Introduction
Pancreatic cancer is the fourth most common cause of
cancer-related mortality worldwide (1). The overall prognosis for patients
with pancreatic cancer remains poor; less than 5% of patients with
adenocarcinoma of the pancreas survive for more than 5 years
following diagnosis (2). Surgical
resection is currently the only potentially curative treatment for
pancreatic cancer. However, 80–85% patients have advanced
unresectable disease at diagnosis (2). Chemotherapy is a very important
therapeutic strategy for unresectable cancer patients. Gemcitabine
(GEM) and 5-fluorouracil (5-FU) are the common chemotherapy drugs
used to treat advanced pancreatic cancer. Gemcitabine
(2′-deoxy-2′-difluorodeoxycytidine: Gemzar; GEM) is a deoxycytidine
analogue with structural and metabolic similarities to cytarabine.
Currently, this nucleoside analogue appears to be the only
clinically effective drug for pancreatic cancer, and has shown
improvement in overall survival and quality of life (3–6).
However, many clinical trials have shown that GEM alone or GEM in
combination with other drugs is not likely to achieve marked
success and the acquisition of resistance during chemotherapy may
further limit the therapeutic success (7,8).
Even if a potentially curative surgical resection can be performed,
the five-year overall survival is low at 10–25%, mostly due to an
almost complete chemoresistance which may be inherent and/or
acquired (9,10). Many studies have been directed
towards understanding the mechanism of chemotherapy response and
resistance, with the ultimate goal of identifying molecular markers
that allow the prediction of chemotherapy response. For pancreatic
adenocarcinoma, the process of finding new chemotherapeutics has
been exceedingly slow and disappointing. To date, there has been no
significant breakthrough in the treatment of pancreatic cancer.
Therefore, identifying novel molecular markers related to GEM
resistance would be helpful to further understand the exact
mechanisms of chemoresistance and produce more effective
chemotherapy strategies for patients with pancreatic cancer in the
future.
Due to intrinsic chemoresistance, chemotherapeutic
drugs usually fail to kill all tumor cells at clinically tolerated
doses. The presence of surviving cells above a certain threshold
following primary therapy increases the relapse occurrence, which
may then generate acquired and more complex resistance phenotypes
as a result of sequential genetic changes over long periods of time
(11,12). These surviving cells may either be
pre-existent or may be generated as a direct result of the
chemotherapy itself. Recently, we introduced a new model for the
study of de novo drug resistance of pancreatic cancer, which
involves the utilization of surviving cancer cells following
primary drug treatment. The surviving cells are potentially
drug-resistant and exhibit similar growth and tumorigenic
potentials to their untreated parental cells (13). In this study, we investigated the
differential expression of proteins in these stably surviving cells
and cells with or without GEM treatment to screen candidate
molecules related to intrinsic chemoresistance. In addition, we
studied whether these proteins may be used clinically as biomarkers
of gemcitabine sensitivity in tumor specimens.
Materials and methods
Cell lines and materials
The pancreatic cancer cell line Capan-1 (ATCC,
Manassas, VA, USA) was used in this study. Capan-1 was cultured in
IMDM (Gibco, Carlsbad, CA, USA) containing 20% fetal calf serum
(Gibco), 100 U/ml penicillin and 100 U/ml streptomycin. The cells
were cultured at 37°C, in 5% CO2 and 95% relative
humidity. The drug used in this study was GEM (Lilly, France).
Sample preparation and protein
extraction
Capan-1 cells were exposed to GEM at a concentration
of 2,000 μg/ml (to obtain a cell survival rate of <15%) for 72
h. The cells were then washed twice with phosphate-buffered saline
(PBS) solution, and cultured in drug-free media for another 7 days.
The surviving cells were then washed twice with PBS, trypsinized
and counted. Non-viable cells were excluded using Trypan blue
staining. Control cells were counted when the drug treatment was
completed. The viable cell rate was calculated as the number of
surviving cells divided by the number of control cells ×100%. A
ReadyPrep™ Protein Extraction kit (total protein) (Bio-Rad,
Hercules, CA, USA) was used for control and surviving cells
according to the manufacturer's instructions. The cells were washed
3 times with PBS, centrifuged at 800 rpm for 5 min, the supernatant
was discarded, and the cells were lysed to produce protein lysates.
The mixture was then sonicated for 30 sec, 3 times (pulse duration
of 30 sec on and 15 sec off) in an ice bath sonicator. Finally, the
sample was centrifuged at 12,000 rpm at 20°C for 30 min, and the
supernatant was collected. The protein cleanup was performed
according to the manufacturer's instructions using the ReadyPrep™
2-D Cleanup kit (Bio-Rad) and the dry proteins were diluted with
rehydration buffer (8 M urea, 4% CHAPS). The concentration of the
prepared protein samples was determined by the Shanghai Generay
Biotech Co., Ltd. (China) BCA protein assay according to the
manufacturer's instructions. The samples were stored at −80°C until
further analysis.
Two-dimensional gel electrophoresis
(2-DE) and silver staining
Firstly, 250 μg protein was diluted with rehydration
buffer [8 M urea, 4% CHAPS, 65 mM DTT, 0.2% (w/v) Bio-Lyte
(Bio-Lyte 3/10 ampholyte, 40%, Bio-Rad) and 0.001% bromophenol
blue] to 350 μl for isoelectric focusing (IEF). IEF was applied on
IPG strips (ReadyStrip IPG Strips, pH 3–10, 17 cm, Bio-Rad) with a
Protean IEF system (Bio-Rad) according to the following protocol:
active hydration (50 V for 14 h), higher voltage liner (250 V for
30 min), rapid cleanup (1,000 V for 1 h), boost voltage (liner,
10,000 V for 5 h) and isoelectric focus (rapidly, 10,000 V for a
total 60,000 kVh). Strips were then equilibrated at room
temperature for 15 min in 10 ml equilibration solution [6 M urea,
0.375 M pH 8.8 Tris-HCl, 20% glycerol, 2% sodium dodecyl sulfate
(SDS), 2% DTT] and incubated for another 15 min in an equilibration
solution that was the same as previously used, with the exception
of replacement of DTT with 2.5% iodoacetamide. The two-dimensional
electrophoresis was performed on 12% SDS gels with a Protean II xi
system (Bio-Rad). The SDS-PAGE gels were run at 5 mA/gel (until the
proteins had run out of the strips) and then at 25 mA/gel until the
bromophenol blue dye front reached the bottom of the gels. Three
2-D gels per sample were run to guarantee reproducibility.
Following 2-DE, proteins were visualized by silver-staining
according to the manufacturer's instructions (Bio-Rad): Fixing
(acetic acid 100 ml, ethanol 400 ml and pure water up to 1,000 ml)
for 120 min; incubation (150 ml ethanol, 34 g Na-acetate, 1 g
Na2S2O3·5H2O and pure
water to 500 ml) for 30 min; washing (pure water) 3×5 min;
silvering (AgNO3 0.8 g, add pure water to 500 ml) for 20
min; washing (pure water) 2×1 min; development (12.5 g
Na2CO3, 200 μl formaldehyde and pure water to
500 ml) 8–10 min; stopping (200 ml acetic acid and pure water to
500 ml); washing (pure water) 3×5 min; and preserving (4°C,
packaged by preservative film).
Visualization and image analysis
The silver-stained gels were scanned using a GS-800™
calibrated densitometer (Bio-Rad) with 300 dpi and analyzed using
PDQuest software (version 7.0, Bio-Rad). Protein spots were
detected automatically, and manual spot editing or deleting was
performed when required. Only those protein spots for which the
greatest alterations were >1.5-fold between control cells and
the surviving cells after GEM treatment were selected for further
characterization using mass spectrometry. The protein spots that
were selected were sent to Shanghai Unlimit Biotechnology Co., Ltd.
(China) for mass spectrometric analysis.
In-gel digestion
Briefly, spots of interest were excised from the
2-DE gels and were placed in deionized water to soak. After washing
with deionized water, the spots of interest were centrifuged,
supernatant was absorbed, calcium ammonium nitrate (CAN) was used
to condense the gel, and samples were vacuum dried. Then, 45 mM
DTT/25 mM NH4HCO3 was added and the mixture
was left to react for 1 h at 56°C. Subsequently, the mixture was
cooled to room temperature, centrifuged, the supernatant was
discarded and the sample was reacted in the dark for 45 min with
100 mM iodoacetoamide/25 mM NH4HCO3. The
mixture was then centrifuged, the supernatant was discarded, 25 mM
NH4HCO3 was added, then the sample was
vibrated for 3–5 min, then centrifuged, the supernatant was
discarded, acetonitrile (ACN) was added to condense the gel, the
sample was vibrated and vacuum dried. The proteins in-gel were then
digested with a tryptic enzyme, firstly at 4°C for 30 min, secondly
at 37°C for 12–16 h, and lastly were sonicated for 10 min with 50%
ACN/5% formic acid. The sample was then centrifuged, the
supernatant was collected in another eppendorf pipe and 50% ACN/5%
formic acid was added to the pre-pipe. The sample was sonicated,
centrifuged, the supernatant of the pre-pipe was collected and the
supernatants were mixed. The supernatants were condensed to 10 μl
in a speed-vacuum drier for mass spectrometric analysis.
Liquid chromatography-mass spectrometry
detection and analysis
The tryptic peptides were added to an opposite
enriching column [(C18, 5 μm, 300 Å, 300-μm inner diameter ×5 mm;
Dionex/LC Packings), velocity (10 μl/min), mobile phase (3% ACN,
0.1% FA)]. The enriching column was in series with another opposite
analytical column (Dionex/LC Packings, C18, 75-μm inner diameter,
150 mm length). The peptides were eluted using a QStar-XL mass
spectrograph (MDS Sciex/Applied Biosystems) with NanoESI ion source
at a velocity of 250 μl/min. The liquid phase system of Nano UPLC
(Waters) provided gradient elution: mobile phase A, 0.1% formic
acid/H2O; and mobile phase B, 0.1% formic
acid/acetonitrile. The gradient was 5–50% B (60 min), 50–90% B (30
min) and 90% B (15 min).
The parameter settings of the ion source were
voltage 3,000 V, assisting nitrogen 0.2 L/h and temperature 100°C.
Chromatography-cascade spectrum acquisition was performed in data
dependent acquisition (DDA) mode. There were 8 highest-ionic
strength at first level of the spectrum and the 2–4 electrically
charged peptides had been selected for cracking. The scanning area
of first level of the spectrum was 450–1,800 m/z and the scanning
area of the chromatography-cascade spectrum was 50–1,800 m/z.
The chromatography-cascade spectrum searched human
protein databases (ftp://ftp.ebi.ac.uk/pub/databases/IPI/Current, version
ipi.HUMAN.v3.57.fasta), and the software used for research was
Mascot (http://www.matrixscience.com). The
quality tolerance of the chromatography-cascade spectrum was 0.3
Da, the maximal enzyme digestion omissive spot was 2, and potential
modifiers were carbamoylmethylation of cysteine, oxidize of
methionine and phosphatase of serine and tyrosine.
Western blot analysis
Equivalent amounts of total protein (80 μg) and the
nuclear and cytoplasm proteins (40 μg respectively, according to
the protocol of the extraction of nuclear and cytoplasm protein
kit, KeyGen Biotech, China) were loaded in each lane and were
fractioned by electrophoresis on 12% (w/v) SDS-PAGE gels
(Mini-Protean3, Bio-Rad), then transferred onto a PVDF membrane
using Mini Trans-Blot (Bio-Rad) and blocked with TBST containing 5%
BSA at 4°C overnight. The PVDF membrane was probed with a 1:2,000
dilution of rabbit anti-heat shock protein B1 (HSP27) polyclonal
antibody (ab5579, Abcam, Cambridge, UK). Horseradish
peroxidase-conjugated goat anti-rabbit IgG at a dilution of 1:5,000
was used as a secondary antibody. The protein bands were visualized
using the ECL system. The same membrane was reblotted with mouse
affinity purified anti-GAPDH antibody (Shanghai Kangcheng Biotech
Co. Ltd., China) for total protein and cytoplasm protein, and
anti-lamin B1 antibody (KeyGen Biotech) for nuclear protein, at a
dilution of 1:2,000, to confirm equal loading.
Patients and specimens
The pancreatic ductal adenocarcinoma (PDAC)
specimens were collected with informed consent from 47 patients
undergoing radical resection of primary PDAC in Changhai Hospital,
Second Military Medical University during 2003–2007. The patients
were selected based on a distinctive pathological diagnosis of PDAC
according to the World Health Organization classification of tumors
of the digestive system (2010). All the experimental procedures
were approved by the Research Ethics Committee of the Second
Military University. The medical records of the patients were
retrospectively reviewed, and the demographic, clinicopathological
and treatment data were also collected. The tumor location and size
were obtained from the surgical report. Patients receiving
preoperative chemotherapy or radiotherapy were excluded from the
study.
Immunohistochemistry analysis
Tissue microarrays were constructed using the 47
paraffin-embedded tumor tissue specimens by a precision arraying
instrument (Beecher Instruments, Silver Spring, MD, USA). In each
case, three tumor cores and two surrounding non-tumor tissues were
selected. The sections from the tissue microarray blocks were
deparaffinized, rehydrated and then heated in 0.01 mol/l sodium
citrate buffer (pH 6.0) for 8 min at 95°C. Following incubation
with 0.3% hydrogen peroxide in methanol for 15 min at room
temperature and treatment with normal goat serum (Invitrogen,
Carlsbad, CA, USA), the sections were incubated overnight at 4°C
with rabbit anti-HSP27 polyclonal antibody (ab5579, Abcam) at a
dilution of 1:250 (Zymed Labroatories, Invitrogen). Slides were
rinsed for 10 min in PBS wash solution and incubated for 30 min
with the HRP-labeled polymer conjugated secondary antibody
(EnVision+; DakoCytomation, Carpintera, CA, USA) was applied
according to the manufacturer's instructions. A known positive
endometrial cancer tissue biopsy sample was used as the positive
control. PBS and non-immune serum were used instead of the primary
antibody for the blank control and negative control samples,
respectively.
HSP27 protein was stained brown in the cytoplasm of
cancer cells. The evaluation of HSP27 staining was carried out by
two independent observers, and the staining area and intensity were
scored separately. Specifically, a score of 0 was assigned to a
staining area with ≤10% of the tumor cells, 1 for an area with
11–25% of the tumor cells, 2 for 26–50% tumor cells, and 3 for
>51% tumor cells. For the staining intensity, a score of 0 was
assigned for absent/weak staining (negative control), 1 for weak
staining markedly stronger than the negative control level, 2 for
moderately intense staining, and 3 for intense staining. The final
grade of the section was derived from the sum of the staining area
and intensity scores. A final score ≥3 was recognized to indicate
positive expression. Equivocal stains were considered negative.
Results
Sensitivity of Capan-1 to GEM
As in our previous study, it was necessary to
identify doses of GEM (2,000 μg/ml) in vitro that had fewer
than 15% viable cells after treatment, and the cell viability rate
was 6.03±3.26%. After 7 days of culturing cells in drug-free
medium, there was no significant cell mortality. The cells had
similar morphology and comparable growth and tumorigenic potential
to their untreated parental cells. Repeated subculture affected the
cell-cycle profile and growth characteristics of the surviving
cells (13). Surviving cells were
harvested for protein extraction, at which time the cells were
viable, stable, and in some cases, exhibited growth.
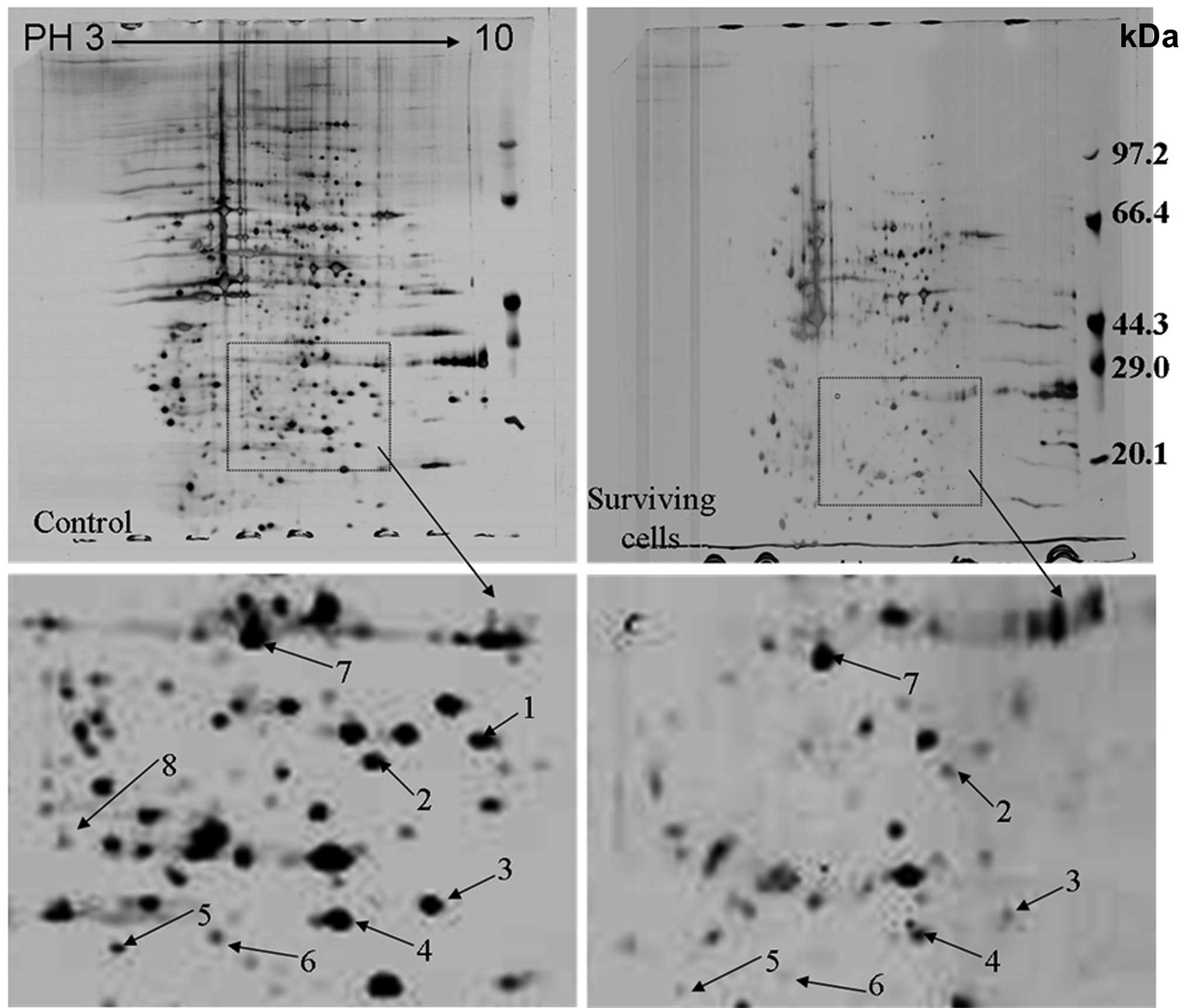
Differentially expressed proteins
downregulated in the Capan-1 cell line and in stable surviving
cells following primary GEM treatment by proteomic analysis
Protein expression was assessed using three samples
each of Capan-1 cells cultured under the same conditions. More than
1,000 protein spots were visualized on the 2-DE gels. We adopted a
2-DE analysis to quantitatively compare the protein profiles of the
Capan-1 cell line and its stable surviving cells following primary
GEM treatment. The differently expressed proteins with more than
1.5-fold changes between the two groups were selected to perform
protein identification by liquid chromatography-mass spectrometry
analysis. A total of 9 downregulated proteins were successfully
identified (Fig. 1). Information
on the 9 proteins identified is shown in Table I. Based on their functions, these
proteins are mainly involved in signal transduction (guaniune
nucleotide-binding protein subunit β-2-like 1 and annexin A1),
transport (galectin-3 and hydroxysteroid 17β dehydrogenase-10),
metabolism (proline synthetase co-transcribed bacterial homolog
protein, proteasome subunit α type-2 and lysophospholipase 1) and
as chaperones (heat shock protein B1). There was also one protein
with an unknown protein. In this study, we selected HSP27 for
further study of intrinsic chemoresistance in pancreatic
cancer.
 | Table IDifferential expression of proteins
between the surviving cells of Capan-1 after primary GEM treatment
compared with Capan-1 cells without GEM treatment on proteomic
analysis. |
Table I
Differential expression of proteins
between the surviving cells of Capan-1 after primary GEM treatment
compared with Capan-1 cells without GEM treatment on proteomic
analysis.
| Spot No | Protein | MW (Da) | pI (PH) | Protein ID | Protein function |
|---|
| 1 | Guaniune
nucleotide-binding protein subunit β-2-like 1 | 35054 | 7.5645 | 56605 | Signal
transduction |
| 2 | Galectin-3 | 26136 | 8.8755 | 31258 | Transport
protein |
| 3 | Proline synthetase
co-transcribed bacterial homolog protein | 30324 | 7.2759 | 4351 | Metabolism |
| 4 | Hydroxysteroid 17β
dehydrogenase-10 | 25967 | 7.025 | 21856 | Transport
protein |
| 5 | Proteasome subunit α
type-2 | 25882 | 7.2979 | 16711 | Metabolism |
| 6 | cDNA FLJ52710 | 19784 | 5.9381 | 71428 | Unknown |
| 7 | Lysophospholipase
1 | 22860 | 6.0478 | 25964 | Metabolism |
| 8 | Annexin A1 | 38689 | 6.6359 | 16324 | Signal
transduction |
| 9 | Heat shock protein
B1(HSP27) | 22768 | 5.9588 | 6789 | Chaperone |
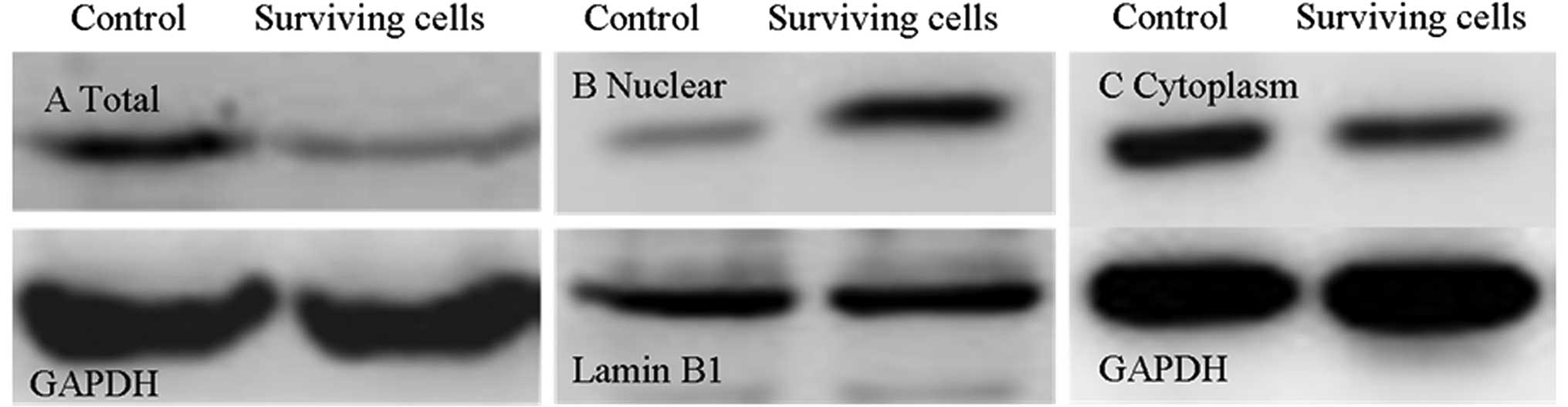
Verification of protein expression by
western blot analysis
The differential expression of the Capan-1 cell line
and its stable surviving cells following primary GEM treatment
identified in the proteomic study were further validated by western
blot analysis. As shown in Fig. 2,
the surviving cells had a decreased expression level of HSP27
compared to Capan-1 cells, which is consistent with the findings of
the proteomic analysis (Fig. 2).
The changes of the location of HSP27 indicated that HSP27 decreased
in the cytoplasm but increased in the nucleus, compared to Capan-1
cells (Fig. 2).
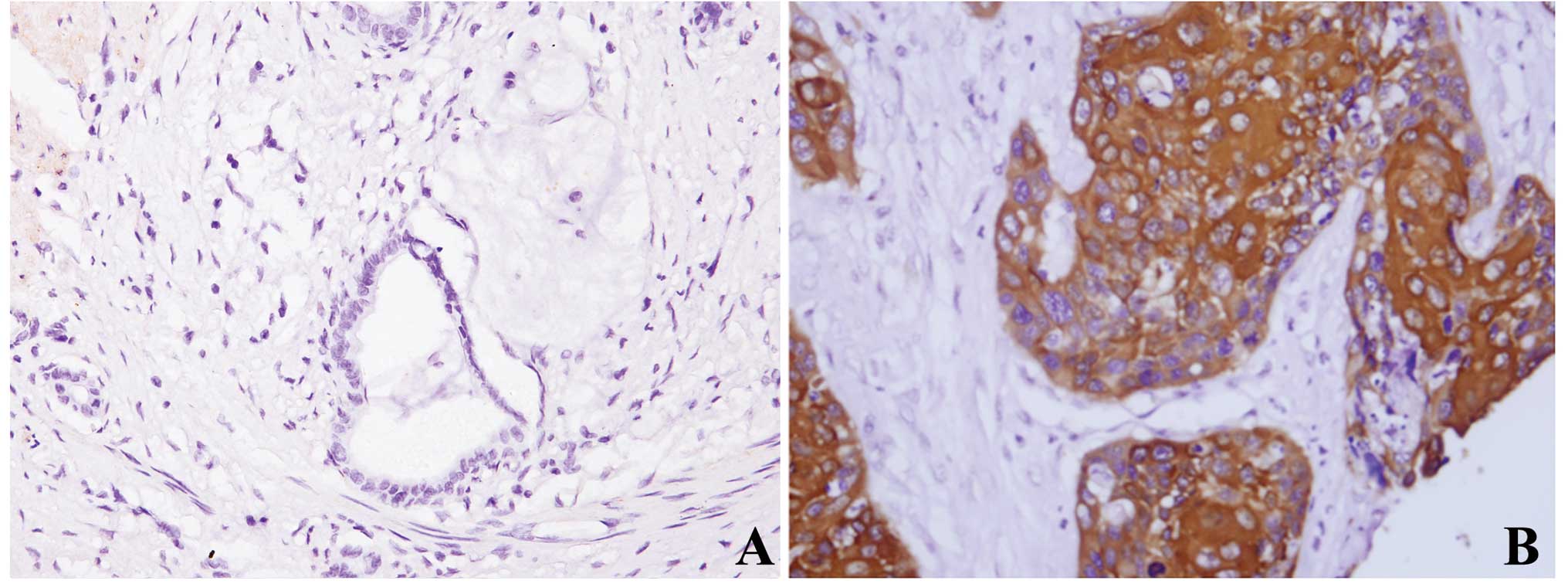
Immunohistochemistry of HSP27 in
pancreatic cancer tissues and the correlation with GEM effects and
survival
We further validated the data by examining
expression levels of HSP27 at the protein level. Immunoreactivity
was detected in the cytoplasm. Adjacent benign pancreatic
parenchyma was negative or weakly focally expressed (13%, 6/47). Of
the 47 PDAC specimens, 38 (81%) exhibited HSP27 immunoreactivity
(Fig. 3). The results of the
correlation between tumor HSP27 expression and tumor pathological
features are shown in Table II.
HSP27 was aberrantly overexpressed in PDACs relative to adjacent
non-tumor tissues. However, there was no significant difference
between HSP27 levels and other clinicopathological findings such as
age, gender, location of tumor, histological grade and perineural
invasion in PDAC.
| Table IICorrelation of HSP27 expression with
clinicopathological features of pancreatic ductal adenocarcinoma
patients. |
Table II
Correlation of HSP27 expression with
clinicopathological features of pancreatic ductal adenocarcinoma
patients.
| | HSP27
immunoreactivity | | |
|---|
| |
| | |
|---|
| Variables | n | − | ± | + | ++ | r value | P-value |
|---|
| Gender |
| Male | 28 | 6 | 10 | 9 | 3 | 0.146 | 0.326 |
| Female | 19 | 3 | 6 | 5 | 5 | | |
| Age (yrs) |
| ≤55 | 23 | 4 | 9 | 8 | 2 | 0.090 | 0.548 |
| >55 | 24 | 5 | 7 | 6 | 6 | | |
| Location of
tumor |
| Head | 29 | 6 | 9 | 12 | 2 | 0.106 | 0.479 |
| Body/tail | 18 | 3 | 7 | 2 | 6 | | |
| Histological
grade |
|
Well-differentiated | 5 | 1 | 1 | 2 | 1 | −0.136 | 0.361 |
| Moderately
differentiated | 37 | 6 | 14 | 10 | 7 | | |
| Poorly
differentiated | 5 | 2 | 1 | 2 | 0 | | |
| Perineural
invasion |
| Present | 25 | 6 | 7 | 8 | 4 | 0.044 | 0.768 |
| Absent | 22 | 3 | 9 | 6 | 4 | | |
Discussion
In this study, we applied 2-DE-based proteomics to
identify the differentially expressed proteins of pancreatic
carcinoma cells that survived GEM primary treatment in
vitro. We successfully identified 9 proteins with significantly
altered expression levels. Based on their functions, these proteins
are mainly involved in signal transduction, transport, metabolism,
and as chaperones. One protein has an unknown function. The
expression of HSP27 was decreased in a gemcitabine-resistant
pancreatic cancer cell line. HSP27 was aberrantly overexpressed in
PDACs relative to adjacent nontumor tissues.
HSP27, an ATP-independent chaperone, is involved in
a number of processes such as apoptosis, DNA repair and
recombination (14,15). HSP27 is upregulated in many types
of cancer, including colorectal (16), breast (17), prostate (18) and pancreatic cancer (19). HSP27 may be expressed in a wide
range of human cancers and could be a prognostic marker in many
cancers (20–22). HSP27 is associated with poor
prognosis in non-small cell lung carcinoma (23) and gastric cancer (24), as well as pancreatic ductal
adenocarcinoma (22,25). However, HSP27 is associated with
good prognosis in esophageal cancer, oral squamous cell carcinoma
and malignant fibrous histiocytoma (23,26).
These results suggest that HSP27 may have different functions in
various types of cancer. There is evidence that HSP27 is involved
in anti-cancer resistance, which is related to its location in many
cancer cells. Vargas-Roig et al (27) found that, following drug
administration, cytoplasmic HSP27 decreased or disappeared in 5
cases (36%), increased in 5 cases and remained unchanged in 4 cases
(28%). Following chemotherapy, HSP27 was expressed in the nuclear
compartment in 10 cases (71%); this increase was statistically
significant (P=0.007). Increased HSP27 expression is correlated
with higher rates of GEM resistance in patients with pancreatic
cancer. Furthermore, knock-out of HSP27 reduces chemoresistance
(28,29). RP101 (brivudine) may bind to heat
shock protein HSPB1 (HSP27) and enhances survival in animals and
pancreatic cancer patients (30).
As previously shown, HSP27 may be phosphorylated and translocated
to the nucleus under different circumstances and exposed to
different stresses regulated by multiple steps (31). The phosphorylation status of HSP27
plays a key role in GEM-induced growth suppression of pancreatic
cancer, and it could also be a possible biomarker for predicting
the response of pancreatic cancer patients to treatment with GEM
(32,33). The combination of HSP27 knockdown
with OGX-427 and chemotherapeutic agents such as GEM may be a novel
strategy to inhibit the progression of pancreatic cancer (34). In our study, we found the
downregulation of total HSP27 in pancreatic carcinoma cells
survived from GEM treatment. Furthermore, we found that HSP27 was
decreased in the cytoplasm and increased in the nucleus, and the
nuclear expression of HSP27 was always low in immunostaining of
pancreatic cancers, which demonstrated that the expression of HSP27
in pancreatic cancer occurs mainly in the cytoplasm. The results
indicated that researching the location of HSP27 was more
significant for total HSP27 and the different locations of HSP27
had a correlation with cancer chemotherapy resistance. However, the
expression of HSP27 in the nucleus was related to the intrinsic
chemoresistance of pancreatic cancer cells.
In conclusion, HSP27 may be important in the
intrinsic chemoresistance of pancreatic cancer cells and the
location of HSP27 in pancreatic cancer cells was more significant
compared to the cytoplasmic location. For therapeutic application,
a targeted therapy against HSP27 in the nuclear signaling pathway
could be applied to overcome GEM resistance and may be beneficial
in the treatment of pancreatic cancer. The mechanisms of HSP27 in
pancreatic cancer require further research.
Acknowledgements
This project was supported by the National Key
Project of Scientific and Technical Supporting Programs of China
(No. 2006BAI02A14) and the National Natural Science Foundation of
China (No. 30770996 and No. 81172310).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar
|
|
3
|
Kawanami T, Takiguchi S, Ikeda N and
Funakoshi A: A humanized anti-IGF-1R monoclonal antibody (R1507)
and/or metformin. Oncol Rep. 27:867–872. 2012.PubMed/NCBI
|
|
4
|
Kasuya K, Tsuchida A, Nagakawa Y, et al:
Hypoxia-inducible factor-1alpha expression and gemcitabine
chemotherapy. Oncol Rep. 26:1399–1406. 2011.PubMed/NCBI
|
|
5
|
Chen H, Wei W, Guo Y, et al: Enhanced
effect of gemcitabine by emodin against pancreatic cancer in vivo
via cytochrome C-regulated apoptosis. Oncol Rep. 25:1253–1261.
2011.PubMed/NCBI
|
|
6
|
Di Marco M, Di Cicilia R, Macchini M, et
al: Metastatic pancreatic cancer: Is gemcitabine still the best
standard treatment? (Review). Oncol Rep. 23:1183–1192.
2010.PubMed/NCBI
|
|
7
|
Burris HA 3rd, Moore MJ, Andersen J, et
al: Improvements in survival and clinical benefit with gemcitabine
as first-line therapy for patients with advanced pancreas cancer: a
randomized trial. J Clin Oncol. 15:2403–2413. 1997.PubMed/NCBI
|
|
8
|
Neoptolemos JP, Stocken DD, Bassi C, et
al: Adjuvant chemotherapy with fluorouracil plus folinic acid vs
gemcitabine following pancreatic cancer resection: a randomized
controlled trial. Jama. 304:1073–1081. 2010. View Article : Google Scholar
|
|
9
|
Banerjee D, Mayer-Kuckuk P, Capiaux G,
Budak-Alpdogan T, Gorlick R and Bertino JR: Novel aspects of
resistance to drugs targeted to dihydrofolate reductase and
thymidylate synthase. Biochim Biophys Acta. 1587:164–173. 2002.
View Article : Google Scholar
|
|
10
|
Muerkoster S, Wegehenkel K, Arlt A, et al:
Tumor stroma interactions induce chemoresistance in pancreatic
ductal carcinoma cells involving increased secretion and paracrine
effects of nitric oxide and interleukin-1beta. Cancer Res.
64:1331–1337. 2004. View Article : Google Scholar
|
|
11
|
Yamauchi K, Yang M, Hayashi K, et al:
Induction of cancer metastasis by cyclophosphamide pretreatment of
host mice: an opposite effect of chemotherapy. Cancer Res.
68:516–520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu YJ, Muldoon LL, Dickey DT, Lewin SJ,
Varallyay CG and Neuwelt EA: Cyclophosphamide enhances human tumor
growth in nude rat xenografted tumor models. Neoplasia. 11:187–195.
2009.PubMed/NCBI
|
|
13
|
Liu QH, Zhang J, Zhao CY, et al: Surviving
cells after treatment with gemcitabine or 5-fluorouracil for the
study of de novo resistance of pancreatic cancer. Cancer Lett.
314:119–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mosser DD and Morimoto RI: Molecular
chaperones and the stress of oncogenesis. Oncogene. 23:2907–2918.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Garrido C, Brunet M, Didelot C, Zermati Y,
Schmitt E and Kroemer G: Heat shock proteins 27 and 70:
anti-apoptotic proteins with tumorigenic properties. Cell Cycle.
5:2592–2601. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao L, Liu L, Wang S, Zhang YF, Yu L and
Ding YQ: Differential proteomic analysis of human colorectal
carcinoma cell lines metastasis-associated proteins. J Cancer Res
Clin Oncol. 133:771–782. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang SH, Kang KW, Kim KH, et al:
Upregulated HSP27 in human breast cancer cells reduces Herceptin
susceptibility by increasing Her2 protein stability. BMC Cancer.
8:2862008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Larkin SE, Holmes S, Cree IA, et al:
Identification of markers of prostate cancer progression using
candidate gene expression. Br J Cancer. 106:157–165. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xia Y, Liu Y, Wan J, et al: Novel triazole
ribonucleoside down-regulates heat shock protein 27 and induces
potent anticancer activity on drug-resistant pancreatic cancer. J
Med Chem. 52:6083–6096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ciocca DR and Calderwood SK: Heat shock
proteins in cancer: diagnostic, prognostic, predictive, and
treatment implications. Cell Stress Chaperones. 10:86–103. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Soti C, Nagy E, Giricz Z, Vigh L, Csermely
P and Ferdinandy P: Heat shock proteins as emerging therapeutic
targets. Br J Pharmacol. 146:769–780. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schafer C, Seeliger H, Bader DC, et al:
Heat shock protein 27 as a prognostic and predictive biomarker in
pancreatic ductal adenocarcinoma. J Cell Mol Med. View Article : Google Scholar : 2011.PubMed/NCBI
|
|
23
|
Berrieman HK, Cawkwell L, O'Kane SL, Smith
L and Lind MJ: Hsp27 may allow prediction of the response to
single-agent vinorelbine chemotherapy in non-small cell lung
cancer. Oncol Rep. 15:283–286. 2006.PubMed/NCBI
|
|
24
|
Yang YX, Xiao ZQ, Chen ZC, et al: Proteome
analysis of multidrug resistance in vincristine-resistant human
gastric cancer cell line SGC7901/VCR. Proteomics. 6:2009–2021.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liao WC, Wu MS, Wang HP, Tien YW and Lin
JT: Serum heat shock protein 27 is increased in chronic
pancreatitis and pancreatic carcinoma. Pancreas. 38:422–426. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leonardi R, Pannone G, Magro G, Kudo Y,
Takata T and Lo Muzio L: Differential expression of heat shock
protein 27 in normal oral mucosa, oral epithelial dysplasia and
squamous cell carcinoma. Oncol Rep. 9:261–266. 2002.PubMed/NCBI
|
|
27
|
Vargas-Roig LM, Gago FE, Tello O, Aznar JC
and Ciocca DR: Heat shock protein expression and drug resistance in
breast cancer patients treated with induction chemotherapy. Int J
Cancer. 79:468–475. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mori-Iwamoto S, Kuramitsu Y, Ryozawa S, et
al: Proteomics finding heat shock protein 27 as a biomarker for
resistance of pancreatic cancer cells to gemcitabine. Int J Oncol.
31:1345–1350. 2007.PubMed/NCBI
|
|
29
|
Mori-Iwamoto S, Kuramitsu Y, Ryozawa S, et
al: A proteomic profiling of gemcitabine resistance in pancreatic
cancer cell lines. Mol Med Report. 1:429–434. 2008.PubMed/NCBI
|
|
30
|
Heinrich JC, Tuukkanen A, Schroeder M,
Fahrig T and Fahrig R: RP101 (brivudine) binds to heat shock
protein HSP27 (HSPB1) and enhances survival in animals and
pancreatic cancer patients. J Cancer Res Clin Oncol. 137:1349–1361.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arrigo AP: Hsp27: novel regulator of
intracellular redox state. IUBMB Life. 52:303–307. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nakashima M, Adachi S, Yasuda I, et al:
Phosphorylation status of heat shock protein 27 plays a key role in
gemcitabine-induced apoptosis of pancreatic cancer cells. Cancer
Lett. 313:218–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Taba K, Kuramitsu Y, Ryozawa S, et al:
Heat-shock protein 27 is phosphorylated in gemcitabine-resistant
pancreatic cancer cells. Anticancer Res. 30:2539–2543.
2010.PubMed/NCBI
|
|
34
|
Baylot V, Andrieu C, Katsogiannou M, et
al: OGX-427 inhibits tumor progression and enhances gemcitabine
chemotherapy in pancreatic cancer. Cell Death Dis. 2:e2212011.
View Article : Google Scholar : PubMed/NCBI
|