ca | (157) |
| euL1db | A curated database
of human-specific L1 insertion polymorphisms identified in healthy
or pathological human samples. It facilitates the understanding of
the link between L1 retrotransposon insertion polymorphisms and
phenotype or disease. | Mir et al,
2015 | www.eul1db.ircan.org | (158) |
|
| B, piRNA
databases |
|
|
Database |
Description | First author/s,
year | Web
page | (Refs.) |
|
| piRNAdb | Storage and search
system that includes information on the alignments, clusters,
datasets and targets of piRNAs, providing information on >27,000
piRNAs from six model | Piuco and Galante,
2021 | www.pirnadb.org | (159) |
| piRBase | Organisms. | Wang et al,
2019 | www.regulatoryrna.org/database/piRNA | (58) |
| piRNABank | Information on
piRNAs in human, mouse, rat and Drosophila that compiles
piRNA clusters and depicts piRNAs along with the associated genomic
elements. | Lakshmi and
Agrawal, 2008 | www.pirnabank.ibab.ac.in | (160) |
| piRNAQuest | Annotation of
human, mouse and rat piRNAs based on their genomic location.
Information on all possible piRNA clusters along with significant
motifs, piRNA expression and several analysis tools (Homology
search, Dynamic piRNA Clusters, Pattern Search and AT-GC%
calculator). The results can be browsed using piBROWSE and
piSynBrowse. | Sarkar et
al, 2014 | www.bicresources.jcbose.ac.in/zhumur/pirnaquest | (161) |
| piRNA cluster
database | Data on piRNA
clusters in multiple species, tissues and developmental stages
based on small RNA sequence data deposited in the NCBI Sequence
Read Archive. | Rosenkranz,
2016 | www.smallrnagroup-mainz.de/piRNAclusterDB.html | (162) |
Detection of piRNAs requires additional specialized
methodology that is different from optimized techniques used for
the analysis of miRNAs (81). To
profile piRNAs on a genome-wide level, an NGS variant modified for
the detection of small RNAs, such as small RNA-seq, is utilized
(82). However, 2′-O-methylation of
the 3′-terminal nucleotide within piRNA molecules may hinder
adaptor ligation during library preparation, causing the
underrepresentation of piRNA species in the data output (83). This was previously confirmed by
Leshkowitz et al (84), who
modified the 3′ end of synthetic miRNAs with an O-methyl group and
compared the results of the analysis with their unmodified
counterparts, which revealed that this modification resulted in a
reduction in sequence detection (84). A number of solutions have been
proposed to avoid this bias and improve library preparation, where
several ‘low bias’ kits have been designed (85). These include the use of randomized
adapters (86) and polyethylene
glycol for ligation reactions (87)
or avoiding the ligation step altogether (85). After library sequencing, small RNAs
that are 24-32 nucleotides long were extracted from the raw reads
and computationally processed (88,89).
Bioinformatic analysis of piRNAs within the sequencing data can
also be challenging, since a large number of piRNAs may be rare or
expressed at low levels, such that single counts can result in
technical artifacts (90).
Therefore, the presence of small RNAs should be checked in public
databases (Table I), where a huge
number of piRNA sequences discovered by previous large-scale
sequencing studies have been deposited.
As new TE and piRNA molecules accumulate, there
remains to be insufficient understanding in their activities,
interactome with other molecules or functions in cellular
processes. Therefore, physiological studies of these newly
discovered molecules are essential. In particular, the expression
of individual TE/piRNA transcripts should be validated by reverse
transcription-quantitative PCR (RT-qPCR), in situ
hybridization or Northern blotting. Western blotting and
immunofluorescence detection of the TE-derived proteins, such as
Orf2 protein from LINE-1, can help to validate the expression of
the TE mobilization machinery itself. Similarly, purification or
direct visualization (such as by using electron microscopy) of
replication complexes, such as ribonucleoprotein particles or
virus-like particles, can also facilitate the detection of the
assembled replication intermediates. This can consequently unravel
information on the functionality of TEs (91). Furthermore, the ability of TEs to
induce double-stranded breaks can be monitored using the
immunofluorescence imaging of the γ-H2AX foci (92) or by using single-cell gel
electrophoresis analysis (93). In
addition, co-immunoprecipitation of the piRNAs with Piwi proteins
is an important method for the verification of any small RNA
detected as a true piRNA. Since piRNAs are defined as Piwi-binding
RNAs (55), the Piwi complex should
be isolated using a Piwi-specific antibody by immunoprecipitation.
Subsequently, RNAs associated with Piwi protein should be extracted
and sequenced or quantified by RT-qPCR to validate their
involvement in the piRNA/Piwi pathway (94). The mechanism underlying the function
of individual piRNAs or TEs can be evaluated by modulating their
expression levels, by either overexpression using their
corresponding mimics or suppression of their transcription using
RNA interference or CRISPR-Cas9 approaches, before assessing
changes in cell physiology. However, it is also important to note
that the repetitive nature of TE sequences must be considered
during experimental design and analysis. In particular, short DNA
oligonucleotide sequences for PCR, short-hairpin RNAs or
CRISPR-Cas9 need to be carefully designed and validated to ensure
their specificity for a unique target.
In addition to wet laboratory methods,
identification of TE/piRNA feature, functions and exploration of
underlying molecular mechanisms have become important areas of
research in computational biology (95,96). A
number of bioinformatics methods have been proposed for the deeper
characterization of these molecules, where a number of analytical
software programs are readily available to the scientific community
in the form of the databases aforementioned (Table I). Furthermore, integrative data
analysis protocols that can simultaneously process multiple
heterogeneous datasets facilitate the discovery of complex
relationships between different types of piRNAs and TEs by
assembling various piRNA-TE-mRNA coexpression networks (97). Based on the knowledge obtained from
target prediction algorithms and information on protein-protein
interactions within these networks, computational analysis can be
used to predict novel TE/piRNA regulatory functions (98). This approach has already been
applied, for example, by Liu et al (99). Based on network construction, they
predicted cancer-associated piRNA-mRNA and piRNA-lncRNA
interactions as well as piRNA regulatory functions in breast, head
and neck, kidney and lung cancer (99). However, functional analyzes can also
be enabled by using different freely accessible tools. For example,
the piPipes (88) software includes
various independent tools to analyze different types of datasets,
including small RNA sequencing, RNA sequencing, degradome- and
CAGE-sequencing, chromatin immunoprecipitation- sequencing and
genomic DNA sequencing, to provide information on TE expression, TE
insertions, structural variation and piRNA-induced cleavage product
data for comprehensively studying piRNA-TE interactions. Similarly,
TEtools (100) maps unassembled
reads of RNA sequencing data obtained from previous classical mRNA
and small RNA analyzes, which has been used by Lerat et al
(100) to show the association in
expression between TEs and piRNA precursor genes. In conclusion,
computational biology has become an integral part of large-scale,
big-data studies due to the enhanced supercomputing and data
storage capabilities, coupled with the continuously optimized
algorithms.
TEs and piRNAs in somatic and cancer
cells
Accumulating evidence has documented the important
roles of TEs and piRNAs in human carcinogenesis (6,66,101,102). Mobile TEs are considered to be
highly mutagenic, where their transposition can cause mutational
events that are associated with cancer development and progression
(102). Transposition can induce
the rearrangement of host genes or their regulatory elements by
creating frameshifts and premature stop codons or by exon skipping.
For example, TE insertions into DNA repair-associated genes, such
as breast cancer type 1/2 susceptibility protein (103) or retinoblastoma 1 (104), can cause genomic instability and
activate a number of oncogenic pathways. Increased transcription of
TEs has also been reported in various of types of cancers,
including lymphoma (105),
colorectal cancer (106) and
bladder cancer (107). Therefore,
it is generally considered to be detrimental. Oncogenic processes
associated with TE reactivation include the expression of
TE-derived endonucleases, which can induce DNA breaks and genomic
instability (108), whereas the
activation of TE promoters that can also lead to oncogene
activation (109). For example,
reactivation of ancient LTRs has been associated with the aberrant
oncogenic transcription of the Erb-B2 receptor tyrosine kinase
4 gene in anaplastic large-cell lymphoma (110). In addition, previous studies have
revealed that the accumulation of RNA transcripts and
extrachromosomal DNA copies derived from TEs may trigger an innate
immune response leading to autoimmune diseases and inflammation
(111–113). Chiappinelli et al (111) showed that the presence of TE
copies in the cytoplasm can activate cellular viral defense
responses and activate interferon signaling in cancer cells. This
activation could induce lymphocyte recruitent, reversing immune
tolerance in tumors (111). An
important consequence of this process is that the increased
transcription of TEs can mediate not only deleterious but also
beneficial effects, because this increased transcription can also
lead to the activation of the viral recognition pathway and immune
system-mediated cancer cell death (111,112).
Although piRNAs were originally described in
germline cells as suppressors of TEs that function to preserve
genome integrity during development, the roles of piRNAs in somatic
and cancer cells have also been previously documented (6,114,115). Somatic functions of the piRNA
pathway are associated not only with TE silencing but also with
genome rearrangement and epigenetic programming, with biological
roles in stem cell functions, differentiation and malignant
transformation. Despite the high number of piRNAs encoded in the
human genome, Martinez et al (116) demonstrated that only a small
fraction of piRNAs are consistently expressed in both somatic
non-malignant and tumor tissues derived from 10 different
anatomical sites: Bladder, breast, colon, head and neck, kidney,
lung, prostate, stomach, thyroid and uterine corpus available from
The Cancer Genome Atlas (TCGA) consortium. However, the expression
patterns of these piRNAs differed sufficiently in that they can be
applied to identify their corresponding tissue of origin and
differentiate tumors from non-malignant tissues in a cancer-type
specific manner (116). A number
of studies have shown that differential expression of piRNAs can be
detected in various human cancers, including multiple myeloma (MM),
breast, lung and gastric cancers (6,90,117,118). At present, the role of piRNAs in
cancer has been extensively reviewed elsewhere (6,90,117,118). However, piRNAs have been defined
to be either oncogenes or tumor suppressors. For example, piRNA-823
is among the most extensively studied of piRNA molecules in the
context of human cancer (119).
Reduced piRNA-823 expression was observed in gastric cancer
(118) or renal cell carcinoma
(120). Overexpression of this
piRNA using piRNA mimics in gastric cancer cell lines was shown to
inhibit cell proliferation, the results of which were supported by
similar inhibitory effects of tumor growth in a xenograft nude
mouse model (118). However,
piR-823 expression was found to be increased in patients with
breast cancer (120) and MM
(121,122), where it contributes to
tumorigenesis. This suggests that the role of piR-823 is rather
disease-specific (119). The
expression profile of particular piRNAs can be applied to delineate
clinical features, including histological subgroups, disease stages
or survival, which was also shown by Martinez et al
(116) in 12 different tumor
types. The findings that piRNA expression can associate with poorer
clinical outcomes, particularly in breast cancer, suggest that
these molecules can be utilized as a novel class of cancer
biomarkers (116).
Similar to piRNAs, a number of studies have also
documented the aberrant expression of Piwi proteins in a variety of
somatic cancers. Hiwi was demonstrated to be overexpressed in
testicular tumors (121) and
gastric cancer (122,123), whereas Hili expression was found
to be increased in breast cancer, colon cancer, gastrointestinal
stromal tumors, renal cell carcinoma and endometrial carcinoma
(124). In particular, as
diagnostic procedures transition from biopsies towards less
invasive methods, extracellular piRNAs may serve to be an
attractive alternative biomarker of cancer. The majority of studies
in this field have been mainly focused on the application of
measuring miRNAs in the blood circulation. Similar to miRNAs,
piRNAs remain largely stable in the blood plasma and have the
ability to survive a wide range of incubation and storage
conditions regularly used in the laboratory (125). A previous study of piRNAs in
gastric cancer found that, compared with existing miRNA-based
biomarkers (miR-106a and miR-17), piRNAs showed higher sensitivity
and specificity, particularly piR-823 and piR-651 (126).
Although the number of studies on dysregulated
expression of TEs and piRNAs in cancer is rapidly accumulating,
conclusions remain preliminary and specific functions of these
molecules in cancer physiology remain poorly understood. In
addition, the majority of the reported findings is only
correlative, whereby altered expression does not necessarily mean
that they have a causative role in oncogenesis. Instead, it may
merely be a consequence of other processes associated with
oncogenesis.
TEs in leukemia and MDS
The role of TEs in hematopoiesis is slowly becoming
unraveled (113,127). Although evidence in support of a
role for TE in leukemia is gradually increasing, its functional
consequence in MDS remains scarce and existing studies generally
focused on AML rather than MDS.
The expression of TEs in leukemia has been recently
measured by Colombo et al (113,127), who investigated the mobilization
of TEs in AML. They first measured TE expression in pre-leukemic
HSCs, leukemic stem cells (LSCs) and leukemic blasts from seven
patients with AML in freely-available sequencing data produced by
Corces et al (128). From
RNA sequencing data obtained from Wang et al (129), significant downregulation of a
number of TEs, including the SINE Alu and LTRs ERV1, ERVL and ERVK)
retrotransposons, was reported in LSCs compared with that in
preleukemic HSCs and blasts (130). Furthermore, they also showed that
the expression of TEs, especially that of LTR retrotransposons ERV3
and ERVL, is significantly reduced in high-risk MDS (n=6) compared
with that in patients with low-risk MDS (n=6) (130). Since this suppression of TEs was
associated with the significant reduction in the activities of
several immune system, interferon and inflammation-related
pathways, induction of TE expression in low-risk cases was proposed
to be a potential mechanism for immune system-mediated clearance of
cancer cells through the viral recognition pathway (130). By contrast, TE suppression in
high-risk cases of AML may enable the AML cells to escape immune
surveillance (113).
In a subsequent study, Colombo et al
(127) explored the transcriptome
data of 178 patients with AML from The Cancer Genome Atlas (TCGA)
with respect to the mutation-specific dysregulation of TEs and
correlated the TE expression profile to the respective
transcription networks. However, since the sequencing libraries in
TCGA were prepared using the polyA selection method, a number of
TEs, which are mainly comprised of SINEs without polyA tails,
remained undetectable from the dataset (127). The highest numbers of altered TE
transcripts were associated with promyelocytic leukemia/retinoic
acid receptor-α (PML-RARα) translocation and mutations in
mitochondrially encoded cytochrome c oxidase II (MTCO2),
nucleophosmin 1 (NMP1), structural maintenance of chromosomes 1a
and runt-related transcription factor 2, with only minimal overlap
(127). PML-RARα, MTCO2 and NMP1
were generally associated with the upregulation of TEs, whereas
FMS-like tyrosine kinase 3, isocitrate dehydrogenase 1, TP53 and
neuroblastoma-RAS were predominantly or uniquely associated with
the downregulation of TEs (127).
Although CpG methylation has been shown to regulate TE expression
(111,112,130), DNA methyltransferase (DNMT)3A and
tet methylcytosine dioxygenase 2 mutations were associated with
only minimal TE dysregulation (127). In addition, a TE expression
signature was found to accurately predict AML prognosis independent
of mutation-based and coding gene expression-based risk
stratification (127). In total,
14 candidate prognostic TE transcripts were identified, namely TEs
from Alu (AluJo and AluSq2), ERV1 (LTR24C, LTR53, MER101B, MER31-1
and LTR45B), ERVK (MER11A and LTR14A), ERVL (MER77), L1 (putative
monooxygenase locus 2) and Tc/mariner (MER44C, Tigger5A and
Tigger9b) subfamilies, all of which were associated with AML
prognosis (127). These 14 TEs
constituted a diverse group of transcripts, some of which were
classified as low risk and some as high risk, likely due to their
naturally diverse functions (127). The validity of their prognostic
power was successfully demonstrated in two independently downloaded
datasets consisting of cohorts of 284 pediatric patients with AML
(131) and 19 adult patients with
relapsed AML (132). From this
analysis, an improved prognostic algorithm utilizing a
comprehensive model that includes the mutational status,
cytogenetic status, coding gene expression and expression profiles
of the 14 TEs was proposed for the prognosis of AML (127).
Recently, Zeng et al (133) explored the presence of TEs in the
open chromatin regions of genomes of patients with AML produced
using assay for transposase-accessible chromatin with HTS data
performed by Corces et al (128). A cluster of ~21,000 TEs,
consisting primarily of MIRs and LINE-2, were found to be more
abundant in AML cells compared with those in normal blood cells
(133). Subsequent pathway
enrichment analysis revealed that nearby genes, such as FBJ
Murine Osteosarcoma Viral Oncogene Homolog B (FOSB), FBJ Murine
Osteosarcoma Viral Oncogene Homolog (FOS) and V-Jun Avian
Sarcoma Virus 17 Oncogene Homolog (JUN), in the open chromatin
regions of these MIRs were involved in leukemogenesis (133). Using the AML cell line Kasumi-1,
several MIRs (particularly MIR9 and MIR18) were validated to be
functional enhancers (133).
Although MIRs are potentially involved in myeloid leukemogenesis,
the specific mechanism for MIR involvement in AML remains unknown.
However, it was speculated that MIRs can function as independent
enhancers through interaction with other important upstream
regulators to alter gene expression in AML cells (133).
A recent study by Deniz et al (134) revealed ERVs to be potentially
oncogenic enhancers and regulators in AML. They identified six ERV
families (LTR2B, LTR2C, LTR5B, LTR5_Hs, LTR12C and LTR13A) with
AML-associated enhancer chromatin signatures which are enriched in
the binding of key regulators of hematopoiesis and AML pathogenesis
(134). Furthermore, correlations
were found between differential chromatin accessibility at 20 ERVs
and the expression of nearby genes, some of which were linked to
AML prognosis (134). CRISPR-based
experiments additionally identified five ERVs that can function as
enhancers in leukemia cells and 13 different elements, the
silencing of which led to the downregulation of nearby genes
(134). Similar to observations by
Colombo et al (127),
variations in ERV activity were observed among patients with AML,
which appeared to be partly driven by their unique mutational
profiles (134).
Because genomic regions that contain TEs are highly
methylated and silenced by heterochromatin in somatic cells
(135), hypomethylating agents can
increase the expression of TEs in cancer cells. Since
hypomethylating compounds are used for the treatment of AML and
high-risk MDS (8,9), these compounds may at least partially
operate via the demethylation of TE promoters, which may in turn
induce ‘viral mimicry’ and interferon signaling to mediate leukemic
cell clearance (111,112). In addition, there has even been
speculation that the ‘viral mimicry’ pathways are likely to be
activated endogenously without hypomethylation therapy in low-risk
MDS to enable immune system-mediated control of low-risk AML
(113). Despite this clinically
proven efficacy of hypomethylating agents against MDS, only ~50%
patients benefit from this type of therapy (9). The majority of patients eventually
develop therapy resistance or disease relapse whilst still under
treatment (10,11). Therefore, understanding the
regulatory mechanism of TEs in MDS pathogenesis and during disease
treatment would facilitate the discovery of novel biomarkers for
response prediction and novel strategies for preventing relapse in
patients treated with hypomethylating agents.
piRNAs in leukemia and MDS
Germ cells, stem cells and cancer cells share key
biological characteristics, including the ability to rapidly
proliferate and self-renew (136).
Because the piRNA pathway maintains germline stem cells by
preserving the self-renewal mechanism, the same pathway may also
serve similar roles in the self-renewal of rapidly-dividing
hematopoietic stem and leukemic cells. However, reports on the
transcription and exact role of piRNAs in blood cells remain
scarce.
The first study on the function of the piRNA
pathway in hematopoiesis and leukemia was reported in 2001 by
Sharma et al (137), who
demonstrated that the Piwi protein is expressed in human
CD34+ hematopoietic stem and progenitor cells but not in
differentiated hematopoietic cell populations. In addition, it was
shown that transient overexpression of Piwi in human leukemia cells
resulted in a potent reduction in cell proliferation and activation
of programmed cell death, suggesting that Piwi functions as an
important negative developmental regulator, where they regulate
cell ‘stemness’ (137). However,
another previous study where all three mouse Piwi genes were
knocked out revealed no detectable effects on hematopoiesis
(138). This lead to the
conclusion that Piwi expression in HSCs is not required for normal
adult hematopoiesis or that Piwi function is redundant with other
proteins, possibly those in the Ago subfamily (138).
Over the past decade, piRNA dysregulation has been
repeatedly reported in various hematological malignancies,
including MM (139–141) and classical Hodgkin lymphoma (cHL)
(142). Yan et al (139) previously measured the expression
of piRNA-823 in MM and showed that it was upregulated in bone
marrow biopsies from patients with MM and four MM-derived cell
lines compared to samples from healthy individuals, which was in
turn positively associated with clinical stages. The silencing of
piRNA-823 also dysregulated the expression of cell cycle regulators
and apoptosis-related proteins, which was accompanied by the
inhibition of cell tumorigenicity (139). In addition, suppression of
piRNA-823 expression in MM cells resulted in the reduction of de
novo expression of DNA methyltransferases DNMT3A and 3B at both
mRNA and protein levels, which in turn led to a decrease in global
DNA methylation and reactivation of the tumor suppressor p16 INK4A,
which was previously inhibited by methylation (139). piRNA-823 accumulation was also
found in MM-derived extracellular vesicles, which transported
piRNA-823 to endothelial cells effectively and promoted their
malignant transformation (140).
Cordeiro et al (142)
previously investigated the Piwi/piRNA pathway in cHL by measuring
the expression of Piwi proteins and three selected piRNAs piR-651,
piR-20365 and piR-20582. Piwi proteins were found to be expressed
mainly in cells derived from patients with cHL and cell lines,
suggesting that this pathway was active (142). At diagnosis, piR-651 expression
was found to be reduced in the serum of patients with cHL, whilst
after complete remission, piR-651 levels were increased to levels
similar to those of healthy individuals (142). Furthermore, lower levels of
piR-651 were found to be associated with the lack of complete
response to first line treatment, shorter disease-free and overall
survival (142).
To date, the function of the piRNA/Piwi axis in
leukemia has only been partially explored, with the majority of the
emphasis on Piwi proteins rather than piRNAs. Bamezai et al
(143) showed that Piwil4 showed
aberrantly high expression in >72% patients with AML. Depletion
of Piwil4 in mixed-lineage leukemia (MLL) rearranged AML cell lines
was found to impair cell proliferation and clonogenic growth whilst
delaying the onset of leukemia in mice (143). This depletion was found to be
associated with a global reduction in the levels of repressive
H3K9me3 signatures and an increase in the levels activating H3K4me3
signatures (143). In addition, it
was demonstrated that Piwil4 depletion altered the piRNA expression
profile in AML cells with rearranged MLL (143). The role of Piwi has also been
previously studied in chronic myelogenous leukemia (CML) (144). A lentiviral expression vector was
utilized for the overexpression of Hiwi in a CML cell line, which
was shown to inhibit CML cell proliferation and migration,
suggesting that Hiwi is a negative regulator of leukemogenesis
(144).
Unlike miRNAs, for which there is a large body of
information regarding their role in MDS and has been
comprehensively reviewed elsewhere (145,146), information on the role of other
types of sncRNAs remain limited. In terms of piRNA expression in
MDS, only preliminary data have been reported to date (147,148). Using sequencing of the small
RNAome, Beck et al (147)
demonstrated the enrichment of piRNAs in unsorted bone marrow cells
from patients with low-risk MDS, refractory anemia (RA). Because
this previous study was focusing on miRNAs, piRNA expression was
reported to be increased in patients with the RA status and
accounted for ~9% of total small RNA counts, compared with ~2% in
those with high-risk MDS and 1% in healthy individuals, without
studying which piRNA further (147). Additionally, transcription of
Piwil1 and Piwil2 was significantly upregulated in patients with RA
compared with that in healthy individuals and patients with
high-risk MDS (147). It was
concluded that piRNA enrichment may potentially protect DNA from
mutations in low-risk MDS cells, a mechanism that is not observed
in cells of high-risk MDS (147).
In a more recent study (148), the
extracellular small RNAome was explored by sequencing in the plasma
samples of patients with MDS. Although miRNAs was focused upon, a
number of piRNAs were also found to be dysregulated in MDS,
including the upregulation of hsa_piR_019914 and hsa_piR_020450,
and those associated with specific characteristics of the disease,
namely the upregulation of hsa_piR_000805 and downregulation of
hsa_piR_019420 in low-risk MDS (148). These early findings suggest that
piRNAs are dysregulated in MDS and might be informative as MDS
biomarkers. However, further studies of their transcription profile
and roles in MDS pathogenesis are warranted.
Conclusions
TE mobilization and piRNA expression are generally
increased in advanced malignancies and are considered deleterious.
Analogous to other types of cancers, these pathways were
anticipated to be endogenously activated in MDS and may contribute
to genome instability, where they in turn induce harmful changes
during disease progression and leukemic transformation. However,
early studies by Colombo et al (113) and Beck et al (147) reported clear enrichment of TEs and
piRNAs during early MDS and subsequent suppression as the disease
progressed (Fig. 2). These findings
led to an opposing hypothesis, which assumes that the induction of
TEs is a potential mechanism for the immune system-mediated
elimination of leukemic cells during early phases of the
disease.
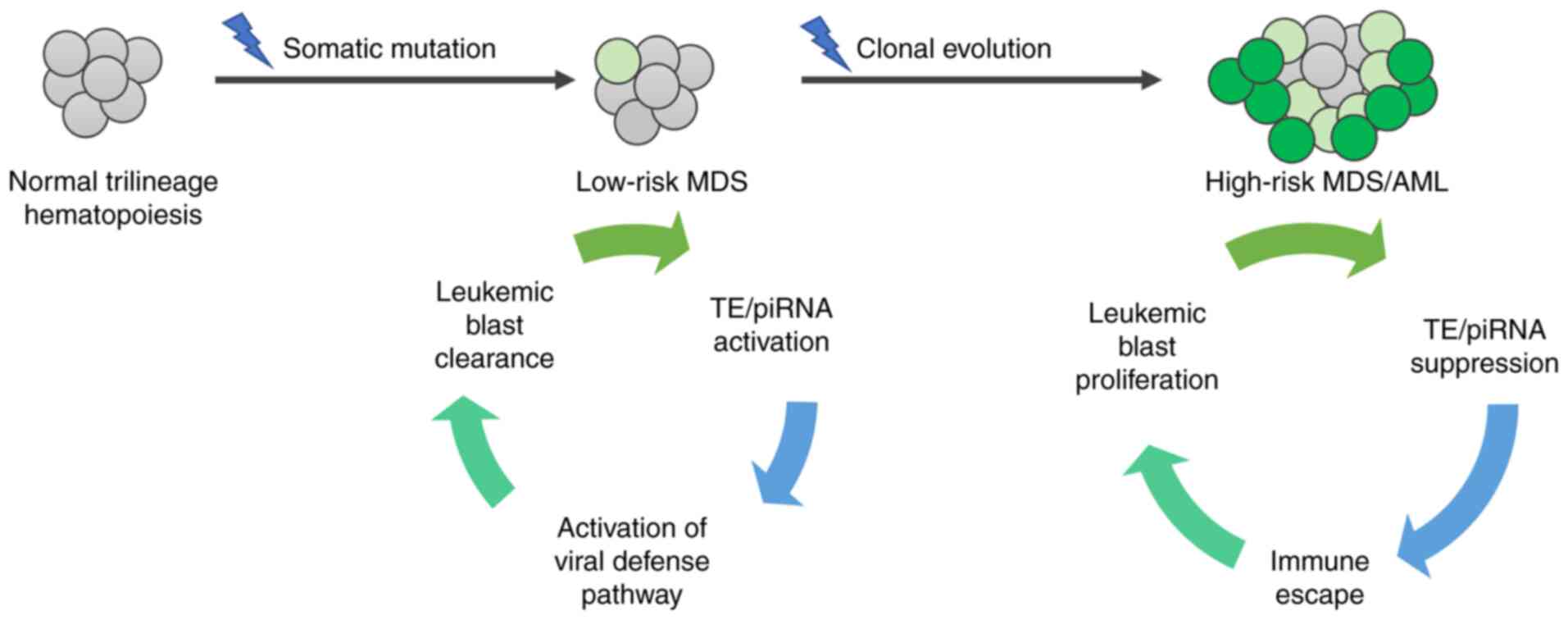 | Figure 2.TEs and piRNAs in MDS. A somatic
mutation in the genome of a healthy cell leads to the development
of myelodysplasia. In low-risk MDS, activated transcription of TEs
and piRNAs has been described by Colombo et al (113) and Beck et al (147), respectively. Increased TE
transcription induces the viral defense pathway, which may
facilitate the elimination of leukemic blasts and maintain the
non-proliferative status of the disease. However, subsequent
somatic mutations can mediate clonal evolution and the development
of high-risk MDS. In this stage, TE/piRNA expression is suppressed,
which leads to immune escape, leukemic blast proliferation and
further progression of the disease. PIWI, P-element-induced WImpy
testis; piRNA, PIWI-interacting RNA; TE, transposable element; MDS,
myelodysplastic syndrome. |
MDS is a largely heterogeneous disease that is
mainly classified on the clinicopathological criteria (149). Therefore, aberrant transcription
of novel classes of molecules, such as TEs and piRNAs, may be
potentially exploited as novel molecular markers of disease
progression or to individualize patient management. In addition,
because hypomethylating therapy has been associated with the
induction of TE expression in colorectal cancer cells (112), understanding the regulation of TEs
during MDS treatment by this type of compound may assist in the
definition of novel predictive markers for therapy response and
prevention of relapse.
Beyond their possible role as biomarkers, a number
of early studies have also demonstrated the potential benefits of
using piRNA molecules for therapy (90). Since piRNA dysregulation can alter
the biological features of cancer cells through various regulatory
mechanisms, modulating their expression may reverse certain cancer
phenotypes. Several therapeutic strategies can be designed, the
most appealing of which involves the use of synthetic piRNAs
capable of blocking the synthesis of cancer-related proteins by
binding to mRNAs. Previously, artificial piRNAs introduced to male
embryonic germ cells were reported to be sufficient to induce
piRNA-dependent gene silencing (150). Although enthusiasm for
sncRNA-based cancer therapy is on the rise, it is necessary to
unravel the precise molecular mechanisms and specific downstream
targets of these agents. The current state of knowledge, evolving
principles and challenges associated with the delivery and
mitigation of the off-target effects of sncRNA-based therapeutic
approaches have been previously summarized elsewhere (151,152).
Whilst investigations of TEs and piRNAs in leukemia
remain in its infancy, further progress in the field of disease
classification and monitoring, in addition to the area of
therapeutic intervention is expected in the future. This is due to
the continuous improvements in the understanding of the mechanisms
and functions underlying TE/piRNA-associated processes in normal
and leukemic cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by AZV CR (grant no.
NU20-03-00412) and MH CZ-DRO (UHKT; grant no. 00023736).
Availability of data and materials
Not applicable.
Authors' contributions
MDM: conceptualization, writing of original draft,
revision and funding acquisition. ZK: writing of original
draft.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cheng H, Zheng Z and Cheng T: New
paradigms on hematopoietic stem cell differentiation. Protein Cell.
11:34–44. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Haas S, Trumpp A and Milsom MD: Causes and
consequences of hematopoietic stem cell heterogeneity. Cell Stem
Cell. 22:627–638. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sperling AS, Gibson CJ and Ebert BL: The
genetics of myelodysplastic syndrome: From clonal haematopoiesis to
secondary leukaemia. Nat Rev Cancer. 17:5–19. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aguilera A and García-Muse T: Causes of
genome instability. Annu Rev Genet. 47:1–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ali A, Han K and Liang P: Role of
transposable elements in gene regulation in the human genome. Life
(Basel). 11:1182021.PubMed/NCBI
|
|
6
|
Liu Y, Dou M, Song X, Dong Y, Liu S, Liu
H, Tao J, Li W, Yin X and Xu W: The emerging role of the piRNA/piwi
complex in cancer. Mol Cancer. 18:1232019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garcia-Manero G, Chien KS and
Montalban-Bravo G: Myelodysplastic syndromes: 2021 Update on
diagnosis, risk stratification and management. Am J Hematol.
95:1399–1420. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feld J, Belasen A and Navada SC:
Myelodysplastic syndromes: A review of therapeutic progress over
the past 10 years. Expert Rev Anticancer Ther. 20:465–482. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bewersdorf JP, Carraway H and Prebet T:
Emerging treatment options for patients with high-risk
myelodysplastic syndrome. Ther Adv Hematol.
11:20406207209550062020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeidan AM, Stahl M, Deveaux M, Giri S,
Huntington S, Podoltsev N, Wang R, Ma X, Davidoff AJ and Gore SD:
Counseling patients with higher-risk MDS regarding survival with
azacitidine therapy: Are we using realistic estimates? Blood Cancer
J. 8:552018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Santini V: How I treat MDS after
hypomethylating agent failure. Blood. 133:521–529. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferrando AA and López-Otín C: Clonal
evolution in leukemia. Nat Med. 23:1135–1145. Oct 6–2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen J, Kao YR, Sun D, Todorova TI,
Reynolds D, Narayanagari SR, Montagna C, Will B, Verma A and Steidl
U: Myelodysplastic syndrome progression to acute myeloid leukemia
at the stem cell level. Nat Med. 25:103–110. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hosono N: Genetic abnormalities and
pathophysiology of MDS. Int J Clin Oncol. 24:885–892. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shallis RM, Ahmad R and Zeidan AM: The
genetic and molecular pathogenesis of myelodysplastic syndromes.
Eur J Haematol. 101:260–271. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bejar R, Stevenson K, Abdel-Wahab O,
Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine
RL, Neuberg D and Ebert BL: Clinical effect of point mutations in
myelodysplastic syndromes. N Engl J Med. 364:2496–2506. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Papaemmanuil E, Gerstung M, Malcovati L,
Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC,
Pellagatti A, et al: Clinical and biological implications of driver
mutations in myelodysplastic syndromes. Blood. 122:3616–3627, 3699.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Haferlach T, Nagata Y, Grossmann V, Okuno
Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T,
et al: Landscape of genetic lesions in 944 patients with
myelodysplastic syndromes. Leukemia. 28:241–247. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Spaulding TP, Stockton SS and Savona MR:
The evolving role of next generation sequencing in myelodysplastic
syndromes. Br J Haematol. 188:224–239. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Klein SJ and O'Neill RJ: Transposable
elements: Genome innovation, chromosome diversity, and centromere
conflict. Chromosom Res. 26:5–23. 2018. View Article : Google Scholar
|
|
21
|
McClintock B: The origin and behavior of
mutable loci in maize. Proc Natl Acad Sci USA. 36:344–355. 1950.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kojima KK: Structural and sequence
diversity of eukaryotic transposable elements. Genes Genet Syst.
94:233–252. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barabas O: Snapshots of a genetic
cut-and-paste. Nature. 575:447–448. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Muñoz-López M and García-Pérez J: DNA
transposons: Nature and applications in genomics. Curr Genomics.
11:115–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wicker T, Sabot F, Hua-Van A, Bennetzen
JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O,
et al: A unified classification system for eukaryotic transposable
elements. Nat Rev Genet. 8:973–982. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grandi N and Tramontano E: Human
endogenous retroviruses are ancient acquired elements still shaping
innate immune responses. Front Immunol. 9:20392018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Johnson WE: Origins and evolutionary
consequences of ancient endogenous retroviruses. Nat Rev Microbiol.
17:355–370. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han JS: Non-long terminal repeat (non-LTR)
retrotransposons: Mechanisms, recent developments, and unanswered
questions. Mob DNA. 1:152010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dewannieux M and Heidmann T: LINEs, SINEs
and processed pseudogenes: Parasitic strategies for genome
modeling. Cytogenet Genome Res. 110:35–48. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mita P, Wudzinska A, Sun X, Andrade J,
Nayak S, Kahler DJ, Badri S, LaCava J, Ueberheide B, Yun CY, et al:
LINE-1 protein localization and functional dynamics during the cell
cycle. Elife. 7:e300582018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Martin SL: The ORF1 protein encoded by
LINE-1: Structure and function during L1 retrotransposition. J
Biomed Biotechnol. 2006:456212006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ardeljan D, Wang X, Oghbaie M, Taylor MS,
Husband D, Deshpande V, Steranka JP, Gorbounov M, Yang WR, Sie B,
et al: LINE-1 ORF2p expression is nearly imperceptible in human
cancers. Mob DNA. 11:12019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brouha B, Schustak J, Badge RM,
Lutz-Prigge S, Farley AH, Morant JV and Kazazian HH Jr: Hot L1s
account for the bulk of retrotransposition in the human population.
Proc Natl Acad Sci USA. 100:5280–5285. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Del Re B and Giorgi G: Long INterspersed
element-1 mobility as a sensor of environmental stresses. Environ
Mol Mutagen. 61:465–493. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Okada N, Hamada M, Ogiwara I and Ohshima
K: SINEs and LINEs share common 3′ sequences: A review. Gene.
205:229–243. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Carnevali D, Conti A, Pellegrini M and
Dieci G: Whole-genome expression analysis of mammalian-wide
interspersed repeat elements in human cell lines. DNA Res.
24:59–69. 2017.PubMed/NCBI
|
|
37
|
Cordaux R, Hedges DJ, Herke SW and Batzer
MA: Estimating the retrotransposition rate of human Alu elements.
Gene. 373:134–137. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Piégu B, Bire S, Arensburger P and Bigot
Y: A survey of transposable element classification systems-a call
for a fundamental update to meet the challenge of their diversity
and complexity. Mol Phylogenet Evol. 86:90–109. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bourque G, Burns KH, Gehring M, Gorbunova
V, Seluanov A, Hammell M, Imbeault M, Izsvák Z, Levin HL, Macfarlan
TS, et al: Ten things you should know about transposable elements.
Genome Biol. 19:1992018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Campos-Sánchez R, Cremona MA, Pini A,
Chiaromonte F and Makova KD: Integration and fixation preferences
of human and mouse endogenous retroviruses uncovered with
functional data analysis. PLoS Comput Biol. 12:e10049562016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hancks DC and Kazazian HH Jr: Roles for
retrotransposon insertions in human disease. Mob DNA. 7:92016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang CR, Burns KH and Boeke JD: Active
transposition in genomes. Annu Rev Genet. 46:651–675. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang N, Bao Z, Zhang X, Eddy SR and
Wessler SR: Pack-MULE transposable elements mediate gene evolution
in plants. Nature. 431:569–573. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cowley M and Oakey RJ: Transposable
elements re-wire and fine-tune the transcriptome. PLoS Genet.
9:e10032342013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Clayton EA, Rishishwar L, Huang TC, Gulati
S, Ban D, McDonald JF and Jordan IK: An atlas of transposable
element-derived alternative splicing in cancer. Philos Trans R Soc
Lond B Biol Sci. 375:201903422020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cosby RL, Chang NC and Feschotte C:
Host-transposon interactions: Conflict, cooperation, and cooption.
Genes Dev. 33:1098–1116. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Riordan JD and Dupuy AJ: Domesticated
transposable element gene products in human cancer. Mob Genet
Elements. 3:e266932013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sinzelle L, Izsvák Z and Ivics Z:
Molecular domestication of transposable elements: From detrimental
parasites to useful host genes. Cell Mol Life Sci. 66:1073–1093.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Friedli M and Trono D: The developmental
control of transposable elements and the evolution of higher
species. Annu Rev Cell Dev Biol. 31:429–451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang P, Wang Y and Macfarlan TS: The role
of KRAB-ZFPs in transposable element repression and mammalian
evolution. Trends Genet. 33:871–881. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Imbeault M, Helleboid PY and Trono D: KRAB
zinc-finger proteins contribute to the evolution of gene regulatory
networks. Nature. 543:550–554. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Deniz Ö, Frost JM and Branco MR:
Regulation of transposable elements by DNA modifications. Nat Rev
Genet. 20:417–431. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Virciglio C, Abel Y and Rederstorff M:
Regulatory non-coding RNAs: An overview. Methods Mol Biol.
2300:3–9. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Marzec M: New insights into the function
of mammalian Argonaute2. PLoS Genet. 16:e10090582020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ozata DM, Gainetdinov I, Zoch A, O'Carroll
D and Zamore PD: Piwi-interacting RNAs: Small RNAs with big
functions. Nat Rev Genet. 20:89–108. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tian Y, Simanshu DK, Ma JB and Patel DJ:
Structural basis for piRNA 2′-O-methylated 3′-end recognition by
Piwi PAZ (Piwi/Argonaute/Zwille) domains. Proc Natl Acad Sci USA.
108:903–910. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mei Y, Wang Y, Kumari P, Shetty AC, Clark
D, Gable T, MacKerell AD, Ma MZ, Weber DJ, Yang AJ, et al: A
piRNA-like small RNA interacts with and modulates p-ERM proteins in
human somatic cells. Nat Commun. 6:73162015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang J, Zhang P, Lu Y, Li Y, Zheng Y, Kan
Y, Chen R and He S: PiRBase: A comprehensive database of piRNA
sequences. Nucleic Acids Res. 47:D175–D180. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yamanaka S, Siomi MC and Siomi H: PiRNA
clusters and open chromatin structure. Mob DNA. 5:222014.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sato K and Siomi H: Piwi proteins and
their slicer activity in piRNA biogenesis and transposon silencing.
Enzymes. 32:137–162. 2012. View Article : Google Scholar
|
|
61
|
Czech B and Hannon GJ: One loop to rule
them all: The ping-pong cycle and piRNA-guided silencing. Trends
Biochem Sci. 41:324–337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Han YN, Li Y, Xia SQ, Zhang YY, Zheng JH
and Li W: Piwi proteins and Piwi-interacting RNA: Emerging roles in
cancer. Cell Physiol Biochem. 44:1–20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Grimson A, Srivastava M, Fahey B,
Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS and Bartel
DP: Early origins and evolution of microRNAs and Piwi-interacting
RNAs in animals. Nature. 455:1193–1197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Beyret E, Liu N and Lin H: piRNA
biogenesis during adult spermatogenesis in mice is independent of
the ping-pong mechanism. Cell Res. 22:1429–1439. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Czech B, Munafò M, Ciabrelli F, Eastwood
EL, Fabry MH, Kneuss E and Hannon GJ: piRNA-guided genome defense:
From biogenesis to silencing. Annu Rev Genet. 52:131–157. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Balmeh N, Mahmoudi S and
Karabedianhajiabadi A: piRNAs and Piwi proteins: From biogenesis to
their role in cancer. Gene Rep. 22:1010132021. View Article : Google Scholar
|
|
67
|
Peng JC, Valouev A, Liu N and Lin H: Piwi
maintains germline stem cells and oogenesis in Drosophila
through negative regulation of polycomb group proteins. Nat Genet.
48:283–291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sadoughi F, Mirhashemi SM and Asemi Z:
Epigenetic roles of Piwi proteins and piRNAs in colorectal cancer.
Cancer Cell Int. 21:3282021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tóth KF, Pezic D, Stuwe E and Webster A:
The pirna pathway guards the germline genome against transposable
elements. Adv Exp Med Biol. 886:51–77. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Iwasaki YW, Murano K, Ishizu H, Shibuya A,
Iyoda Y, Siomi MC, Siomi H and Saito K: Piwi modulates chromatin
accessibility by regulating multiple factors including histone H1
to repress transposons. Mol Cell. 63:408–419. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Faulkner GJ, Kimura Y, Daub CO, Wani S,
Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T,
et al: The regulated retrotransposon transcriptome of mammalian
cells. Nat Genet. 41:563–571. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
72
|
Marinov GK, Wang J, Handler D, Wold BJ,
Weng Z, Hannon GJ, Aravin AA, Zamore PD, Brennecke J and Toth KF:
Pitfalls of mapping high-throughput sequencing data to repetitive
sequences: Piwi's genomic targets still not identified. Dev Cell.
32:765–771. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Amarasinghe SL, Su S, Dong X, Zappia L,
Ritchie ME and Gouil Q: Opportunities and challenges in long-read
sequencing data analysis. Genome Biol. 21:302020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Goerner-Potvin P and Bourque G:
Computational tools to unmask transposable elements. Nat Rev Genet.
19:688–704. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Tarailo-Graovac M and Chen N: Using
RepeatMasker to identify repetitive elements in genomic sequences.
Curr Protoc Bioinforma Chapter 4. Unit 4.10. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bao W, Kojima KK and Kohany O: Repbase
update, a database of repetitive elements in eukaryotic genomes.
Mob DNA. 6:112015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wheeler TJ, Clements J, Eddy SR, Hubley R,
Jones TA, Jurka J, Smit AF and Finn RD: Dfam: A database of
repetitive DNA based on profile hidden Markov models. Nucleic Acids
Res. 41:D70–D82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Flynn JM, Hubley R, Goubert C, Rosen J,
Clark AG, Feschotte C and Smit AF: RepeatModeler2 for automated
genomic discovery of transposable element families. Proc Natl Acad
Sci USA. 117:9451–9457. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Novák P, Neumann P and Macas J:
Graph-based clustering and characterization of repetitive sequences
in next-generation sequencing data. BMC Bioinformatics. 11:3782010.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Jin Y, Tam OH, Paniagua E and Hammell M:
TEtranscripts: A package for including transposable elements in
differential expression analysis of RNA-seq datasets.
Bioinformatics. 31:3593–3599. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ye J, Xu M, Tian X, Cai S and Zeng S:
Research advances in the detection of miRNA. J Pharm Anal.
9:217–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ray R and Pandey P: piRNA analysis
framework from small RNA-Seq data by a novel cluster prediction
tool-PILFER. Genomics. 110:355–365. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Munafó DB and Robb GB: Optimization of
enzymatic reaction conditions for generating representative pools
of cDNA from small RNA. RNA. 16:2537–2552. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Leshkowitz D, Horn-Saban S, Parmet Y and
Feldmesser E: Differences in microRNA detection levels are
technology and sequence dependent. RNA. 19:527–538. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Dard-Dascot C, Naquin D, d'Aubenton-Carafa
Y, Alix K, Thermes C and van Dijk E: Systematic comparison of small
RNA library preparation protocols for next-generation sequencing.
BMC Genomics. 19:1182018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Fuchs RT, Sun Z, Zhuang F and Robb GB:
Bias in ligation-based small RNA sequencing library construction is
determined by adaptor and RNA structure. PLoS One. 10:e01260492015.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Song Y, Liu KJ and Wang TH: Elimination of
ligation dependent artifacts in T4 RNA ligase to achieve high
efficiency and low bias microRNA capture. PLoS One. 9:e946192014.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Han BW, Wang W, Zamore PD and Weng Z:
piPipes: A set of pipelines for piRNA and transposon analysis via
small RNA-seq, RNA-seq, degradome- and CAGE-seq, ChIP-seq and
genomic DNA sequencing. Bioinformatics. 31:593–595. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Li J, Kho AT, Chase RP, Pantano L, Farnam
L, Amr SS and Tantisira KG: COMPSRA: A COMprehensive platform for
small RNA-Seq data analysis. Sci Rep. 10:45522020. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yu Y, Xiao J and Hann SS: The emerging
roles of PIWI-interacting RNA in human cancers. Cancer Manag Res.
11:5895–5909. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lanciano S and Cristofari G: Measuring and
interpreting transposable element expression. Nat Rev Genet.
21:721–736. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lee Y, Wang Q, Shuryak I, Brenner DJ and
Turner HC: Development of a high-throughput γ-H2AX assay based on
imaging flow cytometry. Radiat Oncol. 14:1502019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Langie SA, Azqueta A and Collins AR: The
comet assay: Past, present, and future. Front Genet. 6:2662015.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zuo L, Wang Z, Tan Y, Chen X and Luo X:
piRNAs and their functions in the brain. Int J Hum Genet. 16:53–60.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
da Costa AH, dos Santos RAC and Cerri R:
Investigating deep feedforward neural networks for classification
of transposon-derived piRNAs. Complex Intell Syst. 1–11. 2021.
|
|
96
|
Giassa IC and Alexiou P: Bioinformatics
and machine learning approaches to understand the regulation of
mobile genetic elements. Biology (Basel). 10:8962021.PubMed/NCBI
|
|
97
|
Stuart JM, Segal E, Koller D and Kim SK: A
gene-coexpression network for global discovery of conserved genetic
modules. Science. 302:249–255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zhao Y, Wang J, Chen J, Zhang X, Guo M and
Yu G: A literature review of gene function prediction by modeling
gene ontology. Front Genet. 11:4002020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Liu Y, Zhang J, Li A, Liu Z, He Z, Yuan X
and Tuo S: Prediction of cancer-associated piRNA-mRNA and
piRNA-lncRNA interactions by integrated analysis of expression and
sequence data. Tsinghua Sci Technol. 23:115–125. 2018. View Article : Google Scholar
|
|
100
|
Lerat E, Fablet M, Modolo L, Lopez-Maestre
H and Vieira C: TEtools facilitates big data expression analysis of
transposable elements and reveals an antagonism between their
activity and that of piRNA genes. Nucleic Acids Res.
45:e172017.PubMed/NCBI
|
|
101
|
Moyano M and Stefani G: PiRNA involvement
in genome stability and human cancer. J Hematol Oncol. 8:382015.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Anwar SL, Wulaningsih W and Lehmann U:
Transposable elements in human cancer: Causes and consequences of
deregulation. Int J Mol Sci. 18:9742017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Teugels E, De Brakeleer S, Goelen G,
Lissens W, Sermijn E and De Grève J: De novo Alu element insertions
targeted to a sequence common to the BRCA1 and BRCA2 genes. Hum
Mutat. 26:2842005. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Rodríguez-Martín C, Cidre F,
Fernández-Teijeiro A, Gómez-Mariano G, de la Vega L, Ramos P,
Zaballos Á, Monzón S and Alonso J: Familial retinoblastoma due to
intronic LINE-1 insertion causes aberrant and noncanonical mRNA
splicing of the RB1 gene. J Hum Genet. 61:463–466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Lamprecht B, Walter K, Kreher S, Kumar R,
Hummel M, Lenze D, Köchert K, Bouhlel MA, Richter J, Soler E, et
al: Derepression of an endogenous long terminal repeat activates
the CSF1R proto-oncogene in human lymphoma. Nat Med. 16:571–579.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lock FE, Babaian A, Zhang Y, Gagnier L,
Kuah S, Weberling A, Karimi MM and Mager DL: A novel isoform of
IL-33 revealed by screening for transposable element promoted genes
in human colorectal cancer. PLoS One. 12:e01806592017. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wolff EM, Byun HM, Han HF, Sharma S,
Nichols PW, Siegmund KD, Yang AS, Jones PA and Liang G:
Hypomethylation of a LINE-1 promoter activates an alternate
transcript of the MET oncogene in bladders with cancer. PLoS Genet.
6:e10009172010. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Hedges DJ and Deininger PL: Inviting
instability: Transposable elements, double-strand breaks, and the
maintenance of genome integrity. Mutat Res. 616:46–59. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Babaian A and Mager DL: Endogenous
retroviral promoter exaptation in human cancer. Mob DNA. 7:242016.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Scarfò I, Pellegrino E, Mereu E, Kwee I,
Agnelli L, Bergaggio E, Garaffo G, Vitale N, Caputo M, Machiorlatti
R, et al: Identification of a new subclass of ALK-negative ALCL
expressing aberrant levels of ERBB4 transcripts. Blood.
127:221–232. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Chiappinelli KB, Strissel PL, Desrichard
A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et
al: Inhibiting DNA methylation causes an interferon response in
cancer via dsRNA including endogenous retroviruses. Cell.
162:974–986. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Roulois D, Loo Yau H, Singhania R, Wang Y,
Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al:
DNA-demethylating agents target colorectal cancer cells by inducing
viral mimicry by endogenous transcripts. Cell. 162:961–973. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Colombo AR, Zubair A, Thiagarajan D,
Nuzhdin S, Triche TJ and Ramsingh G: Suppression of transposable
elements in leukemic stem cells. Sci Rep. 7:70292017. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ross RJ, Weiner MM and Lin H: Piwi
proteins and Piwi-interacting RNAs in the soma. Nature.
505:353–359. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Cheng Y, Wang Q, Jiang W, Bian Y, Zhou Y,
Gou A, Zhang W, Fu K and Shi W: Emerging roles of piRNAs in cancer:
Challenges and prospects. Aging (Albany NY). 11:9932–9946. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Martinez VD, Vucic EA, Thu KL, Hubaux R,
Enfield KS, Pikor LA, Becker-Santos DD, Brown CJ, Lam S and Lam WL:
Unique somatic and malignant expression patterns implicate
Piwi-interacting RNAs in cancer-type specific biology. Sci Rep.
5:104232015. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ng KW, Anderson C, Marshall EA, Minatel
BC, Enfield KS, Saprunoff HL, Lam WL and Martinez VD:
Piwi-interacting RNAs in cancer: Emerging functions and clinical
utility. Mol Cancer. 15:52016. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Cheng J, Deng H, Xiao B, Zhou H, Zhou F,
Shen Z and Guo J: piR-823, a novel non-coding small RNA,
demonstrates in vitro and in vivo tumor suppressive activity in
human gastric cancer cells. Cancer Lett. 315:12–17. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Ding X, Li Y, Lü J, Zhao Q, Guo Y, Lu Z,
Ma W, Liu P, Pestell RG, Liang C and Yu Z: piRNA-823 is involved in
cancer stem cell regulation through altering DNA methylation in
association with luminal breast cancer. Front Cell Dev Biol.
9:6410522021. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Iliev R, Fedorko M, MacHackova T,
Mlcochova H, Svoboda M, Pacik D, Dolezel J, Stanik M and Slaby O:
Expression levels of Piwi-interacting RNA, piR-823, are deregulated
in tumor tissue, blood serum and urine of patients with renal cell
carcinoma. Anticancer Res. 36:6419–6423. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Hempfling AL, Lim SL, Adelson DL, Evans J,
O'Connor AE, Qu ZP, Kliesch S, Weidner W, O'Bryan MK and Bergmann
M: Expression patterns of HENMT1 and Piwil1 in human testis:
Implications for transposon expression. Reproduction. 154:363–374.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Gao CL, Sun R, Li DH and Gong F: Piwi-like
protein 1 upregulation promotes gastric cancer invasion and
metastasis. Onco Targets Ther. 11:8783–8789. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Shi S, Yang ZZ, Liu S, Yang F and Lin H:
Piwil1 promotes gastric cancer via a piRNA-independent mechanism.
Proc Natl Acad Sci USA. 117:22390–22401. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Hu W, Sun X, Ye T, Feng S, Ruan Q, Xi M,
Zhou X, Li M, Ye Z, Cheng X and Xie W: Piwil2 may serve as a
prognostic predictor in cancers: A systematic review and
meta-analysis. J BUON. 25:2721–2730. 2020.PubMed/NCBI
|
|
125
|
Qu A, Wang W, Yang Y, Zhang X, Dong Y,
Zheng G, Wu Q, Zou M, Du L, Wang Y and Wang C: A serum piRNA
signature as promising non-invasive diagnostic and prognostic
biomarkers for colorectal cancer. Cancer Manag Res. 11:3703–3720.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Cui L, Lou Y, Zhang X, Zhou H, Deng H,
Song H, Yu X, Xiao B, Wang W and Guo J: Detection of circulating
tumor cells in peripheral blood from patients with gastric cancer
using piRNAs as markers. Clin Biochem. 44:1050–1057. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Colombo AR, Triche T Jr and Ramsingh G:
Transposable element expression in acute myeloid leukemia
transcriptome and prognosis. Sci Rep. 8:164492018. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Corces MR, Buenrostro JD, Wu B, Greenside
PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A,
Greenleaf WJ, et al: Lineage-specific and single-cell chromatin
accessibility charts human hematopoiesis and leukemia evolution.
Nat Genet. 48:1193–1203. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wang H, Wen J, Chang CC and Zhou X:
Discovering transcription and splicing networks in myelodysplastic
syndromes. PLoS One. 8:e791182013. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Leonova KI, Brodsky L, Lipchick B, Pal M,
Novototskaya L, Chenchik AA, Sen GC, Komarova EA and Gudkov AV: p53
cooperates with DNA methylation and a suicidal interferon response
to maintain epigenetic silencing of repeats and noncoding RNAs.
Proc Natl Acad Sci USA. 110:E89–E98. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Bolouri H, Farrar JE, Triche T Jr, Ries
RE, Lim EL, Alonzo TA, Ma Y, Moore R, Mungall AJ, Marra MA, et al:
The molecular landscape of pediatric acute myeloid leukemia reveals
recurrent structural alterations and age-specific mutational
interactions. Nat Med. 24:103–112. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Li S, Garrett-Bakelman FE, Chung SS,
Sanders MA, Hricik T, Rapaport F, Patel J, Dillon R, Vijay P, Brown
AL, et al: Distinct evolution and dynamics of epigenetic and
genetic heterogeneity in acute myeloid leukemia. Nat Med.
22:792–799. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Zeng Y, Cao Y, Halevy RS, Nguyen P, Liu D,
Zhang X, Ahituv N and Han JJ: Characterization of functional
transposable element enhancers in acute myeloid leukemia. Sci China
Life Sci. 63:675–687. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Deniz Ö, Ahmed M, Todd CD, Rio-Machin A,
Dawson MA and Branco MR: Endogenous retroviruses are a source of
enhancers with oncogenic potential in acute myeloid leukaemia. Nat
Commun. 11:35062020. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Gao Y, Yu XF and Chen T: Human endogenous
retroviruses in cancer: Expression, regulation and function. Oncol
Lett. 21:1212021. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Rossi F, Noren H, Jove R, Beljanski V and
Grinnemo KH: Differences and similarities between cancer and
somatic stem cells: Therapeutic implications. Stem Cell Res Ther.
11:4892020. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Sharma AK, Nelson MC, Brandt JE, Wessman
M, Mahmud N, Weller KP and Hoffman R: Human CD34(+) stem cells
express the hiwi gene, a human homologue of the Drosophila gene
piwi. Blood. 97:426–434. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Nolde MJ, Cheng EC, Guo S and Lin H: Piwi
genes are dispensable for normal hematopoiesis in mice. PLoS One.
8:e719502013. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Yan H, Wu QL, Sun CY, Ai LS, Deng J, Zhang
L, Chen L, Chu ZB, Tang B, Wang K, et al: piRNA-823 contributes to
tumorigenesis by regulating de novo DNA methylation and
angiogenesis in multiple myeloma. Leukemia. 29:196–206. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Li B, Hong J, Hong M, Wang Y, Yu T, Zang S
and Wu Q: piRNA-823 delivered by multiple myeloma-derived
extracellular vesicles promoted tumorigenesis through re-educating
endothelial cells in the tumor environment. Oncogene. 38:5227–5238.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Ai L, Mu S, Sun C, Fan F, Yan H, Qin Y,
Cui G, Wang Y, Guo T, Mei H, et al: Myeloid-derived suppressor
cells endow stem-like qualities to multiple myeloma cells by
inducing piRNA-823 expression and DNMT3B activation. Mol Cancer.
18:882019. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Cordeiro A, Navarro A, Gaya A, Díaz-Beyá
M, Gonzalez-Farré B, Castellano JJ, Fuster D, Martínez C, Martínez
A and Monzó M: PiwiRNA-651 as marker of treatment response and
survival in classical Hodgkin lymphoma. Oncotarget. 7:46002–46013.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Bamezai S, Mulaw MM, Zhou F, Rohde C,
Muller-Tidow C, Dohner K, Dohner H, Buske MF, Buske C and Rawat
VPS: Knockdown of the Piwi-like protein 4 (PIWIL4) delays leukemic
growth and is associated with gross changes in the global histone
methylation marks in human MLL-rearranged AML. Blood. 122:5972013.
View Article : Google Scholar
|
|
144
|
Wang Y, Jiang Y, Ma N, Sang B, Hu X, Cong
X and Liu Z: Overexpression of Hiwi inhibits the growth and
migration of chronic myeloid leukemia cells. Cell Biochem Biophys.
73:117–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Veryaskina YA, Titov SE, Kovynev IB,
Fedorova SS, Pospelova TI and Zhimulev IF: MicroRNAs in the
myelodysplastic syndrome. Acta Naturae. 13:4–15. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Kuang X, Chi J and Wang L: Deregulated
microRNA expression and its pathogenetic implications for
myelodysplastic syndromes. Hematology. 21:593–602. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Beck D, Ayers S, Wen J, Brandl MB, Pham
TD, Webb P, Chang CC and Zhou X: Integrative analysis of next
generation sequencing for small non-coding RNAs and transcriptional
regulation in myelodysplastic syndromes. BMC Med Genomics.
4:192011. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Hrustincova A, Krejcik Z, Kundrat D,
Szikszai K, Belickova M, Pecherkova P, Klema J, Vesela J, Hruba M,
Cermak J, et al: Circulating small noncoding RNAs have specific
expression patterns in plasma and extracellular vesicles in
myelodysplastic syndromes and are predictive of patient outcome.
Cells. 9:7942020. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Arber DA, Orazi A, Hasserjian R, Thiele J,
Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M and Vardiman JW:
The 2016 revision to the World Health Organization classification
of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Itou D, Shiromoto Y, Yukiho SY, Ishii C,
Nishimura T, Ogonuki N, Ogura A, Hasuwa H, Fujihara Y,
Kuramochi-Miyagawa S and Nakano T: Induction of DNA methylation by
artificial piRNA production in male germ cells. Curr Biol.
25:901–906. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Toden S, Zumwalt TJ and Goel A: Non-coding
RNAs and potential therapeutic targeting in cancer. Biochim Biophys
Acta Rev Cancer. 1875:1884912021. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Setten RL, Rossi JJ and Han SP: The
current state and future directions of RNAi-based therapeutics. Nat
Rev Drug Discov. 18:421–446. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Levy A, Sela N and Ast G: TranspoGene and
microTranspoGene: Transposed elements influence on the
transcriptome of seven vertebrates and invertebrates. Nucleic Acids
Res. 36:D47–D52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Kim DS, Kim TH, Huh JW, Kim IC, Kim SW,
Park HS and Kim HS: Line fusion genes: A database of LINE
expression in human genes. BMC Genomics. 7:1392006. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Navarro FC and Galante PA: RCPedia: A
database of retrocopied genes. Bioinformatics. 29:1235–1237. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Lappalainen I, Lopez J, Skipper L,
Hefferon T, Spalding JD, Garner J, Chen C, Maguire M, Corbett M,
Zhou G, et al: DbVar and DGVa: Public archives for genomic
structural variation. Nucleic Acids Res. 41:D936–D941. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Wang J, Song L, Grover D, Azrak S, Batzer
MA and Liang P: dbRIP: A highly integrated database of
retrotransposon insertion polymorphisms in humans. Hum Mutat.
27:323–329. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Mir AA, Philippe C and Cristofari G:
euL1db: The European database of L1HS retrotransposon insertions in
humans. Nucleic Acids Res. 43:D43–D47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Piuco R and Galante PAF: piRNAdb: A
piwi-interacting RNA database. bioRxiv: 2021.09.21.461238. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Sai Lakshmi S and Agrawal S: piRNABank: A
web resource on classified and clustered Piwi-interacting RNAs.
Nucleic Acids Res. 36:D173–D177. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Sarkar A, Maji RK, Saha S and Ghosh Z:
PiRNAQuest: Searching the piRNAome for silencers. BMC Genomics.
15:5552014. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Rosenkranz D: piRNA cluster database: A
web resource for piRNA producing loci. Nucleic Acids Res.
44:D223–D230. 2016. View Article : Google Scholar : PubMed/NCBI
|














