Introduction
Primary liver cancer can be divided into
hepatocellular carcinoma (HCC), bile duct cell carcinoma and mixed
cell carcinoma, among which HCC accounts for >80% of primary
liver cancer cases (1). Globally,
liver cancer is the fourth most common cause of cancer-related
death and ranks sixth in cancer incidence (2). According to the annual forecast by
the World Health Organization, it is estimated that >1 million
patients will die of liver cancer in 2030 (3). In the United States, from 2000 to
2016, the mortality rate of liver cancer increased by 43% (from 7.2
cases per 100,000 to 10.3 cases per 100,000), and the 5-year
survival rate was 18% (4). Liver
cancer has become the fourth most lethal cancer, after pancreatic
cancer (5).
HCC can occur in patients with chronic liver
diseases, such as hepatitis B or hepatitis C or those patients
suffering from alcohol abuse (6).
Patients with chronic liver diseases exhibit persistent liver
inflammation, progressive liver fibrosis and abnormal regeneration
of hepatocytes (6). These abnormal
physiological processes eventually lead to cirrhosis and a series
of genetic and epigenetic alterations, which ultimately contribute
to the formation of dysplastic nodules (precancerous lesions)
(7). Another abnormal
physiological processes can then enable dysplastic cells to gain
the advantages of proliferation, invasion and survival, and
completely transform into mature HCC (8). Somatic mutation analysis can be used
to determine the target pathway of telomerase, which can maintain
telomeres by increasing telomerase activity and preventing telomere
shortening and replication attenuation (7). These changes occur in the early stage
of tumor occurrence. Hot spot mutations in the telomerase reverse
transcriptase (TERT) promoter have been found in dysplastic nodules
of liver cirrhosis (8). With the
increase in the degree of atypical hyperplasia, the TERT mutation
frequency has also increased: 6% in low grade dysplastic nodules,
19% in high grade dysplastic nodules and 60% in early HCC. In
chronic liver disease, inflammation is related to the increase of
oxidative stress in liver parenchyma. The activation of nuclear
factor erythroid 2 related factor 2 and kelch-like ECH-related
protein 1 signaling pathways stimulate the protective effect from
oxidation at the cellular level (7,8).
Inactivating mutations in the tumor suppressor gene, TP53, occur in
20-50% of HCC cases and these mutations may interfere with the cell
cycle (7,8). At present, non-alcoholic fatty liver
disease (NAFLD) has become the main cause of HCC in many countries
(9). The majority of risk factors
associated with NAFLD are also independently associated with HCC
including obesity, diabetes and genetic polymorphisms of
patatin-like phospholipase domain containing 3, transmembrane 6
superfamily 2 and glucokinase regulator. Fat toxicity and DNA
oxidative damage associated with steatosis can induce liver
carcinogenesis (10). Therefore,
it is urgent to explore the key factors that regulate the malignant
progression of HCC.
Ubiquitin is a small protein composed of 76 amino
acids with a molecular weight of ~8.5 kDa (11), and the highly conserved ubiquitin
protein is widely distributed in all eukaryotic cells.
Ubiquitination refers to the process by which ubiquitin is attached
to proteins in cells during catalysis by a series of enzymes, which
selectively modify target proteins (12). This process typically requires the
cooperation of three ubiquitination enzymes: E1 ubiquitin
activating enzyme, E2 ubiquitin binding enzyme and E3 ubiquitin
ligase (13). Similar to other
posttranslational modifications, ubiquitination is reversible and
can be reversed by a large group of proteases termed
deubiquitinases (DUBs) (14). Most
DUBs cleave and release ubiquitin from substrate proteins, edit
ubiquitin chains and process ubiquitin precursors (14). As a ubiquitin-specific protease,
USP35 plays an important role in regulating the deubiquitination of
proteins. USP35 interacts with ferritin (FPN) and functions as a
deubiquitinating enzyme to maintain FPN protein stability and
further regulate ferroptosis in lung cancer cells (15). In ovarian cancer, USP35 can
directly deubiquitinate and inactivate the stimulator of interferon
genes protein, decreasing the sensitivity of tumor cells to
cisplatin (16). However, whether
USP35 plays a role in the malignant biological progression of HCC
remains unknown.
The present study aimed to analyze the expression of
USP35 in patients with HCC and the correlation between USP35
expression and HCC prognosis, to investigate the role of USP35 in
HCC malignant progression, and to preliminarily explore the
underlying molecular mechanism of USP35 in HCC.
Materials and methods
Patients and samples
In total, 96 patients (age range, 29-74 years; mean
age, 53 years old) with HCC were enrolled from the Department of
Liver Surgery at West China Hospital of Sichuan University
(Chengdu, China) from March 2015 to May 2019. All studies were
approved by The Ethics Committee of the West China Hospital
(Chengdu, China; approval no. 20150176). All patients involved in
the present study provided signed written informed consent. The
primary cancer tissues and the paired normal adjacent tissues were
immediately snap-frozen in liquid nitrogen and stored in liquid
nitrogen until use for mRNA and protein analyses.
The specific inclusion criteria were as follows: i)
HCC was confirmed through pathological testing after surgical
resection of the tumor; ii) the patient was diagnosed with HCC for
the first time; iii) the patient did not receive any treatment
before the surgery; iv) the patient had not been diagnosed with
other serious malignant diseases; v) the various clinical data of
the patient was complete; and vi) the prognosis follow-up data of
the patient was available and not missing.
The exclusion criteria were as follows: i) The
patient had a communication disorder and could not communicate
effectively; ii) the heart, brain, lung or kidney of the patient
had organic dysfunction; iii) patients diagnosed with HCC for the
first time but with distant metastasis; iv) the clinical data of
the patient was incomplete; v) the patient had systemic diseases;
vi) the patient had autoimmune dysfunction; and vii) the follow-up
data for patient prognosis was incomplete.
Cell culture
HCC cell lines, Hep3B, Huh-7, MHCC-97H and MHCC-97L,
and the human normal hepatocyte, THLE2, were purchased from The
Cell Bank Type Culture Collection of The Chinese Academy of
Sciences. All cells were cultured in DMEM high glucose medium
(Gibco; Thermo Fisher Scientific, Inc.) with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.). Cells were cultured
in a closed incubator with a constant temperature of 37°C and with
5% CO2.
Cell transfection
USP35 and PKM2 (In-PKM2) overexpressing lentiviruses
were constructed using the lentiviral vector pCDH-EF1α-MCS-T2A-Puro
(cat. no. CD527A-1; System Biosciences, LLC). The 3rd generation
system was used and DNA was transfected into 293T cells (The Cell
Bank Type Culture Collection of The Chinese Academy of Sciences)
for 48 h at 37°C. The ratio used for the lentivirus, packaging and
envelope plasmids was 4:3:1. For transduction, 4 μl of
lentivirus titer (1×108 TU/ml) was added to
1×106 HCC cells at 37°C for 12 h and at three infection
gradients (MOI=10, MOI=20, MOI=30). The time interval between
transfection and subsequent experiments was 3 h. For the negative
control (NC) group, empty vector was used instead of the
overexpression vector.
Short hairpin RNA (shRNA) targeting USP35 for
knockdown (De-USP35; 5′-GCT GAG TTG GGC TCT TCT AGA-3′) and the
respective NC (5′-TTC TCC GAA CGT GTC ACG TAA-3′) (vector set cat.
no. C01001) were purchased from Shanghai GenePharma Co., Ltd. A
total of 5 μg of each plasmid was transfected into HCC cells
using Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) for 48 h at 37°C. The time interval between
transfection and subsequent experiments was 3 h. For
co-transfection, the cell lines transfected with De-USP35 were
selected using 400 μg/ml geneticin. These stable cell lines
were then transfected with In-PKM2, followed by selection using 2.5
μg/ml puromycin. Finally, western blotting was used to
validate the results.
Western blotting
Total protein from the HCC samples from patients and
cell lines was extracted in a solution of RIPA buffer (cat. no.
P0013B; Beyotime Institute of Biotechnology) containing 1:100 PMSF
(Beyotime Institute of Biotechnology). The protein concentration
was quantified using a BCA kit (Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. Equal
amounts of protein (25 μg) were separated via SDS-PAGE on a
10% gel and transferred to PVDF membranes. The membranes were
blocked for 1 h at room temperature with 5% non-fat milk powder
(Beyotime Institute of Biotechnology). The membranes were incubated
at 4°C overnight with the following primary antibodies: USP35
(1:2,000; cat. no. PA5-37232; Thermo Fisher Scientific, Inc.), PKM2
(1:1,500; cat. no, 15822-1-AP; Proteintech Group, Inc.), PKM1
(1:1,500; cat. no. 15821-1-AP; Proteintech Group, Inc.) and β-actin
(1:5,000; cat. no. ab8226; Abcam). The membranes were then
incubated with the appropriate HRP secondary antibodies (1:5,000;
cat. nos. bs-0296G-HRP and bs-0295G-HRP; BIOSS) for 1 h at room
temperature. Finally, protein expression was visualized using
chemiluminescence reagents (Hyperfilm ECL; Cytivia). ImageJ
software (V1.8.0.112; National Institutes of Health) was used to
analyze the western blot results.
Immunohistochemistry
A total of 96 HCC tissues were fixed in 10% formalin
for 12 h at room temperature, embedded in paraffin and cut into
4-μm sections. The tissue sections were then used to
generate tissue microarray cores (1.5-mm diameter). Tissue sections
(slice thickness, 4 μm) were dehydrated in xylene and
alcohol followed by 3% H2O2 for 30 min at
37°C. All sections were incubated for 15 min at room temperature
with 5% goat serum (OriGene Technologies, Inc.) to block
non-specific binding, followed by incubation with USP35 (1:500;
cat. no. ab254939; Abcam) and Ki-67 (1:1,000; cat. no. ab15580;
Abcam) at 4°C overnight. Then, the sections incubated with
anti-rabbit secondary IgG antibody (1:100; cat. no. SAP-9100;
OriGene Technologies, Inc.) at 37°C for 30 min. Signals were
visualized using DAB (Boster Biological Technology). Slides were
visualized using a light microscope (magnification, ×10, ×100 or
×200; Zeiss AG) and ImageJ software (version 1.52; National
Institutes of Health).
The immunohistochemistry scoring method was as
follows: The staining intensity score [according to the degree of
color development of the positive marker: No staining, negative
(score, 0); light yellow staining, weak (score, 1); brown-yellow
staining, moderate (score, 2); and brown-black staining, strong
(score, 3)] plus the percentage of positive cells score (score 0,
≤5%; score 1, 6-25%; score 2, 26-50%; score 3, 51-75%; and score 4,
76-100%). USP35 expression was defined as low (total score <4)
or high (total score ≥4).
Co-immunoprecipitation (Co-IP)
Cells were collected and lysed using RIPA lysis
buffer containing 1:100 PMSF (Beyotime Institute of Biotechnology).
In total, 3 μg USP35 (1:5,000; cat. no. PA5-37232; Thermo
Fisher Scientific, Inc.) or PKM2 (1:500; cat. no. 15822-1-AP;
Proteintech Group, Inc.) antibody was added to the lysate (20
μl) and the mix was incubated overnight at 4°C. Then, 20
μl Protein A/G PLUS-Agarose beads (cat. no. sc-2003; Santa
Cruz Biotechnology, Inc.) was added to the lysate mix, which was
incubated with rotation for 4 h at 4°C. The bead-antibody-protein
complexes were washed with pre-cooled PBS solution three times and
then boiled (10,000 x g centrifugation for 5 sec at room
temperature) for subsequent western blot analysis.
Ubiquitination assay
Prior to cell lysis, HCC cells were treated with
MG132 (10 μM) for 8 h at 37°C. MG132 is a protease inhibitor
that can prevent protease activity during degradation, thereby
promoting the ubiquitination process (17). HCC cell lines were then lysed in 1%
SDS-containing RIPA buffer by sonication (20 KHz, 60 sec) on ice.
Then, lysates (20 μl) were treated with Protein A/G
Plus-Agarose for 1 h at room temperature. After that, each sample
was incubated with 10 μl IgG (cat. no. 30000-0-AP;
Proteintech Group, Inc.) and PKM2 (cat. no, 15822-1-AP; Proteintech
Group, Inc.) overnight at 4°C. Then, the nuclear pellet was
collected by centrifugation at 10,000 x g for 5 min at 4°C and
subsequently washed four times with Protein A/G Plus-Agarose beads
to purify the protein (this was achieved by absorbing the protein
on the beads, then washing the beads to release the protein). The
purified proteins were separated by 10% SDS-PAGE. An anti-PKM2
(1:500; cat. no. ab137852; Abcam) or anti-ubiquitin antibody
(1:500; cat. no. ab7780; Abcam) was used for immunoblotting
according to the protocol for western blotting analysis
aforementioned.
The Cancer Genome Atlas (TCGA)-LIHC data
acquisition and analysis
The TCGA-LIHC RNA sequencing file of expression data
(from TCGA database) was downloaded from the XENA website
(http://xena.ucsc.edu/download-data/)
and the proteomic data of HBV-HCC patients was acquired from the
Clinical Proteomic Tumor Analysis Consortium database (CPTAC;
https://pdc.cancer.gov/pdc/). The
expression of USP35 was selected and analyzed. Paired Student's
t-test was used to compare USP35 expression in HCC and adjacent
tissues using GraphPad software (V8.0.0; Dotmatics).
5-Ethynyl-2′-deoxyuridine (EdU)
assay
After transfection, HCC cells (2.5×104
cells/well) were seeded into 96-well plates and cultured for 24 h
at 37°C. Then, EdU (50 μM) was incubated with the cells for
2 h at 37°C. Following this, the cells were fixed in 4%
formaldehyde for 30 min at room temperature and then permeabilized
with 0.5% Triton X-100 solution for 10 min at room temperature.
ApolloR reaction cocktail (100 μl; cat. no. 100T; Guangzhou
RiboBio Co., Ltd.) was added, and the reaction was allowed to
proceed for 30 min at room temperature in the dark. Cell nuclei
were stained by adding 1X DAPI (100 μl; cat. no. 100T;
Guangzhou RiboBio Co., Ltd.) for 30 min at room temperature. Cell
proliferation was analyzed using the mean number cells in three
fields for each sample. Cell proliferation was analyzed by
assessing the percentage of EdU+ cells in each sample
using a fluorescence microscope (Lionheart; BioTek Instruments,
Inc.; magnification, ×100). ImageJ software (version 1.8.0;
National Institutes of Health) was used for cell counting.
Cell migration and invasion assays
Transwell chambers (Corning, Inc.) were used to
evaluate cell invasion with inserts precoated with Matrigel (1
mg/ml) at 37°C for 30 min. After transfection, HCC cells were
resuspended in high-glucose DMEM containing 1% FBS and seeded into
the upper Transwell chamber at a density of 1×105
cells/well and 500 μl of high-glucose DMEM containing 10%
FBS was added to the corresponding lower chamber. For the migration
assay, Transwell chambers (Corning, Inc.) without inserts precoated
with Matrigel were used, otherwise the same protocol as for the
invasion assay was followed. After incubation for 48 h at 37°C, the
Transwell chambers were fixed with 4% paraformaldehyde at room
temperature for 30 min and stained with 0.5% crystal violet at room
temperature for 20 min. Finally, six fields were randomly selected,
the number of stained cells in the lower chamber was counted
manually in each field and images were captured under a light
microscope (magnification, ×200; Olympus Corporation).
Flow cytometric analysis of
apoptosis
After transfection, HCC cells were seeded into
6-well plates. At 60-70% confluency, cells were collected and
incubated with Annexin V-FITC (5 μl; Biogot Technology Co.,
Ltd.) and propidium iodide solution (5 μl; Biogot Technology
Co., Ltd.) at room temperature for 15 min according to the
manufacturer's instructions. Cells were subsequently resuspended in
300 μl binding buffer (cat. no. BD0073-2; Biogot Technology
Co., Ltd.). Apoptosis progression was analyzed using flow cytometry
(FACSAria; BD Biosciences). All data were analyzed with ModFit
version 4.0 (Verity Software House, Inc.).
Glucose uptake assay
After transfection, HCC cells (1×105
cells/well) were incubated in DMEM (supplemented with 10% FBS)
without L-glucose or phenol red (Gibco; Thermo Fisher Scientific,
Inc.) for 8 h at 37°C. The D-glucose content of the medium was
measured using a Glucose Colorimetric Assay kit (cat. no. K606-100;
BioVision, Inc.). In total, three biological replicates were
performed.
Intracellular pyruvate, lactate and ATP
assays
After transfection, HCC cells (1×105
cells/well) were incubated in phenol red-free DMEM without FBS for
4 h at 37°C. Then, a pyruvate assay kit (cat. no. BC2205; Beijing
Solarbio Science & Technology Co., Ltd.), lactate assay kit
(cat. no. BC2235; Beijing Solarbio Science & Technology Co.,
Ltd.), and ATP assay kit (cat. no. BC0300; Beijing Solarbio Science
& Technology Co., Ltd.) were used to measure intracellular
concentrations of pyruvate, lactate and ATP, respectively,
according to the kit manufacturer's instructions. Relative
absorbance (450 nm) was measured using a spectrophotometer (Thermo
Fisher Scientific, Inc.). The pyruvate, lactate and ATP contents
were calculated according to the product manual.
Mouse xenograft model
Male BALB/c-nu mice (n=12; age, 5 weeks old; weight,
20-25 g) were purchased from Chengdu Dossy Experimental Animals
Co., Ltd. and housed (25°C and 40-70% humidity, with a 12-h
light/dark cycle and free access to food and water)at the
Experimental Animal Center of West China Hospital of Sichuan
University (Chengdu, China). All animal experiments were approved
by The Committee on The Ethics of Animal Experiments of West China
Hospital of Sichuan University (Chengdu, China; approval no.
20220228036). Specifically, HCC cells (5×106, 50
μl) were subcutaneously injected into the left hip flanks of
the mice (n=3 in each group). The health and behavior of the mice
was monitored once a day and the tumor growth was measured once
every 4 days with a caliper. Tumor volume was then calculated
according to the following formula: Volume=(width2 ×
length)/2. The mice were euthanized, and tumors were collected for
analysis 28 days after cell injection. For this, the mice were
anesthetized with 1% pentobarbital sodium (45 mg/kg,
intraperitoneal) and subsequently euthanized by cervical
dislocation (it was determined that the mouse had died when the
heart stopped beating completely). Body weight loss >20% was
considered a humane endpoint for euthanasia, but no mice reached
this humane endpoint.
Statistical analysis
All data are presented as the mean ± SD. Statistical
analysis of data was conducted using GraphPad version 5.0
(Dotmatics) or SPSS 22.0 software (IBM Corp.). Kaplan-Meier
analysis was used to assess overall survival (OS) and
recurrence-free survival (RFS) times, and the log-rank test was
used to analyze the differences between the survival times. The
relationships between USP35 expression and clinicopathological
parameters in HCC was analyzed using χ2 test. Univariate
and multivariate Cox regression analyses were conducted to
determine the prognostic value of USP35. Statistical differences
between two groups were analyzed by unpaired Student's t-test.
Paired Student's t-test was used to compare USP35 expression in HCC
and adjacent tissues. ANOVA was used for multiple group comparisons
and Tukey's test was used as the post hoc test after ANOVA.
P<0.05 was considered to indicate a statistically significant
difference.
Results
USP35 is upregulated in HCC and predicts
poor prognosis
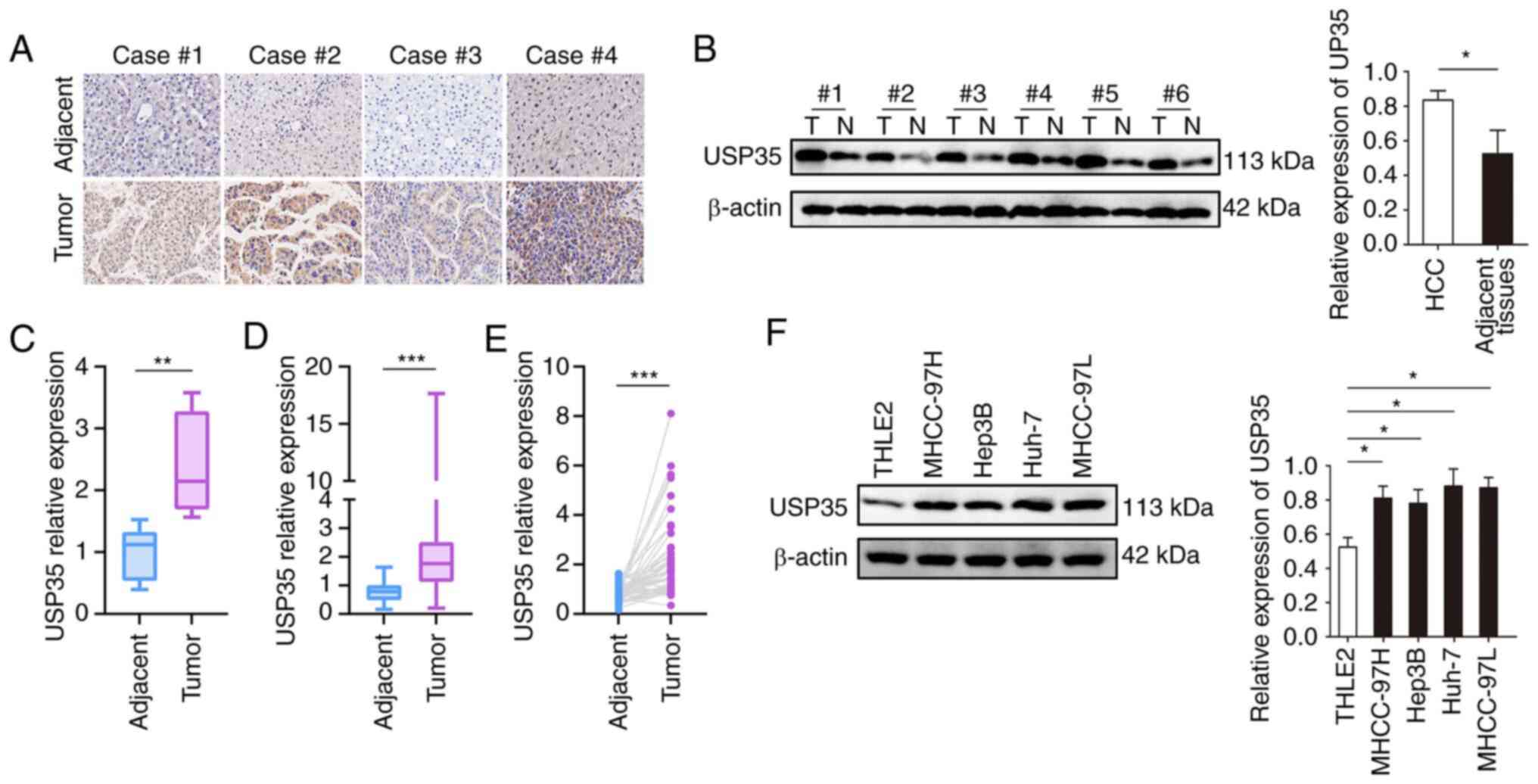
Firstly, the expression of USP35 in paired HCC tumor
and adjacent tissues was detected by immunohistochemistry. As shown
in Fig. 1A, USP35 was highly
expressed in tumor tissues, which was further verified by western
blotting (Fig. 1B). In addition,
the mRNA level of USP35 in the HBV-HCC cohort from the CPTAC
(Fig. 1C) and the TCGA-LIHC cohort
from TCGA (Fig. 1D and E) were
analyzed, and it was found that USP35 mRNA was highly expressed in
tumor tissues. The western blot results also demonstrated that
USP35 was significantly upregulated in HCC cell lines compared with
THLE2 normal liver cells (Fig.
1F).
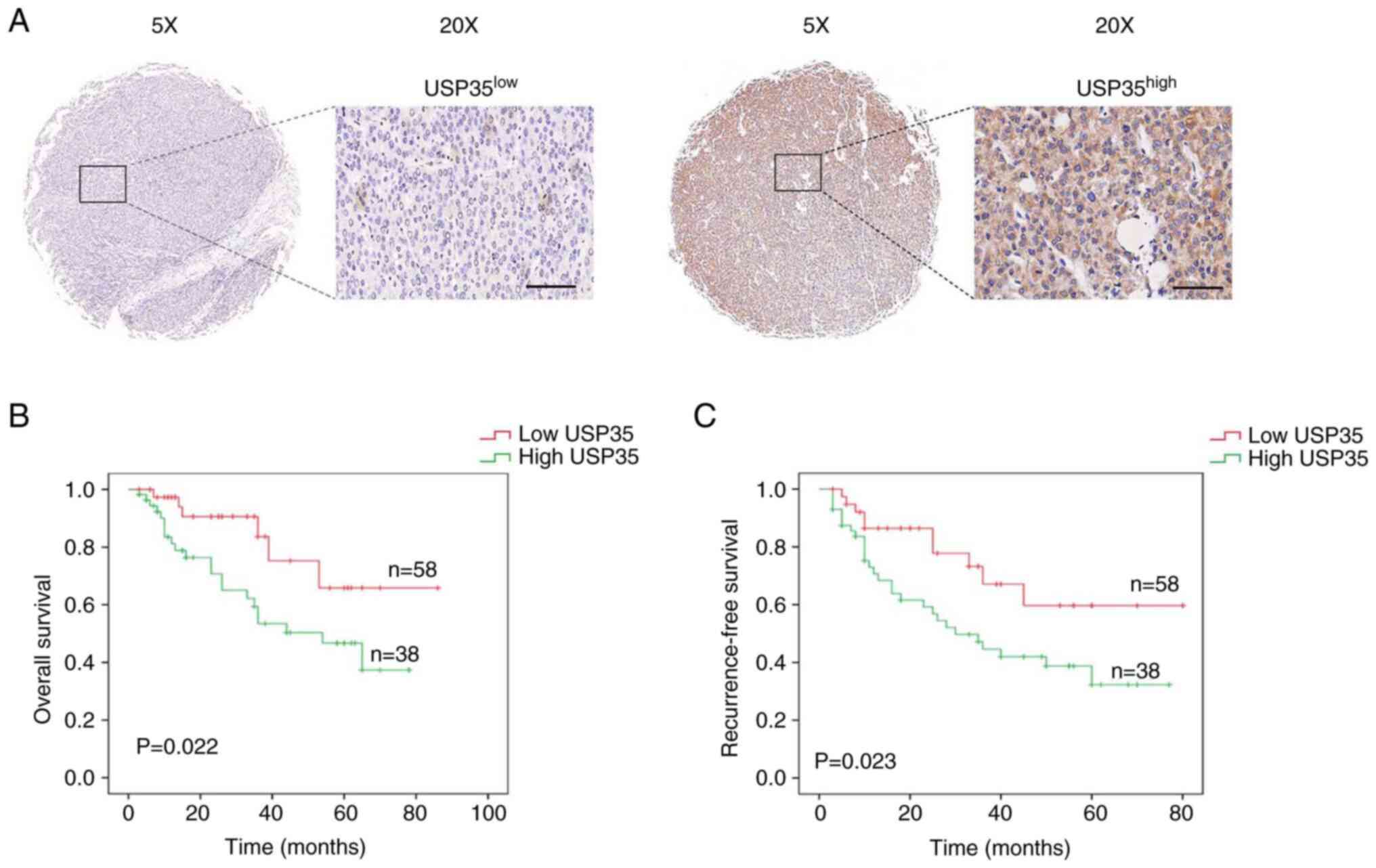
To evaluate the expression of USP35, tissue
microarray was used. The 96 patients with HCC were clustered into
two groups, the USP35 low and USP35 high groups, based on USP35
expression scored by the positive rate and staining intensity
(Fig. 2A). Furthermore, the
association between USP35 expression and clinical parameters were
evaluated. As demonstrated in Table
I, USP35 expression was associated with Tumor-Node-Metastasis
(TNM) stage (P=0.016) [using the Barcelona Clinic Liver Cancer 2022
strategy (18)], microvascular
invasion (P= 0.006) and multiplicity (P= 0.012). Statistical
analysis of prognosis revealed that patients with HCC with higher
USP35 expression had a significantly shorter overall survival time
(Fig. 2B). Univariate analysis
indicated that TNM stage (P=0.017), microvascular invasion
(P=0.023) and USP35 (P=0.011) were significantly associated with OS
time in HCC (Table II). In
addition, statistical analysis of prognosis revealed that patients
with HCC with higher USP35 expression had a significantly shorter
RFS time (Fig. 2C). Univariate
analysis indicated that TNM stage (P=0.014), microvascular invasion
(P=0.010) and USP35 (P=0.012) were significantly associated with
RFS time in HCC (Table III).
 | Table IAssociations between USP35 and the
clinicopathological features of patients with HCC. |
Table I
Associations between USP35 and the
clinicopathological features of patients with HCC.
| Clinicopathological
feature | Cases, n | USP35 expression
| P-value |
|---|
| Low (n=58) | High (n=38) |
|---|
| Sex | | | | 0.560 |
| Male | 49 | 31 | 18 | |
| Female | 47 | 27 | 20 | |
| Age, years | | | | |
| <50 | 45 | 28 | 17 | 0.734 |
| ≥50 | 51 | 30 | 21 | |
| AFP, ng/ml | | | | 0.501 |
| ≤20 | 29 | 19 | 10 | |
| >20 | 67 | 39 | 28 | |
| HBsAg | | | | 0.525 |
| Positive | 62 | 36 | 26 | |
| Negative | 34 | 22 | 12 | |
| TNM stage | | | | 0.016 |
| I/II | 46 | 22 | 24 | |
| III/IV | 50 | 36 | 14 | |
| Tumor size, cm | | | | 0.507 |
| ≤5 | 44 | 25 | 19 | |
| >5 | 52 | 33 | 19 | |
| Multiplicity | | | | 0.012 |
| Single | 48 | 23 | 25 | |
| ≥2 | 48 | 35 | 13 | |
| Microvascular
invasion | | | | 0.006 |
| Presence | 44 | 20 | 24 | |
| Absence | 52 | 38 | 14 | |
| Table IIUnivariate and multivariate analysis
of different prognostic variables influencing overall survival in
patients with HCC. |
Table II
Univariate and multivariate analysis
of different prognostic variables influencing overall survival in
patients with HCC.
| Variables | n | Univariate analysis
| Multivariate
analysis
|
|---|
| HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Sex | | 0.534
(0.345-1.833) | 0.421 | | |
| Male | 49 | | | | |
| Female | 47 | | | | |
| Age, years | | 0.593
(0.487-1.931) | 0.547 | | |
| <50 | 45 | | | | |
| ≥50 | 51 | | | | |
| AFP, ng/ml | | 1.634
(0.208-1.307) | 0.364 | | |
| ≤20 | 29 | | | | |
| >20 | 67 | | | | |
| HBsAg | | 1.744
(0.813-3.175) | 0.364 | | |
| Positive | 62 | | | | |
| Negative | 34 | | | | |
| TNM stage | | 1.117
(0.314-1.311) | 0.017 | 1.174
(0.297-1.473) | 0.005 |
| I/II | 46 | | | | |
| III/IV | 50 | | | | |
| Tumor size, cm | | 0.637
(0.740-1.942) | 0.471 | | |
| ≤5 | 44 | | | | |
| >5 | 52 | | | | |
| Multiplicity | | 1.394
(0.471-1.634) | 0.520 | | |
| Single | 48 | | | | |
| ≥2 | 48 | | | | |
| Microvascular
invasion | | 1.438
(0.604-1.976) | 0.023 | 1.307
(0.573-1.864) | 0.010 |
| Presence | 44 | | | | |
| Absence | 52 | | | | |
| USP35
expression | | 1.084
(0.519-1.843) | 0.011 | 1.128
(0.619-1.784) | 0.013 |
| Low | 58 | | | | |
| High | 38 | | | | |
| Table IIIUnivariate and multivariate analysis
of different prognostic variables influencing recurrence-free
survival in patients with HCC. |
Table III
Univariate and multivariate analysis
of different prognostic variables influencing recurrence-free
survival in patients with HCC.
| Variables | n | Univariate analysis
| Multivariate
analysis
|
|---|
| HR (95% CI) | P-value | HR (95% CI) | P-value |
|---|
| Sex | | 0.237
(0.648-1.178) | 0.348 | | |
| Male | 49 | | | | |
| Female | 47 | | | | |
| Age, years | | 0.573
(0.710-1.686) | 0.513 | | |
| <50 | 45 | | | | |
| ≥50 | 51 | | | | |
| AFP, ng/ml | | 0.713
(0.422-1.571) | 0.484 | | |
| ≤20 | 29 | | | | |
| >20 | 67 | | | | |
| HBsAg | | 1.517
(0.847-1.870) | 0.348 | | |
| Positive | 62 | | | | |
| Negative | 34 | | | | |
| TNM stage | | 1.109
(0.506-1.811) | 0.014 | 1.277
(0.594-2.033) | 0.022 |
| I/II | 46 | | | | |
| III/IV | 50 | | | | |
| Tumor size, cm | | 0.814
(0.664-1.734) | 0. 439 | | |
| ≤5 | 44 | | | | |
| >5 | 52 | | | | |
| Multiplicity | | 1.027
(0.476-1.974) | 0.604 | | |
| Single | 48 | | | | |
| ≥2 | 48 | | | | |
| Microvascular
invasion | | 1.008
(0.307-1.738) | 0.010 | 1.114
(0.411-1.488) | 0.015 |
| Presence | 44 | | | | |
| Absence | 52 | | | | |
| USP35
expression | | 1.375
(0.705-1.881) | 0.012 | 1.512
(0.880-1.947) | 0.015 |
| Low | 58 | | | | |
| High | 38 | | | | |
Decreased USP35 expression inhibits the
malignant biological behavior of HCC
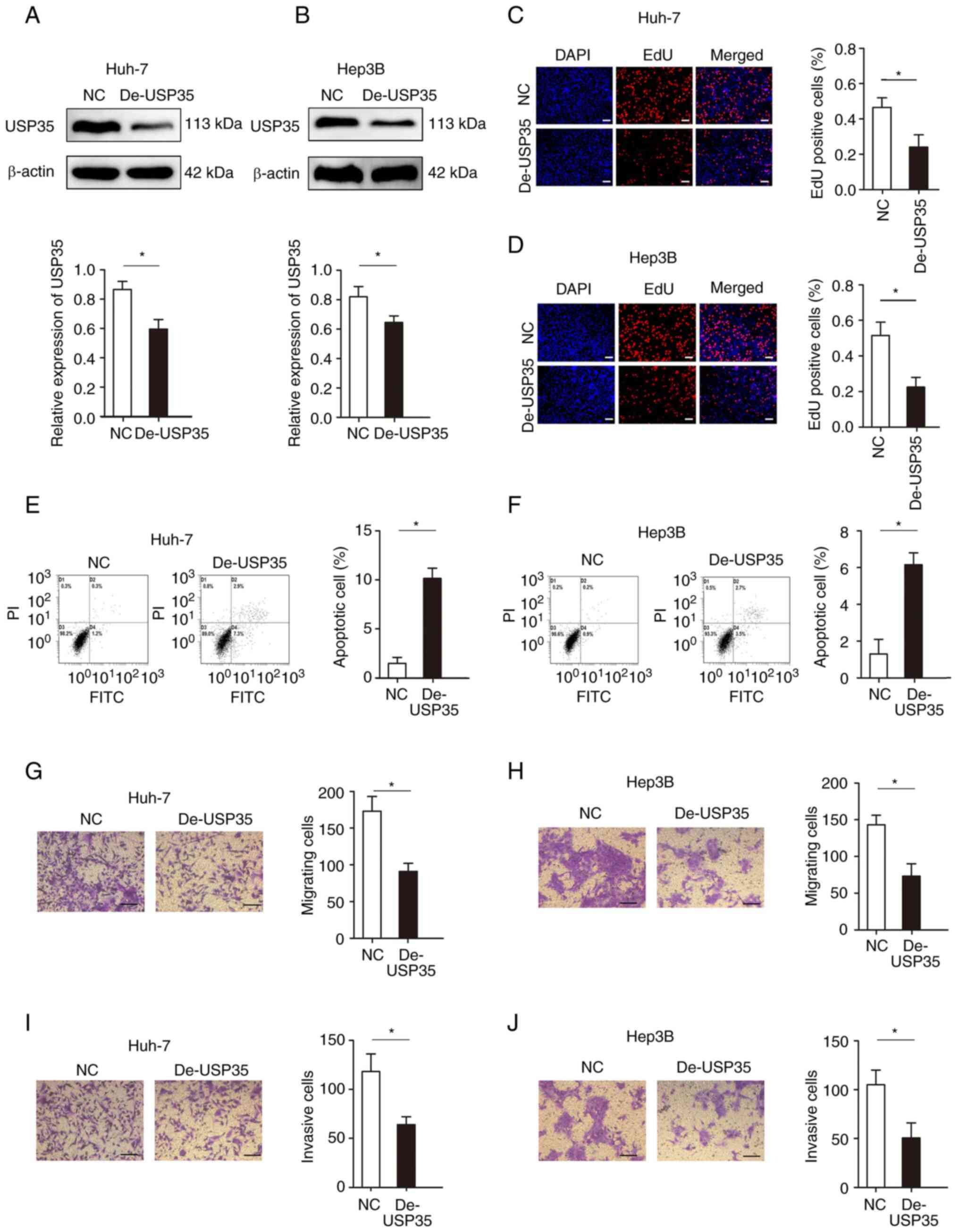
Loss-of-function experiments were performed in Huh-7
and Hep3B cells. The expression of USP35 was effectively knocked
down in Huh-7 and Hep3B cells (Fig. 3A
and B, respectively). Decreased USP35 expression significantly
inhibited cell proliferation, as demonstrated by the EdU assays in
Huh-7 and Hep3B cells (Fig. 3C and
D, respectively). The results from flow cytometry assays
demonstrated that decreased USP35 expression elevated the apoptosis
rate of Huh-7 (1.5 vs. 10.1%; Fig.
3E) and Hep3B (1.1 vs. 6.2%; Fig.
3F) cells. Moreover, the migration and invasion abilities of
Huh-7 and Hep3B cells with decreased USP35 expression were
significantly impaired, as determined by Transwell assays (Fig. 3G-J).
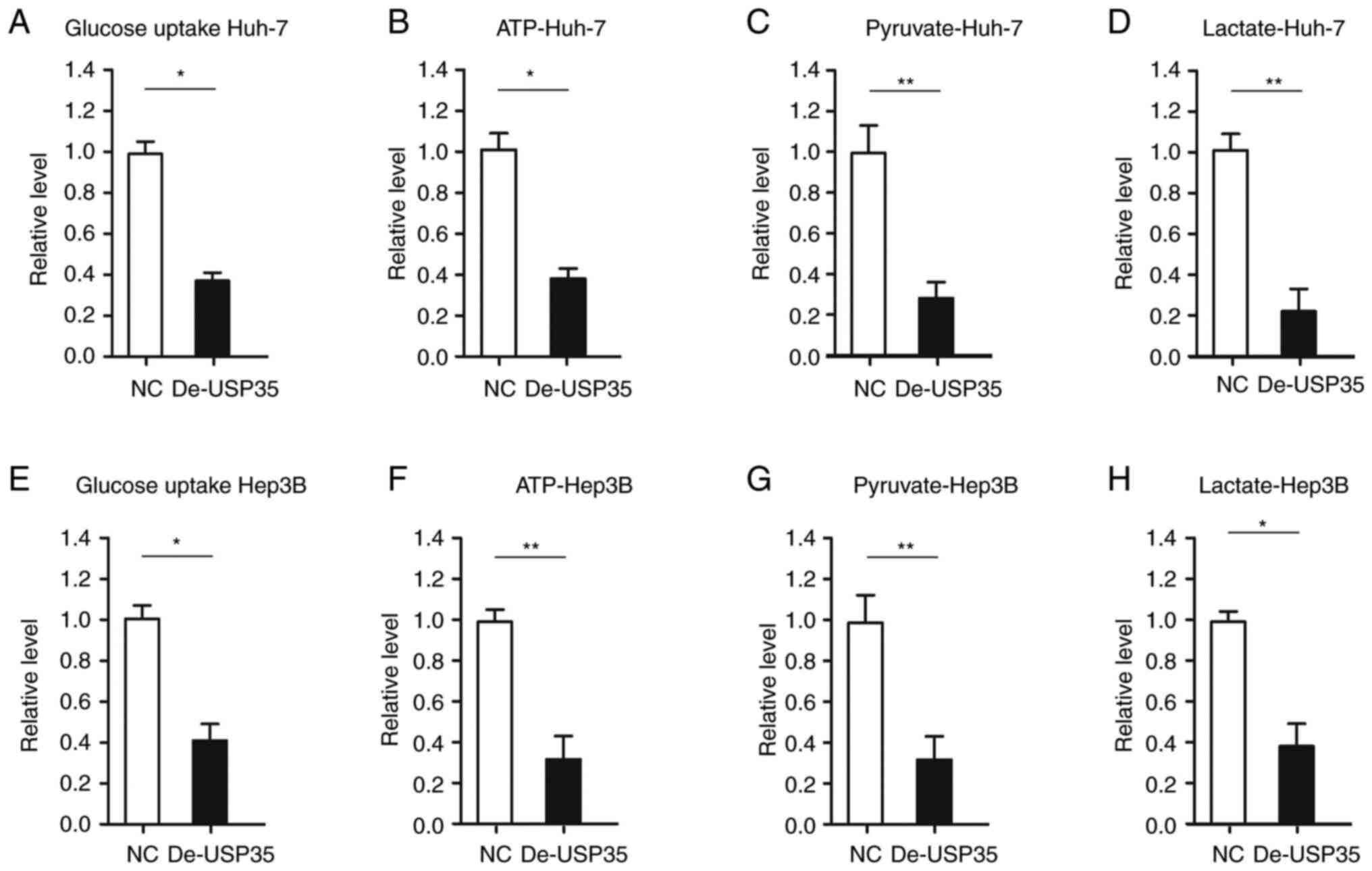
Decreased USP35 expression impedes energy
metabolism by suppressing glycolysis in HCC
The liver is the main organ in which glucose and
lipid metabolism occurs in the body. After the occurrence of liver
cancer, the normal metabolism of glucose and lipids in vivo
markedly changes (19). USP35
subtype 2 is an integral membrane protein of the endoplasmic
reticulum and is also present in lipid droplets (20). Dysregulation of the level of
subtype 2 may lead to rapid endoplasmic reticulum stress and cell
death by interfering with the regulation of lipid homeostasis
(20). In the present study, it
was investigated whether USP35 affects glycolysis in HCC. It was
found that glucose uptake and the production of ATP, pyruvate and
lactate were significantly decreased after USP35 downregulation in
Huh-7 cells (Fig. 4A-D).
Similarly, it was demonstrated that glucose uptake and the
production of ATP, pyruvate and lactate were significantly
decreased upon USP35 downregulation in Hep3B cells (Fig. 4E-H).
USP35 binds to PKM2 and regulates PKM2
expression through ubiquitination in HCC
In the process of glycolysis, PKM1 and PKM2 convert
phosphoenolpyruvate into pyruvate and play critical roles in
regulating the glycolysis process (21). Therefore, in the present study, the
association of USP35 expression with that of PKM1 and PKM2 were
analyzed. It was found that changing the expression level of USP35
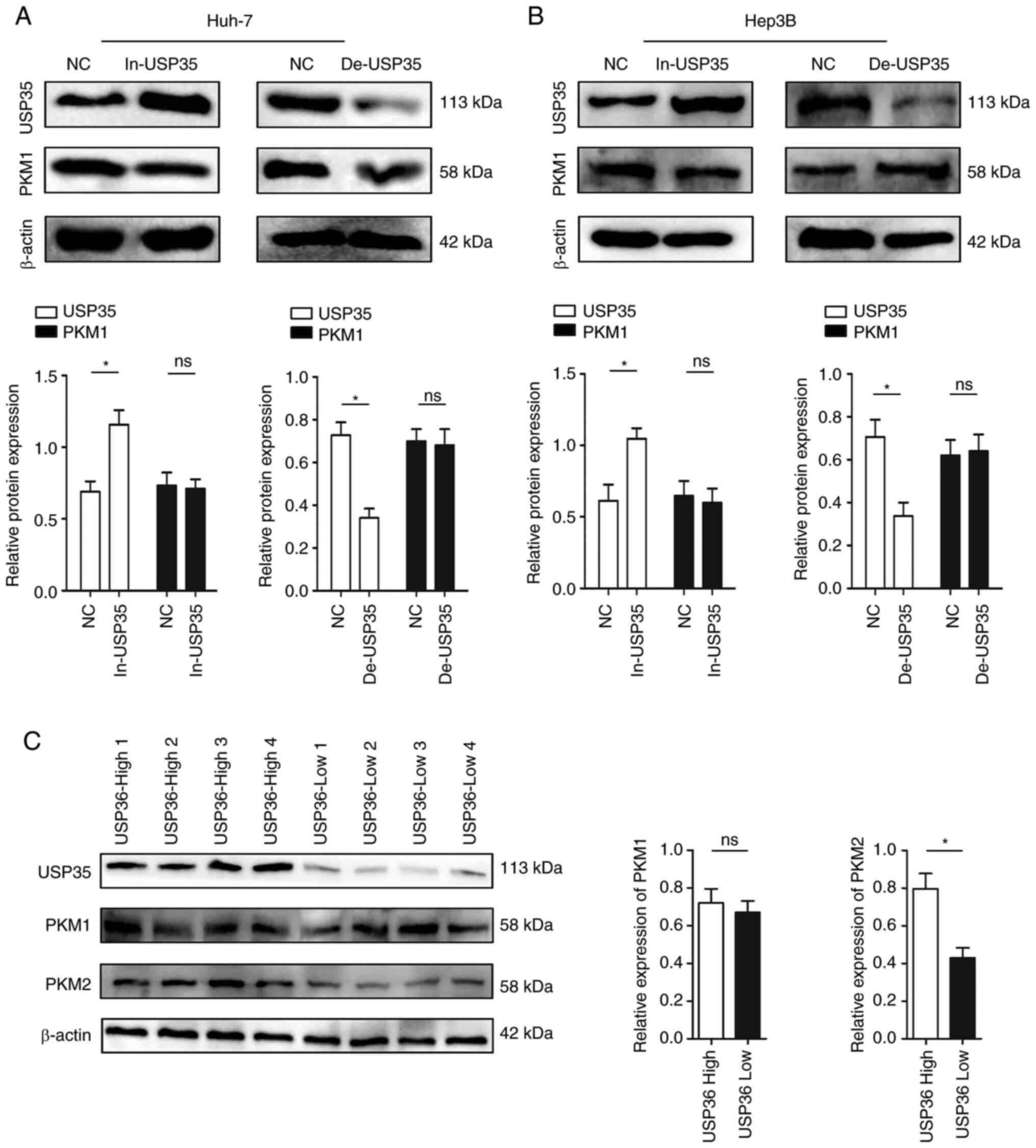
did not affect the expression of PKM1 (Fig. 5A and B). However, PKM2 displayed a
similar expression pattern to USP35 in HCC tissues (for example,
when USP35 was highly expressed, PKM2 was also highly expressed;
Fig. 5C). In addition,
overexpression of USP35 significantly increased the expression of
PKM2, while knock down of USP35 expression significantly decreased
the expression of PKM2 (Fig. 6A and
B). Notably, the Co-IP assay results indicated the existence of
an interaction between endogenous USP35 and PKM2 (Fig. 6C and D). In addition, the
ubiquitination level of PKM2 was decreased in the Huh-7 and Hep3B
cell lines after USP35 overexpression, which indicated that
overexpression of USP35 inhibits PKM2 from entering the degradation
process (Fig. 6E and F).
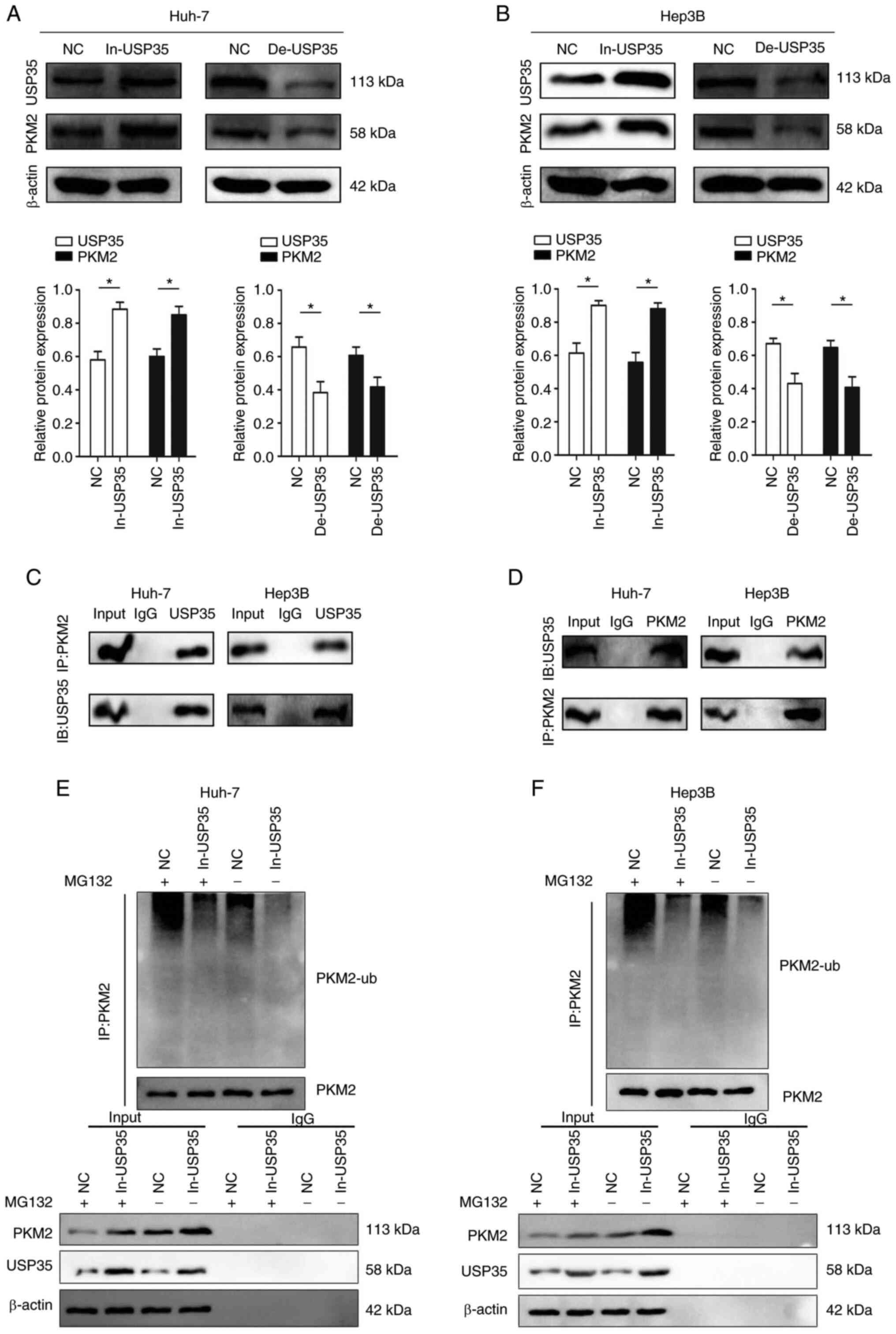 | Figure 6USP35 regulates PKM2 expression by
influencing its ubiquitin level. USP35 and PKM2 protein expression
in (A) Huh-7 and (B) Hep3B cells transfected with In-USP35 or
De-USP35. (C) Co-IP experiments indicated that USP35 interacted
with PKM2 in Huh-7 and Hep3B cell lines (IP:PKM2, IB:USP35). (D)
Co-IP experiments indicated that USP35 interacted with PKM2 in
Huh-7 and Hep3B cell lines (IP:USP35, IB:PKM2). The levels of
PKM2-ub in (E) Huh-7 and (F) Hep3B cells following overexpression
of USP35 were examined by western blotting using an anti-ubiquitin
antibody. *P<0.05. Co-IP, co-immunoprecipitation; NC,
negative control; PKM2, M2 splice isoform of pyruvate kinase;
PKM2-ub, PKM2 ubiquitination; USP35, ubiquitin-specific protease
35; De-USP35, USP35 short hairpin RNA; In-USP35, USP35
overexpression; IB, immunoblotting. |
PKM2 ameliorates the inhibitory effect of
USP35 on HCC progression
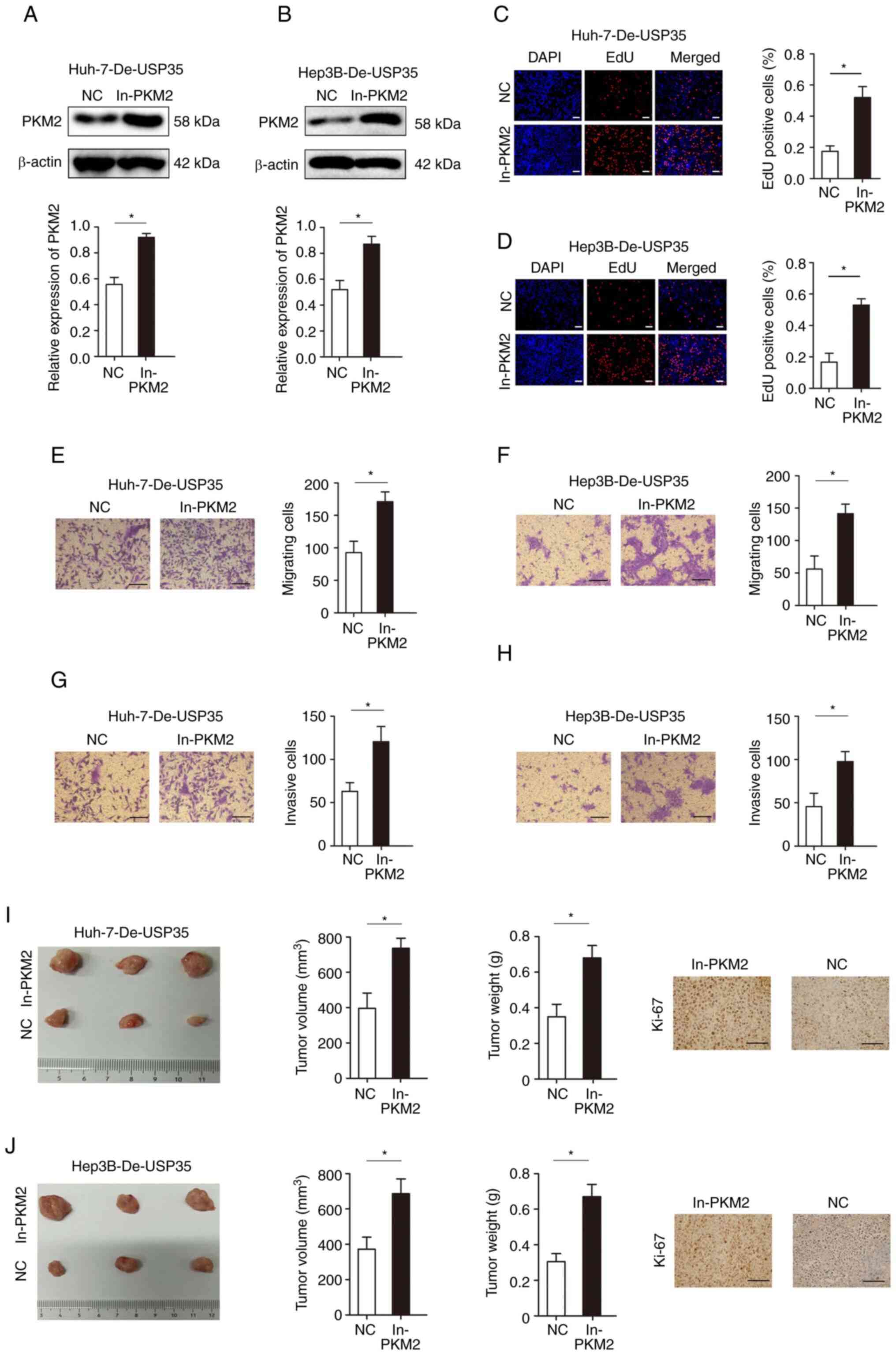
Furthermore, rescue experiments were performed to
investigate whether PKM2 in HCC was mediated by USP35. PKM2
expression was significantly increased when overexpressed in Huh
and Hep3B (Fig. S1), and
significantly increased when overexpressed in Huh-7-De-USP35 and
Hep3B-De-USP35 (Fig. 7A and B).
EdU assays demonstrated that PKM2 overexpression significantly
abolished the inhibitory effects of USP35 knockdown on
proliferation (Fig. 7C and D).
Moreover, increased PKM2 expression significantly decreased the
inhibitory effect of USP35 knockdown on migration and invasion in
Huh-7-De-USP35 and Hep3B-De-USP35 cells (Fig. 7E-H). In addition, PKM2
overexpression significantly rescued the inhibitory effects of
USP35 knockdown on tumor growth in vivo (Fig. 7I and J).
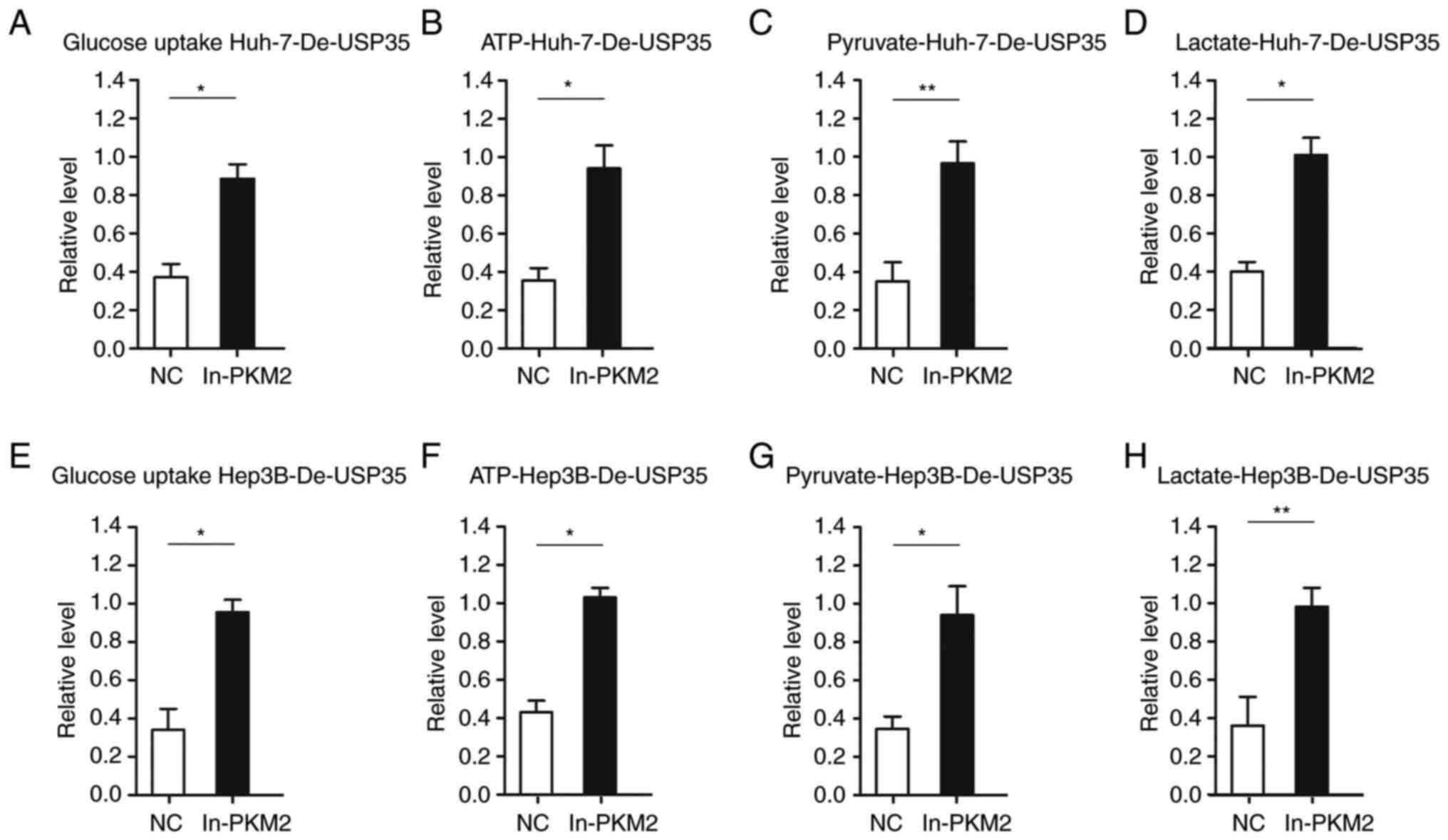
Furthermore, considering the role of PKM2 in
glycolysis, the effect of PKM2 on energy metabolism in HCC was
examined. It was found that increased PKM2 expression significantly
abolished the inhibitory effects of USP35 knockdown on glucose
uptake in Huh-7-De-USP35 and Hep3B-De-USP35 cells (Fig. 8A and E). Additionally, the results
indicated that the levels of pyruvate and lactate, and the
production of ATP were all significantly increased after increasing
of PKM2 expression in Huh-7-De-USP35 and Hep3B-De-USP35 (Fig. 8B-D and Fig. 8F-H, respectively).
Discussion
The ubiquitin-proteasome pathway is one of the
important components in the regulatory system of protein
degradation (22). Through the
multiubiquitination of substrate proteins and the degradation by
proteasomes, various biological processes of intracellular proteins
are regulated, such as the expression of proteins after gene
transcription, regulation of cell growth cycle, immune response,
tumorigenesis and development (22). The ubiquitin-proteasome pathway is
also a dynamic and reversible protein modification process. The
protein substrate in the cell is ubiquitinated by the ubiquitin
ligase system (E1-E2-E3) (23). In
addition, the DUB family removes ubiquitin molecules from
ubiquitin-modified proteins by hydrolyzing ester, peptide or
isopeptide bonds at the carboxyl end of ubiquitin. Members of the
DUB family play a key role in protein deubiquitination and are
involved in various physiological and pathological processes, such
as malignant growth and proliferation of tumors, inflammatory
responses and the immune response (23). The functions of DUBs in cells can
be broadly divided into the following categories: i) Processing
ubiquitin precursors to produce free ubiquitin molecules; ii)
removing a ubiquitin chain on a protein to prevent the protein from
being degraded by the proteasome, thereby stabilizing the protein;
iii) removing the non-degradable ubiquitination signal attached to
a protein; iv) ensuring the homeostasis of ubiquitin molecules in
cells by preventing ubiquitin molecules from being degraded with
substrate proteins; v) participating in the disintegration of
intracellular free ubiquitin chains; and vi) editing the type of
ubiquitin chain by cutting the ubiquitin chain (22,23).
USP35 plays an important role in the deubiquitination of proteins
and in the regulation of tumor progression. USP35 interacts with
estrogen receptor α (ERα) and enhances its stability through
deubiquitination, further enhancing the transcriptional activity of
ERα by interacting with ERα in the DNA region containing the
estrogen response element, prompting the development of endocrine
therapy for resistant ER+ breast cancers (24). USP35 interacts directly with
ribosome binding protein 1 (RRBP1) to prevent its degradation by
proteolytic enzymes (25).
Furthermore, USP35 attenuates endoplasmic reticulum stress-induced
apoptosis by stabilizing RRBP1 in non-small cell lung cancer cells
(25). In addition, USP39
participates in the deubiquitination of SP1 protein and promotes
the proliferation of HCC cells in a SP1-dependent manner (26). Knockdown of USP39 can promote
apoptosis and cell cycle arrest in HCC cells, and SP1 reverses
these effects (26). In HCC, USP10
directly interacts with SMAD4 and stabilizes it through proteolytic
cleavage of its linked ubiquitin, thereby promoting the malignant
progression of HCC (27). In the
present study, the results indicated that USP35 expression was
increased in HCC and was associated with a poor prognosis.
Effectively, decreasing the expression of USP35 may significantly
inhibit the malignant biological behavior of HCC.
Normally differentiated cells mainly rely on
oxidative phosphorylation in mitochondria for energy supply, while
most tumor cells rely on aerobic glycolysis (28). This phenomenon is termed the
Warburg effect. The efficiency of aerobic glycolysis for ATP is
very low, but it endows tumor cells more advantages, such as
providing more energy and metabolic products for the rapid growth
of tumors and helping to maintain cellular redox homeostasis
(28). It has been found that some
cancer-related mutations enable tumor cells to acquire and
metabolize nutrients in a manner conducive to proliferation rather
than efficient production of ATP (29). PKM2 plays a key role in the glucose
metabolism of cancer cells (30).
PK converts phosphoenolpyruvate and ADP into pyruvate and ATP and
is one of the main rate limiting enzymes in glycolysis (31,32).
When the EGF receptor is activated in cells, PKM2 binds to histone
H3 and phosphorylates histone H3 at T11 (33). This phosphorylation is required for
the dissociation of HDAC3 from the cyclin D and Myc promoter
regions and the subsequent acetylation of histone H3 at K9
(33). PKM2-dependent histone H3
modification contributes to EGF-induced cyclin D1 and c-Myc
expression, promoting tumor cell proliferation, cell cycle
progression and tumorigenesis (33). PKM2 translocates to mitochondria
under oxidative stress in tumor cells (33). In mitochondria, PKM2 binds to and
phosphorylates T69 of Bcl2 (34).
This phosphorylation prevents tumor cell apoptosis by preventing
Cul3-E3 ligase-based binding to Bcl2 and subsequent Bcl2
degradation. The results of the present study are consistent with a
previous study where it was demonstrated that PKM2 regulates the
malignant development of tumors (34). Notably, the results of the present
study demonstrated that PKM2 was regulated by ubiquitination by
USP35 and affected the glycolytic process in HCC through the
Warburg effect (as shown by the changes in energy metabolism in HCC
cells). In HCC, zinc finger protein 91 homolog promotes
ubiquitination of the oncoprotein, heterogeneous nuclear
ribonucleoprotein A1, at K48 and its subsequent proteasomal
degradation, and inhibits the splicing of hnRNP Al-dependent PKM
(35). This in turn leads to an
upregulation of PKM1 and downregulation of PKM2, inhibition of the
reprogramming of glucose metabolism and the proliferation and
metastasis of tumor cells (35).
Rho GTPase activating protein 24 (ARHGAP24) recruits the E3 ligase,
WWP1, which promotes the degradation of PKM2 and targets the
ARHGAP24/WWP1/PKM2/β-catenin axis, regulating the malignant
progression of HCC (36).
The present study does have some limitations. First,
the 96 clinical samples of HCC collected constituted a small study
cohort. In a follow-up study, the clinical sample size will be
expanded to provide a reliable clinical basis for further study of
the role of USP35 in HCC. Second, regarding the animal model, USP35
liver-specific knockout mice will be used in future research to
further analyze the molecular mechanisms by which USP35 affects HCC
malignant progression through PKM2. Third, the patients with
multinodular HCC enrolled in the present study were evaluated as
being suitable for hepatectomy, with no more than three nodules,
most of which were <5 cm or were even 3 cm in diameter. In this
case, some patients with HCC were classified as low TNM stage or
early stage. Therefore, although it seems unreasonable, it is
possible for USP35 to be expressed at a low level in these
patients. As patients with HCC with liver cancer nodules >3 were
not included in the study, this is a deficiency of the present
study.
In conclusion, the results of the present study
demonstrated that USP35 was significantly elevated in HCC. USP35
regulated the PKM2 protein level through deubiquitination and
affected the glycolysis process in HCC through the Warburg effect,
which further regulated the malignant progression of HCC. The
present study provides an objective experimental basis for further
study on the role of ubiquitination and the Warburg effect in HCC
and provides a scientific basis for precise treatment of HCC, from
the perspective of energy metabolism.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TL, JY and YZ conceived and designed the study. TL,
BZ, CJ and QZ performed the experiments. TL and YZ analyzed the
data. TL and JY wrote the manuscript. TL, JY and YZ confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by The
Committee on The Ethics of Animal Experiments of West China
Hospital of Sichuan University (Chengdu, China; approval no.
20220228036). The patient study was conducted with the approval of
The Ethics Committee of West China Hospital of Sichuan University
(approval no. 20150176). Written informed consent was obtained from
all patients for the use of their tissue in this study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by grants from the Natural Science
Foundation of China (grant nos. 82070674, 82173255 and 82270691),
the Key R&D projects of Sichuan Provincial Department of
Science and Technology (grant no. 2022YFS0253), the China
Postdoctoral Science Foundation Grant (grant no. 2021M702343), the
'Post-Doctor Research Project' from West China Hospital, Sichuan
University (grant no. 2020HXBH133) and the 'Postdoctoral Cross
Interdisciplinary Innovation Initiation Fund' from Sichuan
University (grant no. 20210317).
References
|
1
|
Couri T and Pillai A: Goals and targets
for personalized therapy for HCC. Hepatol Int. 13:125–137. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chakraborty E and Sarkar D: Emerging
therapies for hepatocellular carcinoma (HCC). Cancers (Basel).
14:27982022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Crocetti L, Bargellini I and Cioni R:
Loco-regional treatment of HCC: Current status. Clin Radiol.
72:626–635. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kulik L and El-Serag HB: Epidemiology and
management of hepatocellular carcinoma. Gastroenterology.
156:477–491.e1. 2019. View Article : Google Scholar
|
|
5
|
De Stefano F, Chacon E, Turcios L, Marti F
and Gedaly R: Novel biomarkers in hepatocellular carcinoma. Dig
Liver Dis. 50:1115–1123. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sayiner M, Golabi P and Younossi ZM:
Disease burden of hepatocellular carcinoma: A global perspective.
Dig Dis Sci. 64:910–917. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fujiwara N, Friedman SL, Goossens N and
Hoshida Y: Risk factors and prevention of hepatocellular carcinoma
in the era of precision medicine. J Hepatol. 68:526–549. 2018.
View Article : Google Scholar :
|
|
8
|
Sahu SK, Chawla YK, Dhiman RK, Singh V,
Duseja A, Taneja S, Kalra N and Gorsi U: Rupture of hepatocellular
carcinoma: A review of literature. J Clin Exp Hepatol. 9:245–256.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sharma D and Mandal P: NAFLD: Genetics and
its clinical implications. Clin Res Hepatol Gastroenterol.
46:1020032022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shah PA, Patil R and Harrison SA:
NAFLD-related hepatocellular carcinoma: The growing challenge.
Hepatology. 77:323–338. 2023. View Article : Google Scholar
|
|
11
|
Popovic D, Vucic D and Dikic I:
Ubiquitination in disease pathogenesis and treatment. Nat Med.
20:1242–1253. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mansour MA: Ubiquitination: Friend and foe
in cancer. Int J Biochem Cell Biol. 101:80–93. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
van Wijk SJ, Fulda S, Dikic I and
Heilemann M: Visualizing ubiquitination in mammalian cells. EMBO
Rep. 20:e465202019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Komander D: The emerging complexity of
protein ubiquitination. Biochem Soc Trans. 37:937–953. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang Z, Jiang W, Mao M, Zhao J, Chen J and
Cheng N: Deubiquitinase USP35 modulates ferroptosis in lung cancer
via targeting ferroportin. Clin Transl Med. 11:e3902021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Chen Y, Chen X, Zhang W, Zhao L,
Weng L, Tian H, Wu Z, Tan X, Ge X, et al: Deubiquitinase USP35
restrains STING-mediated interferon signaling in ovarian cancer.
Cell Death Differ. 28:139–155. 2021. View Article : Google Scholar :
|
|
17
|
Zhong Q, Wang Z, Kang H and Wu R:
Molecular mechanism of FBXW7-mediated ubiquitination modification
in nasopharyngeal carcinoma cell proliferation in vitro and in
vivo. Pathol Res Pract. 244:1540562023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reig M, Forner A, Rimola J, Ferrer-Fàbrega
J, Burrel M, Garcia-Criado Á, Kelley RK, Galle PR, Mazzaferro V,
Salem R, et al: BCLC strategy for prognosis prediction and
treatment recommendation: The 2022 update. J Hepatol. 76:681–693.
2022. View Article : Google Scholar :
|
|
19
|
Beyoğlu D and Idle JR: The metabolomic
window into hepatobiliary disease. J Hepatol. 59:842–858. 2013.
View Article : Google Scholar
|
|
20
|
Leznicki P, Natarajan J, Bader G, Spevak
W, Schlattl A, Abdul Rehman SA, Pathak D, Weidlich S, Zoephel A,
Bordone MC, et al: Expansion of DUB functionality generated by
alternative isoforms-USP35, a case study. J Cell Sci.
131:jcs2127532018. View Article : Google Scholar
|
|
21
|
Zhang Z, Deng X, Liu Y, Liu Y, Sun L and
Chen F: PKM2, function and expression and regulation. Cell Biosci.
9:522019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun T, Liu Z and Yang Q: The role of
ubiquitination and deubiquitination in cancer metabolism. Mol
Cancer. 19:1462020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu Y and Deng J:
Ubiquitination-deubiquitination in the Hippo signaling pathway
(review). Oncol Rep. 41:1455–1475. 2019.PubMed/NCBI
|
|
24
|
Cao J, Wu D, Wu G, Wang Y, Ren T, Wang Y,
Lv Y, Sun W, Wang J, Qian C, et al: USP35, regulated by estrogen
and AKT, promotes breast tumorigenesis by stabilizing and enhancing
transcriptional activity of estrogen receptor α. Cell Death Dis.
12:6192021. View Article : Google Scholar
|
|
25
|
Wang W, Wang M, Xiao Y, Wang Y, Ma L, Guo
L, Wu X, Lin X and Zhang P: USP35 mitigates endoplasmic reticulum
stress-induced apoptosis by stabilizing RRBP1 in non-small cell
lung cancer. Mol Oncol. 16:1572–1590. 2022. View Article : Google Scholar
|
|
26
|
Dong X, Liu Z, Zhang E, Zhang P, Wang Y,
Hang J and Li Q: USP39 promotes tumorigenesis by stabilizing and
deubiquitinating SP1 protein in hepatocellular carcinoma. Cell
Signal. 85:1100682021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yuan T, Chen Z, Yan F, Qian M, Luo H, Ye
S, Cao J, Ying M, Dai X, Gai R, et al: Deubiquitinating enzyme
USP10 promotes hepatocellular carcinoma metastasis through
deubiquitinating and stabilizing Smad4 protein. Mol Oncol.
14:197–210. 2020. View Article : Google Scholar :
|
|
28
|
Liberti MV and Locasale JW: The Warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pascale RM, Calvisi DF, Simile MM, Feo CF
and Feo F: The Warburg effect 97 years after its discovery. Cancers
(Basel). 12:28192020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Israelsen WJ and Vander Heiden MG:
Pyruvate kinase: Function, regulation and role in cancer. Semin
Cell Dev Biol. 43:43–51. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grant MM: Pyruvate kinase, inflammation
and periodontal disease. Pathogens. 10:7842021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zahra K, Dey T, Ashish, Mishra SP and
Pandey U: Pyruvate kinase M2 and cancer: The role of PKM2 in
promoting tumorigenesis. Front Oncol. 10:1592020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang W, Xia Y, Hawke D, Li X, Liang J,
Xing D, Aldape K, Hunter T, Alfred Yung WK and Lu Z: PKM2
phosphorylates histone H3 and promotes gene transcription and
tumorigenesis. Cell. 150:685–696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liang J, Cao R, Wang X, Zhang Y, Wang P,
Gao H, Li C, Yang F, Zeng R, Wei P, et al: Mitochondrial PKM2
regulates oxidative stress-induced apoptosis by stabilizing Bcl2.
Cell Res. 27:329–351. 2017. View Article : Google Scholar :
|
|
35
|
Chen D, Wang Y, Lu R, Jiang X, Chen X,
Meng N, Chen M, Xie S and Yan GR: E3 ligase ZFP91 inhibits
hepatocellular carcinoma metabolism reprogramming by regulating PKM
splicing. Theranostics. 10:8558–8572. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang W, Wang B, Yu Q, Liu T, Li T, Tian T,
Jin A, Ding L, Chen W, Wang H, et al: ARHGAP24 represses β-catenin
transactivation-induced invasiveness in hepatocellular carcinoma
mainly by acting as a GTPase-independent scaffold. Theranostics.
12:6189–6206. 2020. View Article : Google Scholar
|