Introduction
Troponin I (TnI) is a subunit of the troponin
complex that regulates the calcium-dependent activation of
myofilaments in muscle. Cardiac TnI (cTnI) exhibits a unique
N-terminal extension of approximately 30 amino acids. cTnI is not
present in the fast skeletal (fsTnI) or slow skeletal (ssTnI)
isoforms. The N-terminal extension of cTnI contains two protein
kinase A (PKA)-targeted phosphorylation sites [serine (Ser)-23 and
Ser-24] (1,2). Phosphorylation of the cTnI N-terminal
extension enhances contraction and accelerates relaxation during
β-adrenergic (β-A) stimulation by decreased myofibril
Ca2+ sensitivity and increased cross-bridge kinetics
(3). As such, cTnI is a key
regulatory protein in cardiac performance. Hearts of transgenic
mice with ssTnI replacing cTnI show reduced relaxation, blunted
response to β-A stimulation (4)
and protection against ischemia/reperfusion injury (5), while hearts of transgenic mice with
cTnI N-terminal 1–28 amino acid residue deletion show a higher
baseline stroke volume and a relaxation rate, similar to wild-type
mouse hearts under β-A stimulation (6). Furthermore, hearts of mice expressing
cTnI-S23D/S24D (pseudo-phosphorylation at PKA sites) exhibit
constitutive enhancement of rate-dependent increases in systolic
and diastolic function in vivo(7). As well as cTnI phosphorylation, the
allosteric conformation of cTnI N-terminal extension may be
involved in the modulation of cardiac contractile function
(8). Transgenic mice with
postnatal cardiomyocyte-specific overexpression of a truncated cTnI
lacking the acidic N′ region (cTnI-ND2–11) exhibit significantly
reduced rates of cardiac contraction and relaxation under baseline
and β-agonist treatment conditions (9). Overall, these data suggest an
important role of N-terminal cTnI phosphorylation in the regulation
of cardiac function, particularly on the responsiveness to β-A
stimulation.
The tail-suspended rat is a model used to simulate a
cephalic blood shift in microgravity on the ground. The hearts of
rats that have been tail-suspended for 4 weeks exhibit increased
cTnI degradation, with cTnI cleaved at the 26th, 27th and 30th
amino acid residues. Cardiac contractility is also decreased
(10). Reduced response to
isoproterenol (ISO) in the shortening amplitude and relaxation rate
of cardiomyocytes has been demonstrated in 4-week tail-suspended
rats (11) and is suggested to be
modulated by reduced cAMP (12).
However, components of cardiac function regulation on the β-A
receptor (β-AR) signal transduction pathway are unclear in the
tail-suspended rat heart, particularly under β-A stimulation.
Therefore, the aim of the present study was to test
our hypothesis that cTnI N-terminal degradation (cTnI-ND) in 4-week
tail-suspended rats is a major component in the reduction of
cardiac function responsiveness to β-A stimulation in the β-AR
signal transduction pathway.
Materials and methods
Animal model
Two-month-old healthy male Sprague-Dawley rats
weighing 220±10 g were used. Rats were randomly divided into
control (CON) and tail-suspended (SUS) groups. There were 36 rats
in each group. All the rats were housed in a 20±2°C environment
with a 12:12 h light-dark cycle and were fed rat chow and water
ad libitum. Tail suspension was performed using the
Morey-Holton method for 4 weeks (13). Care was taken to protect the tail
tissue and the movement of the rats was not restricted during the
procedure. All animal procedures were approved by the Animal Care
and Use Committee at the Fourth Military Medical University.
Preparation of isolated working
heart
Rats were injected with heparin (100 IU/100 g BW,
i.p.) and anesthetized with pentobarbital sodium (40 mg/kg, i.p.).
The heart was removed and the aorta was cannulated rapidly. The
cannulated hearts were mounted on a heart perfusion apparatus
(Radnoti Glass Technology Inc., Monrovia, CA, USA) and perfused
with an oxygenated (95% O2-5% CO2)
Krebs-Henseleit solution containing (in mM; pH 7.4) 118 NaCl, 4.7
KCl, 2.25 MgSO4, 2.25 CaCl2, 23.8
NaCO3, 1.2 NaH2PO4, 0.32 EDTA and
11.5 D-glucose. Following the establishment of coronary perfusion
in the Langendorff mode, the left atrium was cannulated through the
pulmonary vein with a steel cannula (inner diameter, 1.8 mm; outer
diameter, 2.0 mm). To detect intraventricular pressure, an
ultra-miniature pressure catheter transducer (model SPR-671; Millar
Instruments, Houston, TX, USA) was placed into the left ventricle
through the left atrium. The preload was set at 10 mmHg. The heart
was then switched from the Langendorff to the working mode at an
afterload of 60 mmHg. After the working hearts were equilibrated
for 30 min, they were treated with 1, 10 or 20 nM ISO
(Sigma-Aldrich, St. Louis, MO, USA) for at least 5 min. Before and
after ISO treatment, the aortic flow, coronary flow, heart rate,
left ventricular end-systolic (LVESP) and end-diastolic (LVEDP)
pressure and maximal rate of left ventricular pressure development
(+dP/dtmax) and relaxation (-dP/dtmax) were
measured to evaluate cardiac function of the isolated working
hearts. Cardiac output is equal to aortic flow plus coronary flow.
Data were obtained and analyzed using a PowerLab system and Chart
software (ADInstruments Inc., Sydney, Australia).
Cardiomyocyte isolation and unloaded
contractile function measurement in the single cardiomyocyte
Single ventricular myocyte isolation was performed
as previously described (14). In
brief, the cannulated hearts were mounted on a Langendorff
perfusion apparatus and perfused with Ca2+-free Joklik’s
modified minimum essential medium (Sigma-Aldrich) containing 10 mM
HEPES and 0.1% bovine serum albumin (BSA). After 5 min, the
perfusate was switched to a circulating enzyme solution containing
0.08% collagenase I (Sigma-Aldrich) for 30 min. Perfusion
procedures were performed at 37°C in a constant flow of 10 ml/min
and the perfusion pressure was monitored. The ventricular tissues
were chopped and the cardiomyocytes were dispersed gently by a
wide-tipped pipette. The cell suspension was filtered through a
200-μm nylon mesh. Cells were resuspended in Joklik’s medium
containing 1% BSA after 30 min and the Ca2+
concentration gradually recovered to 1.25 mM.
Measurements of contractile function were performed
in the Edge-Detector system (Crescent Electronics, Sandy, UT, USA)
within 6 h following isolation. Cells were placed into a chamber
situated on the stage of an inverted microscope (Olympus IX71;
Olympus Co., Ltd., Tokyo, Japan). The cells were perfused with
Tyrode’s solution containing (in mM; pH 7.4) 132 NaCl, 4.8 KCl, 1.2
MgCl2, 1.8 CaCl2, 5.0 sodium pyruvate, 10
HEPES and 10 D-glucose, at a flow rate of 0.2 ml/min at 37°C.
Electric field stimulus (15 V, 5 msec, 2.0 Hz) was administered by
the stimulator. The cardiomyocytes were superfused with ISO (1, 5
and 10 nM), forskolin (0.1, 0.5 and 1.0 μM),
N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate sodium
(DB-cAMP; 0.1, 0.5 and 1.0 mM) or 3-isobutyl-1-methylxanthine
(IBMX; 50, 100 and 200 mM). Forskolin is an activator of adenylate
cyclase. DB-cAMP mimics endogenous cAMP by binding to the PKA
regulatory subunit and activates PKA. IBMX is a non-selective
phosphodiesterase (PDE) inhibitor and cAMP is degraded by PDEs.
Forskolin, DB-cAMP and IBMX were purchased from Sigma-Aldrich.
The shortening amplitude and the maximal rates of
shortening (+dL/dtmax) and relaxation
(-dL/dtmax) were measured by Chart software in the
single cardiomyocyte.
Skinned cardiac muscle preparations
Cardiac muscle bundles (~0.2 mm in diameter and
3.6±0.4 mm in length) were dissected from the papillary muscle of
the left ventricle under a dissection microscope. Bundles were
mounted on a fiber apparatus (Aurora Scientific Inc., Aurora, ON,
Canada) for measurement of the isometric force and the sarcomere
length (High-speed Video Sarcomere Length Program, Aurora
Scientific Inc.). Following equilibration for 30 min, resting force
was adjusted to 100 mg, sarcomere length was not different at this
initial load in any of the muscle bundles (CON, 2.01±0.04 vs. SUS,
2.03±0.07 μm). Bundles were then chemically skinned with 1% Triton
X-100 for 60 min in a relaxing solution (pCa 9) containing
(in mM; pH 7.0) 130 potassium acetate, 1 MgCl2, 5 EGTA,
5 Na2ATP, 1X protease inhibitor cocktail (Roche
Diagnostics GmbH, Mannheim, Germany) and 20 imidazole-HCl. A rigor
solution (relaxing solution without ATP) was used to examine
whether the muscle was completely skinned. Phosphorylation was
performed by incubating the muscle bundles with 200 U/ml catalytic
subunit of PKA (Sigma-Aldrich) in relaxing solution for 30 min. The
force-pCa relationship for the cardiac muscle was measured
by adding CaCl2 to the relaxing solution to achieve a
series of pCa (7.0, 6.5, 6.3, 6.0, 5.5, 5.3, 5.0 and 4.5)
(15). All experiments were
performed at ~22°C.
Western blot analysis
PKA, TnI and phospholamban (PLB; total and
Ser16-phosphorylated) proteins were detected by western blot
analysis. Left ventricular myocardium was homogenized in a buffer
containing 50 mM potassium phosphate buffer (pH 7.0), 0.5 mM DTT, 1
mM EDTA, 0.3 mM PMSF and phosphatase inhibitor cocktail (1:100;
Sigma-Aldrich). Samples were subjected to SDS-PAGE in
polyacrylamide gels (12 or 14% depending on protein molecular
weight). Following electrophoresis, proteins were electrically
transferred to nitrocellulose membrane (0.45 μm pore size) using a
Bio-Rad semi-dry transfer apparatus (Bio-Rad, Hercules, CA, USA).
Blotted nitrocellulose membranes were blocked with 1% BSA in
Tris-buffered saline (TBS; 150 mM NaCl, 50 mM Tris-HCl; pH 7.5) and
incubated with rabbit polyclonal anti-PKA C-α [1:10,000; Cell
Signaling Technology (CST) Inc., Danvers, MA, USA], mouse
monoclonal anti-TnI-I (1:4,000) (10), rabbit polyclonal
anti-phosphorylated-Ser23/24 cTnI (1:1,000; CST), rabbit polyclonal
anti-desmin (1:1,000; CST), mouse monoclonal anti-PLB (1:1,000;
CST) and rabbit polyclonal anti-phosphorylated-Ser16 PLB
(Phospho-PLB, 1:1,000; CST) in TBS containing 0.1% BSA at 4°C
overnight. Membranes were incubated in IRDye 680 CW goat anti-mouse
or with IRDye 800 CW goat anti-rabbit secondary antibodies
(1:10,000) for 90 min at room temperature (RT) and visualized using
an Odyssey scanner (LI-COR Biosciences, Lincoln, NE, USA).
Quantification analysis of blots was performed with the NIH Image J
software (available at http://rsbweb.nih.gov/ij/download.html).
Immunofluorescent histochemistry and
confocal analysis
Cardiomyocytes were incubated in a 5% CO2
incubator at 37°C until cells attached to the dish coated with 10
μg/ml laminin (Sigma-Aldrich). The cells were exposed to 10 nM ISO
for 60 min and then fixed in 4% paraformaldehyde for 30 min. The
cells were permeabilized in 0.1% Triton X-100/PBS for 30 min,
blocked with 1% BSA in PBS for 60 min at RT and then incubated with
rabbit polyclonal anti-PKA C-α antibody (1:100) at 4°C overnight.
The slides were rinsed twice in PBS and incubated with
tetramethylrhodamine (TRITC)-labeled goat anti-rabbit IgG (1:400;
Molecular Probes, Eugene, OR, USA) for 60 min. The slides were then
washed in PBS, incubated in Hoechst 33258 (5 μg/ml) for 30 min and
washed twice with PBS. Staining was observed using a laser-scanning
confocal microscope (Olympus FV1000; Olympus Co., Ltd.) equipped
with the FV10-ASW system. TRITC- and Hoechst-labeled signals were
visualized at 555 and 352 nm, respectively. Images were captured at
×60 water objective. Image optical densitometry analysis was
performed using Olympus Fluoview image analysis software (Olympus
Co., Ltd.).
Statistical analysis
Data were presented as the mean ± SEM. Differences
between the two groups were compared by the unpaired Student’s
t-test. For multi-group comparisons, two-way ANOVA followed by
Tukey post-hoc test was performed. P<0.05 was considered to
indicate a statistically significant difference.
Results
Reduced responsiveness of isolated
working heart to ISO in tail-suspended rats
Body weights of tail-suspended rats were similar to
those of age-matched controls (CON, 304.7±11.9 g vs. SUS,
287.5±11.9 g; P>0.05). The wet weight of the hearts in the
tail-suspended rats was also not found to be significantly
different from those in the control rats (CON, 930.2±37.5 mg vs.
SUS, 931.9±24.3 mg; P>0.05).
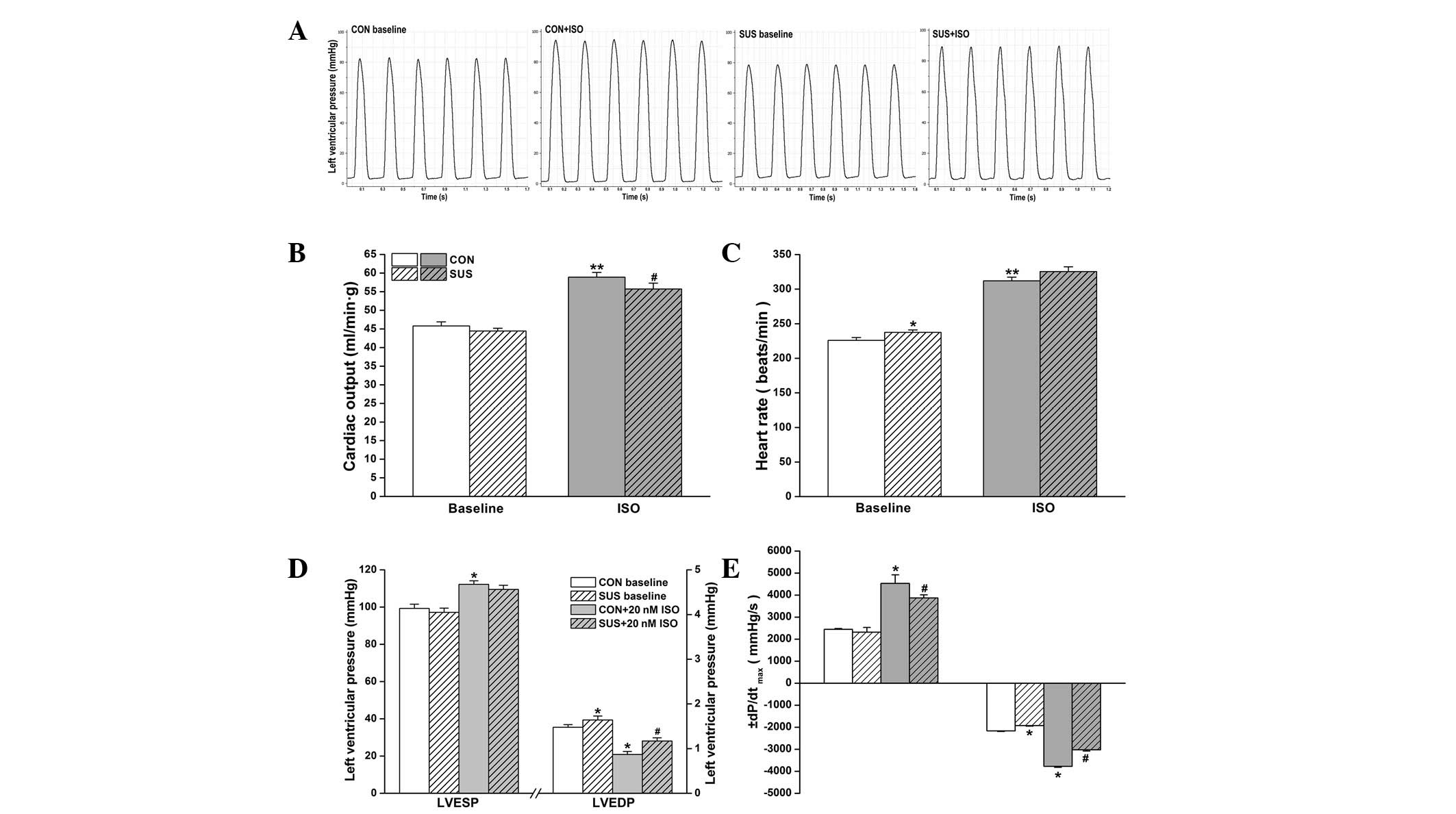
Cardiac function in working heart mode was assessed
at a preload of 10 mmHg and an afterload of 60 mmHg. Basal cardiac
output values were similar between control and tail-suspended rats
(P>0.05). ISO stimulation increased cardiac output in the
control and tail-suspended groups, but there was a higher increase
in cardiac output following 20 nM ISO treatment in the control
compared with the tail-suspended group (Fig. 1B). The intrinsic the heart rate was
225.9±4.1 beats/min in the control group and 237.5±3.5 beats/min in
the tail-suspended group. During 20 nM ISO perfusion, heart rate
increased to 312±5.4 beats/min in the control and to 325.5±6.7
beats/min in the tail-suspended group. The basal heart rate was
found to be significantly higher in the tail-suspended group
compared with the control group (Fig.
1C).
ISO treatment induced a significant increase in
LVESP of the control (P<0.05) and the tail-suspended group
(P<0.05), while no difference in LVESP between the
tail-suspended and control groups with or without ISO treatment was
observed (Fig. 1D). Basal LVEDP
was higher in the tail-suspended group than the control group
(P<0.05). A significant decrease in LVEDP following perfusion
with 20 nM ISO was found in the groups, which was more marked in
the control than in the tail-suspended group (P<0.05, Fig. 1D).
Under baseline conditions, no change in
+dP/dtmax was observed in the tail-suspended group,
while -dP/dtmax was found to be significantly reduced
when compared with the control group (P<0.05; Fig. 1E). Following 20 nM ISO treatment, a
significant increase in +dP/dtmax and
-dP/dtmax was found in the control (P<0.05) and
tail-suspended groups (P<0.05) compared with the values prior to
ISO administration. Increase in ±dP/dtmax in response to
ISO was greater in the control group than the tail-suspended group
(P<0.05, Fig. 1E).
Depressed responsiveness of
cardiomyocytes to β-AR signaling pathway agonists in tail-suspended
rats
To determine the major mediator(s) regulating the
ISO responsiveness in tail-suspended rats, we examined the effects
of the upstream agonists of the β-AR signaling pathway in
cardiomyocytes at a stimulation frequency of 2.0 Hz. The baseline
value in shortening amplitude of cardiomyocytes was lower in the
tail-suspended group than in the control group (Fig. 2). Exposure to ISO, forskolin,
DB-cAMP and IBMX produced dose-dependent positive inotropic
responses in control and tail-suspended rat cardiomyocytes. These
responses were significantly lower in the tail-suspended group than
in the control group (Fig. 2).
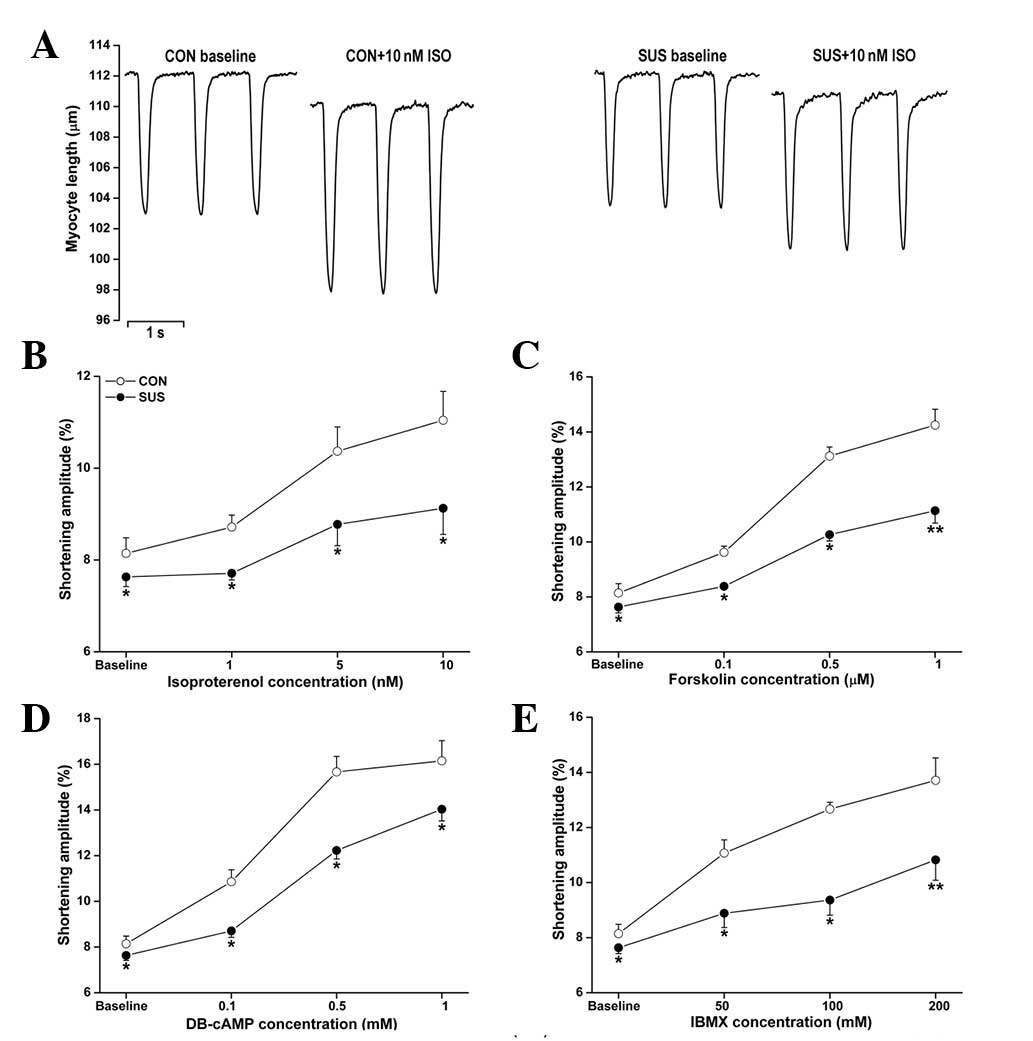 | Figure 2Unloaded contractile functions of
cardiomyocytes with ISO, forskolin, DB-cAMP and IBMX treatments.
Cardiomyocytes were stimulated at 2.0 Hz. The shortening amplitude
of cardiomyocytes was measured in each group. (A) Representative
recordings of unloaded contraction of single cardiomyocytes. (B)
ISO perfusion at 1, 5 and 10 nM. (C) Forskolin perfusion at 0.1,
0.5 and 1.0 μM. (D) DB-cAMP perfusion at 0.1, 0.5 and 1.0 mM. (E)
IBMX perfusion at 50, 100 and 200 mM. Data are the mean ± SEM; n=18
myocytes from six hearts for each agonist in each group.
*P<0.05 or **P<0.01 vs. the control
group. CON, control rats; SUS, tail-suspended rats; ISO,
isoproterenol; DB-cAMP, N6,2′-O-dibutyryladenosine
3′,5′-cyclic monophosphate sodium; IBMX,
3-isobutyl-1-methylxanthine. |
The increase in cardiomyocyte shortening amplitude
following ISO stimulation (1, 5 and 10 nM) was less marked in
tail-suspended rats compared with control rats (Fig. 2B). Forskolin (0.1, 0.5 and 1.0 μM),
an activator of adenylate cyclase, induced a smaller increase in
shortening amplitude in the tail-suspended group (Fig. 2C). DB-cAMP is a cell
membrane-permeable and PDE-resistant cAMP analog. Exposure to 0.1,
0.5 and 1.0 mM DB-cAMP induced a smaller increase in cardiomyocyte
shortening amplitude in tail-suspended rats compared with the
control (Fig. 2D). IBMX (50, 100
and 200 mM), a non-selective inhibitor of PDEs, also induced a
smaller increase in shortening amplitude in the tail-suspended
group (Fig. 2E).
Baseline values of +dL/dtmax of
cardiomyocytes were similar between the control and tail-suspended
groups, while the baseline values of -dL/dtmax decreased
significantly in the tail-suspended compared with the control group
(P<0.05; Fig. 3A). ISO,
forskolin, DB-cAMP and IBMX all enhanced the +dL/dtmax
and -dL/dtmax of cardiomyocytes in the control and
tail-suspended groups (P<0.05; Fig.
3). The increased +dL/dtmax and -dL/dtmax
in response to ISO, forskolin, DB-cAMP or IBMX treatments were
significantly less in the tail-suspended group than the control
(P<0.05; Fig. 3).
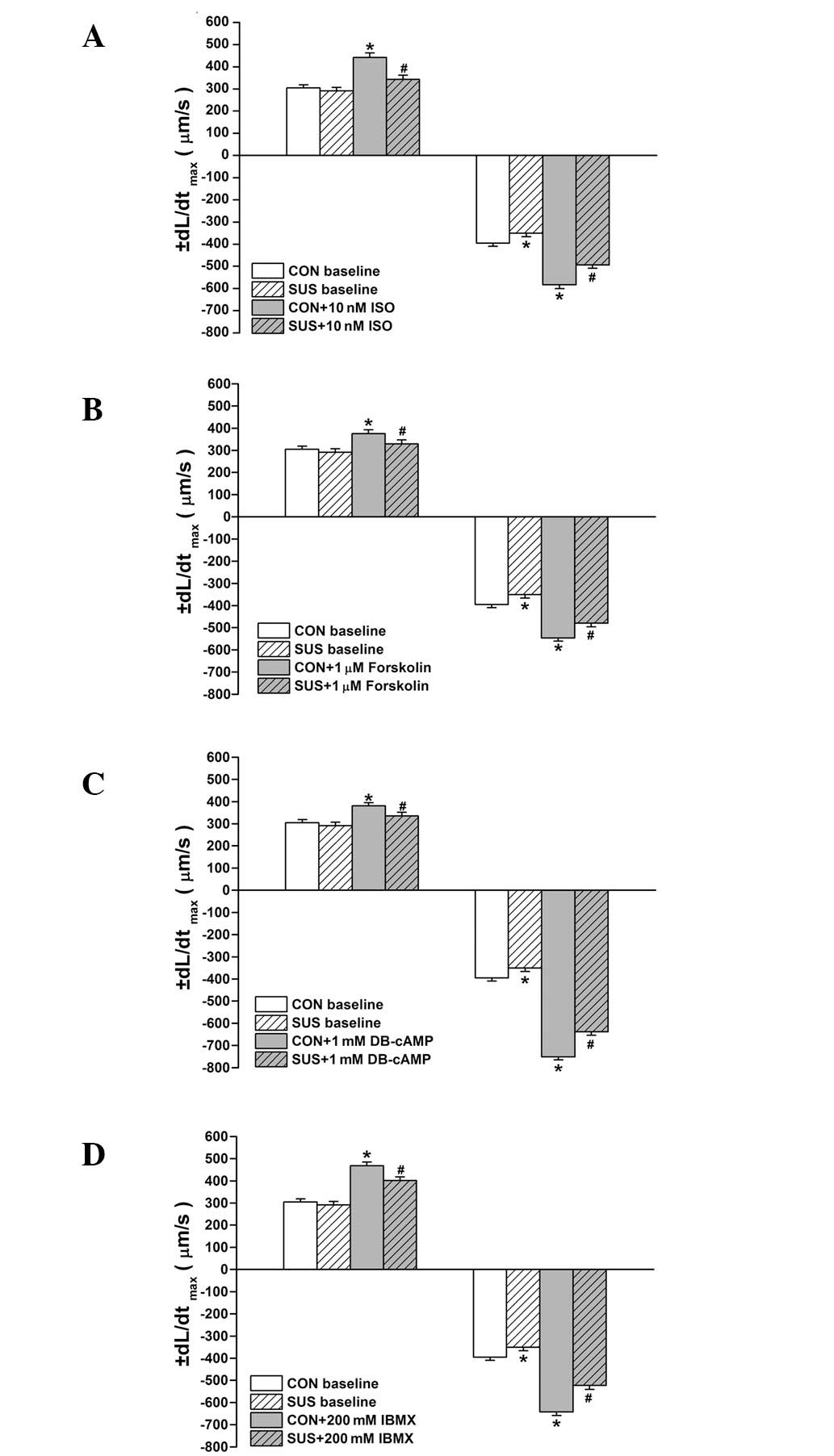 | Figure 3Effects of ISO, forskolin, DB-cAMP and
IBMX on maximal rates of shortening (+dL/dtmax) and
relaxation (-dL/dtmax) in cardiomyocytes. Treatment of
(A) 10 nM ISO, (B) 1.0 μM forskolin, (C) 1 mM DB-cAMP, (D) 200 mM
IBMX is shown. Data are the mean ± SEM; n=18 myocytes from 6 hearts
for each agonist in each group. *P<0.05 vs. baseline
value of the control group. #P<0.05 vs. ISO-treated
control group. CON, control rats; SUS, tail-suspended rats; ISO,
isoproterenol; DB-cAMP, N6,2′-O-dibutyryladenosine
3′,5′-cyclic monophosphate sodium; IBMX,
3-isobutyl-1-methylxanthine. |
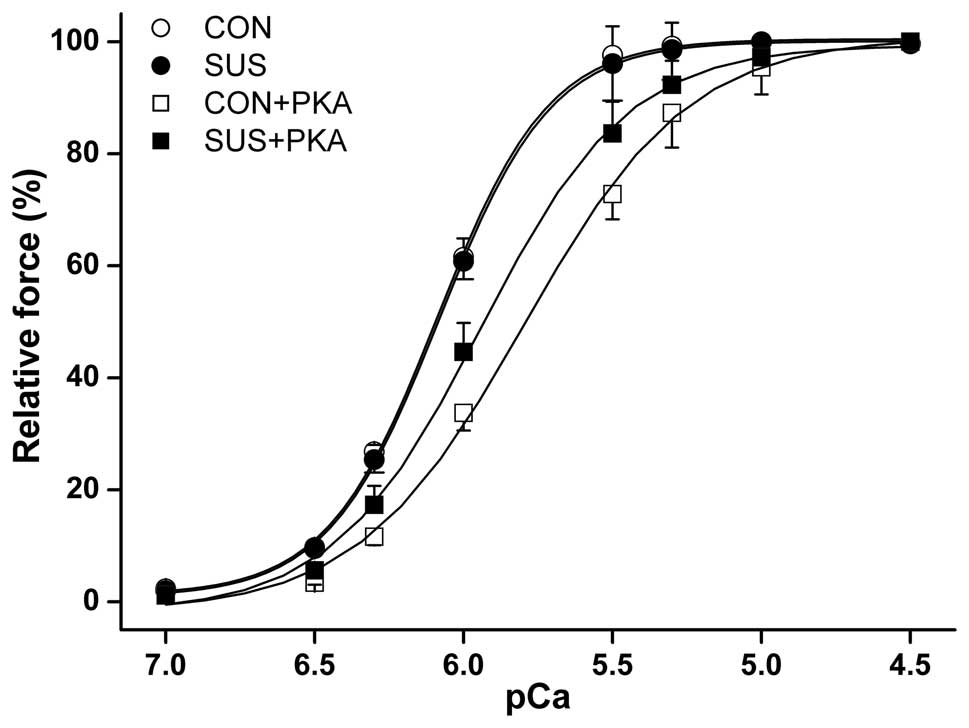
PKA reduced myofibrillar Ca2+
sensitivity in the myocardium
The maximum Ca2+-activated isometric
force of skinned muscle fibers from control rats was significantly
higher than that from tail-suspended rats (10). No difference was observed in the
myocardial isometric force-pCa relationship between the two
groups (Fig. 4). Although PKA
induced a reduction in myofibrillar Ca2+ sensitivity in
the control and tail-suspended groups, the reduction was less
marked in tail-suspended rats. The [Ca2+] required for
50% activation (pCa50) was 6.07±0.17 pCa units at
baseline and 5.93±0.09 following PKA treatment in tail-suspended
rat myofibrils, compared with 6.08±0.12 pCa units at
baseline and 5.79±0.11 following PKA treatment in the control rats
(P<0.05).
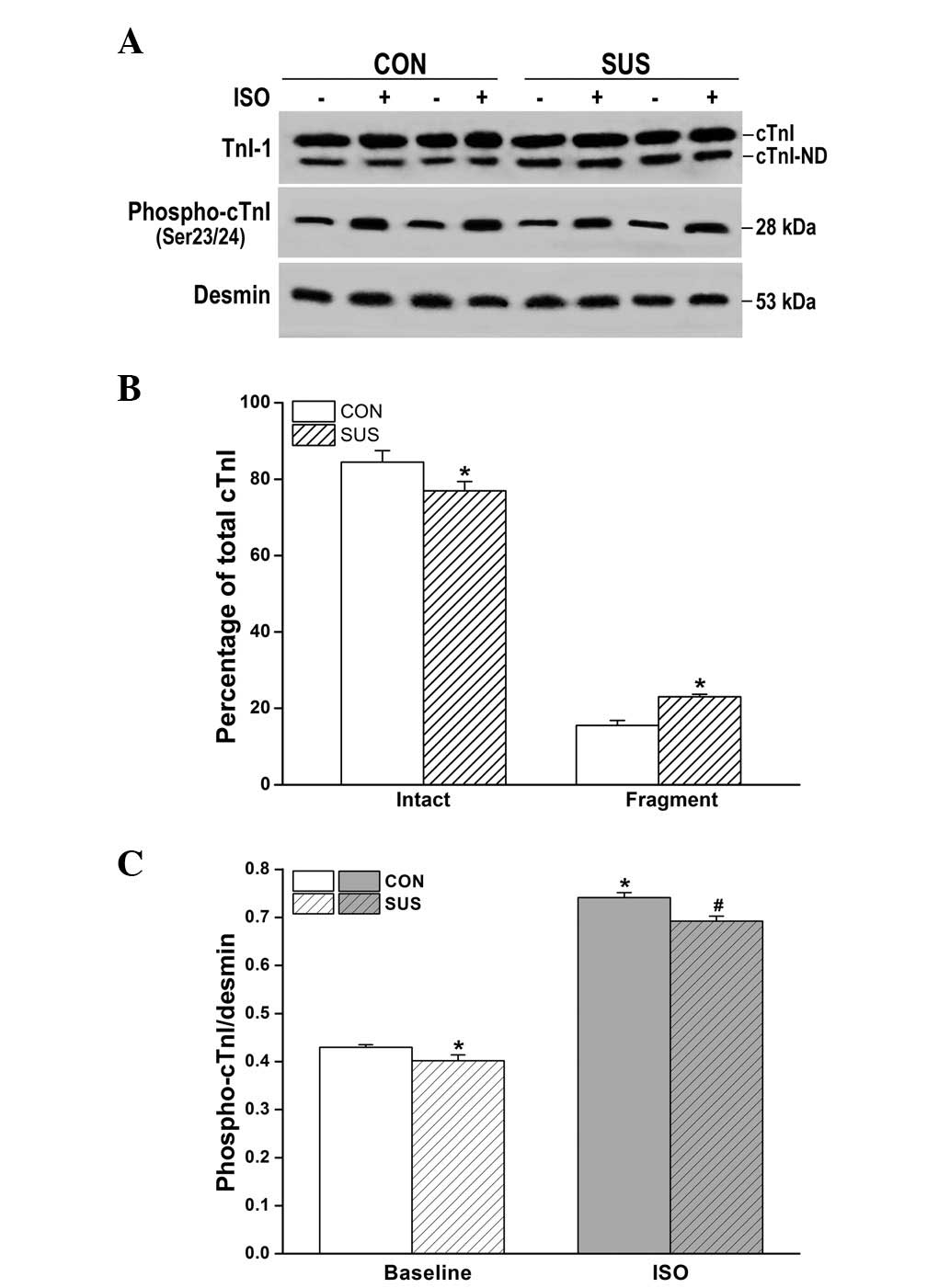
Increased N-terminal degradation of cTnI
and reduced cTnI phosphorylation by ISO in the tail-suspended
group
Western blot analysis revealed two bands of cTnI in
hearts from the control and tail-suspended rats (Fig. 5A). The percentage of intact cTnI
(upper band)/total cTnI (intact cTnI plus cTnI fragment) was
decreased in tail-suspended rat hearts compared with the control
(P<0.05). Optical densitometry of the cTnI fragment (lower band)
was 15.5±1.3% of total cTnI in the control and 23.0±0.7% in the
tail-suspended group (Fig. 5B),
with significantly greater cTnI degradation in the tail-suspended
group versus the control group (P<0.05). The basic
phosphorylation level of cTnI was significantly decreased in the
tail-suspended group compared with the control (P<0.05; Fig. 5A and C). Following ISO stimulation,
phosphorylated cTnI increased in tail-suspended and control groups,
however, the increase was smaller in the tail-suspended group than
that in the control (P<0.05; Fig.
5C).
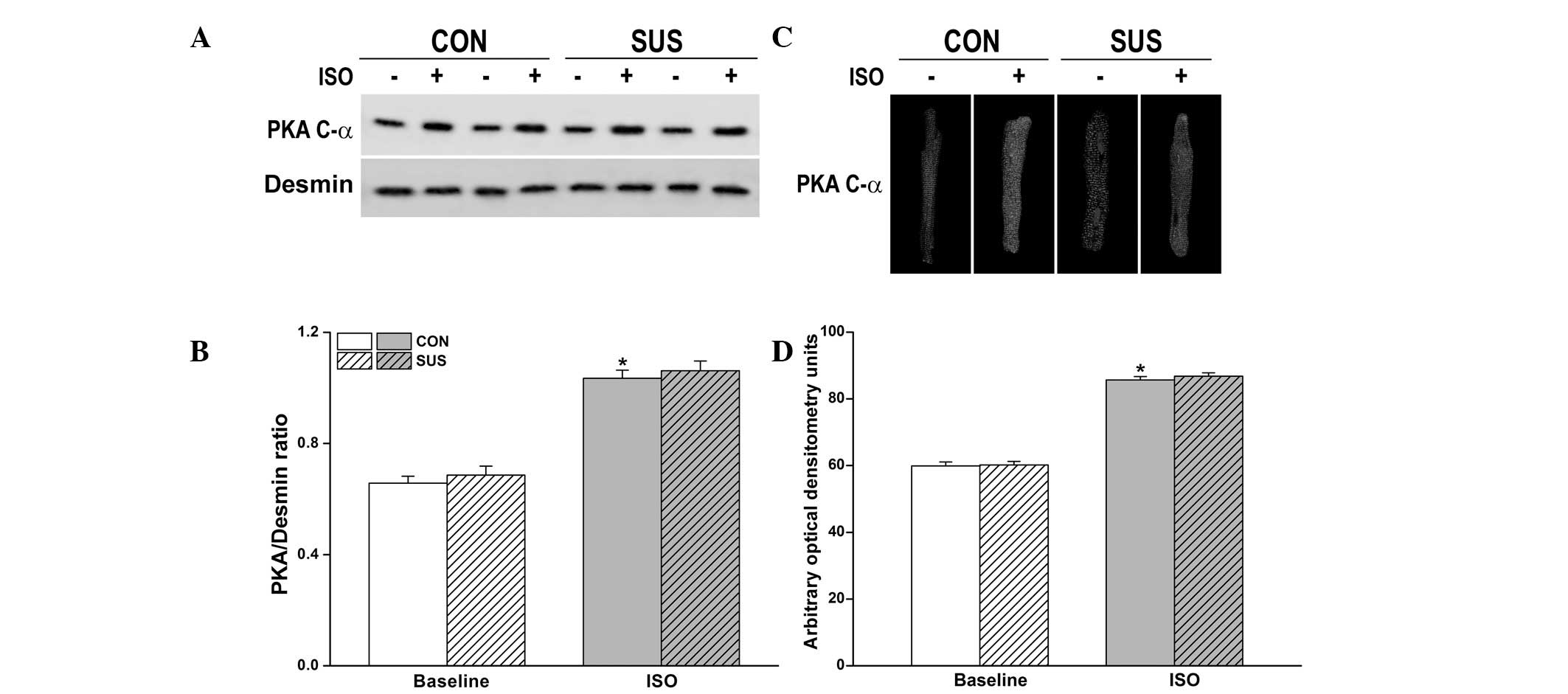
Expression and activation of PKA were
unaltered in the tail-suspended group
Inactive PKA is a heterotetramer composed of a
regulatory subunit (R) dimer and a catalytic subunit (C) dimer. In
its inactive state, pseudosubstrate sequences on the R subunits
block the active sites on the C subunits. When cAMP binds to the R
subunits, the auto-inhibitory contact is relieved and active
monomeric C subunits are released. Three C subunit isoforms (C-α,
-β and -γ) have been identified and C-α is predominantly expressed
in the heart. Thus, using PKA C-α subunit antibody we detected
activated PKA. Compared with desmin, no difference in the
expression of the activated PKA C-α subunit between the control and
tail-suspended groups before and after 10 nM ISO stimulation was
observed (Fig. 6A and B).
Immunofluorescence data revealed a similar degree of PKA
translocation into the sarcolemma of cardiomyocytes following 10 nM
ISO stimulation between the control and tail-suspended groups
(Fig. 6C and D).
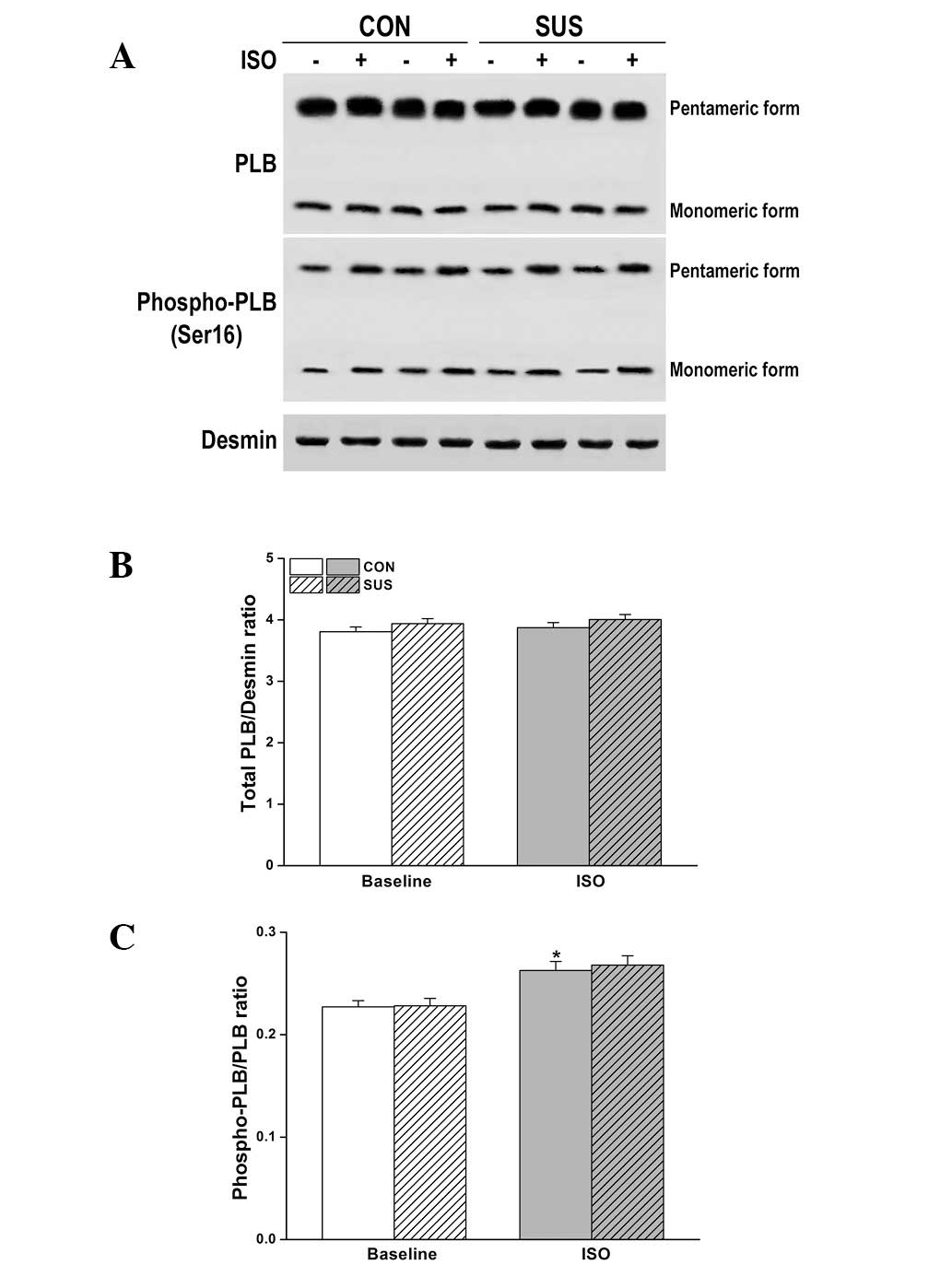
Expression and Ser16 phosphorylation of
PLB were unchanged in the tail-suspended group
The level of total PLB and/or phosphorylation at
Ser16 (the residue phosphorylated by PKA) are important factors
that may alter relaxation function in tail-suspended rat hearts.
However, there was no change in the expression or phosphorylation
of PLB at Ser16 by western blot analysis following ISO stimulation
(Fig. 7).
Discussion
In the present study, the baseline LVEDP of the
working heart was higher in the tail-suspended rats compared with
the control, while a blunted response to ISO in all parameters
measured in the working heart, isolated cardiomyocytes and skinned
fibers was identified in tail-suspended rats. Thus, the decrease in
ISO sensitivity with tail-suspension was associated with changes in
the β-AR signaling pathway at the level of the individual
cardiomyocyte.
Stimulation of β-AR by ISO activates Gs,
which in turn activates adenylate cyclase and increases the
formation of cAMP within cardiomyocytes. Elevated levels of cAMP
increase activation of PKA, which phosphorylates intracellular
targets, including L-type Ca2+ channels, ryanodine
receptor (RyR), PLB, cTnI and myosin-binding protein C (MyBPC).
Therefore, the upstream components in the β-AR signal transduction
pathway include β-AR, Gs, adenylate cyclase, cAMP and
PKA. The downstream components are the targeting proteins of
PKA.
Forskolin, a direct adenylate cyclase agonist,
caused a smaller increase in the shortening amplitude and the
maximal rate of relaxation in tail-suspended rat cardiomyocytes. By
contrast, a reduction in cAMP of cardiomyocytes has been reported
to depress the response to ISO in tail-suspended rats (12). However, in the present study,
response to DB-cAMP was less in the cardiomyocytes of
tail-suspended rats compared with the control. In addition, under
basal conditions and during β-AR stimulation, intracellular cAMP
levels are regulated by PDE, which catalyzes the breakdown of cAMP.
The PDE inhibitor IBMX did not reverse the tail-suspension-related
deficit in positive inotropy caused by β-AR stimulation. PKA
expression and membrane translocation of cardiomyocytes during β-AR
stimulation was similar in both the tail-suspended and control
rats. These findings suggest that events downstream of PKA in the
β-AR signal transduction pathway may be affected by the
tail-suspension.
Phosphorylation of L-type Ca2+ channels
enhances Ca2+ influx, increasing the shortening
amplitude or force of contraction, but does not regulate the
relaxation of cardiomyocytes. A previous study demonstrated that
adrenergic regulation of cardiac contractility does not involve
phosphorylation of the cardiac ryanodine receptor at Ser2808 by PKA
(16). Phosphorylation of MyBPC
does not appear to have any effect on myofibrillar Ca2+
sensitivity of the isometric force, which modulates the relaxation,
but accelerates kinetics of force development (17). Phosphorylated MyBPC is only
involved in myocardial protection during ischemia (18,19).
This indicates that phosphorylation of PLB and cTnI is important in
the regulation of cardiac function, particularly the relaxation
function of cardiomyocytes downstream of the β-AR signal
transduction pathway.
Phosphorylation of PLB ameliorates the inhibition of
SERCA and increases Ca2+ uptake by SERCA. Thus, the rate
of relaxation is enhanced during β-AR stimulation due to increased
sequestration of Ca2+ by increased SERCA activity. In
PLB knockout mice, phosphorylation of PLB has been shown to account
for approximately 50% of the enhanced relaxation rate effect
(20). However, there was no
difference in total PLB and PLB phosphorylation at the PKA-targeted
site Ser16 residue in cardiomyocytes between the tail-suspended and
control rats. cTnI, another substrate phosphorylated by PKA,
exhibited N-terminal degradation in cardiomyocytes of control and
tail-suspended rats, although the N-terminal degradation of cTnI
was more marked in tail-suspended rats. In turn, the increased
N-terminal degradation of cTnI reduced phosphorylation in
tail-suspended rats. Thus, these data strongly suggest that the
blunted cardiac function in β-AR stimulation is correlated with
enhanced cTnI N-terminal degradation in the tail-suspended
rats.
A number of mechanistic systems models have been
developed to analyze the functional roles of PLB, L-type calcium
channel, RyR and cTnI phosphorylation upon β-AR stimulation in rat
ventricular myocytes. The model analysis revealed that the
PKA-mediated phosphorylation of cTnI only exhibits a nominal
lusitropic response during β-AR stimulation (21). However, transgenic mice expressing
ssTnI specifically in cardiomyocytes exhibit less shortening and
prolongation of the half-time of intracellular [Ca2+]
decay, while similar transgenic cardiomyocytes show no enhancement
of the velocity of shortening during isoprenaline treatment
(4). By using PLB knockout
transgenic mice, cTnI phosphorylation has been shown to contribute
14–18% of the lusitropic effect during maximal isometric
contractions (20). Overall, these
data indicate that phosphorylation of cTnI at the N-terminus by PKA
is a key modulator of cardiac function. In addition, the allosteric
conformation of cTnI N-terminal extension also regulates cardiac
contractile function (8).
Degradation of N-terminal extension induces a change in the
allosteric conformation of cTnI. Sadayappan et al(9) have generated transgenic mice with a
truncated cTnI that lacks the acidic N’ region (cTnI-ND2-11). The
acidic N’ region is not involved in the phosphorylated sites of
cTnI, although cTnI-ND2-11 hearts exhibit significantly reduced
rates of contraction and relaxation under baseline and β-agonist
treatment (9).
cTnI has been previously shown to be cleaved at the
26th, 27th and 30th amino acid residues in tail-suspended rat
hearts (10). N-terminal
degradation of cTnI decreased the degree of total cTnI
phosphorylation by PKA in the hearts of tail-suspended rats.
Although cTnI-ND accounted for only approximately 20% of total cTnI
in tail-suspended rats in the present study, this cTnI-ND lacked
the PKA phosphorylation sites, Ser23 and Ser24, changing the
allosteric conformation of cTnI N-terminal extension. These
combined effects may enhance the modulation of the cardiac function
in tail-suspended rat hearts. Therefore, in the present study,
increased cTnI-ND reduced the contraction and relaxation function
of the left ventricle in working hearts and impaired cardiac
function of cardiomyocytes in tail-suspended rats at baseline and
during β-AR stimulation. Barbato et al reported that the
transgenic mouse hearts with the N-terminal truncation of cTnI
reduced LVEDP at the basic level, but the response of
±dP/dtmax to ISO was also lower in transgenic mice than
wild-type mice (6).
Furthermore, cTnI phosphorylation at Ser23 and Ser24
by PKA has been shown to depress the Ca2+ affinity and
Ca2+ off-rate of cTnC in vitro(22). A rapid dissociation of
Ca2+ from cTnC is another important factor that
accelerates the relaxation rate during β-agonist treatment. Removal
of the N-terminal domain of cTnI from 1–28 amino acid residues has
been shown to decrease the Ca2+ sensitivity of
actomyosin ATPase in the transgenic mouse myocardium (6). The lower Ca2+ sensitivity
of the force generated by muscle fibers under β-AR stimulation is
correlated with this more rapid dissociation of Ca2+
from cTnC. Therefore, increased dissociation of Ca2+
from cTnC, coupled with a more rapid uptake of Ca2+ by
the sarcoplasmic reticulum stimulated by PKA phosphorylation of
PLB, may account for the more rapid relaxation observed during the
inotropic response of the heart to ISO. In the present study, we
observed no difference in the Ca2+ sensitivity of
myofibrils between the control and tail-suspended rat hearts, but
the Ca2+ sensitivity of myofibrils exhibited less
reduction following PKA treatment in tail-suspended hearts.
Therefore, the working hearts and cardiomyocytes demonstrated a
slow relaxation and blunted responsiveness to ISO in 4-week
tail-suspended rats.
In summary, N-terminal degradation of cTnI in
tail-suspended rat hearts is a major component to reduce cardiac
function responsiveness to ISO in the β-AR signal transduction
pathway.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 31071044).
References
|
1
|
Layland J, Solaro RJ and Shah AM:
Regulation of cardiac contractile function by troponin I
phosphorylation. Cardiovasc Res. 66:12–21. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marston SB and Redwood CS: Modulation of
thin filament activation by breakdown or isoform switching of thin
filament proteins: physiological and pathological implications.
Circ Res. 93:1170–1178. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Solaro RJ, Rosevear P and Kobayashi T: The
unique functions of cardiac troponin I in the control of cardiac
muscle contraction and relaxation. Biochem Biophys Res Commun.
369:82–87. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fentzke RC, Buck SH, Patel JR, Lin H,
Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL and Leiden
JM: Impaired cardiomyocyte relaxation and diastolic function in
transgenic mice expressing slow skeletal troponin I in the heart. J
Physiol. 517:143–157. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arteaga GM, Warren CM, Milutinovic S,
Martin AF and Solaro RJ: Specific enhancement of sarcomeric
response to Ca2+ protects murine myocardium against
ischemia-reperfusion dysfunction. Am J Physiol Heart Circ Physiol.
289:H2183–H2192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barbato JC, Huang QQ, Hossain MM, Bond M
and Jin JP: Proteolytic N-terminal truncation of cardiac troponin I
enhances ventricular diastolic function. J Biol Chem.
280:6602–6609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takimoto E, Soergel DG, Janssen PM, Stull
LB, Kass DA and Murphy AM: Frequency- and afterload-dependent
cardiac modulation in vivo by troponin I with constitutively active
protein kinase A phosphorylation sites. Circ Res. 94:496–504. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dong WJ, An J, Xing J and Cheung HC:
Structural transition of the inhibitory region of troponin I within
the regulated cardiac thin filament. Arch Biochem Biophys.
456:135–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sadayappan S, Finley N, Howarth JW,
Osinska H, Klevitsky R, Lorenz JN, Rosevear PR and Robbins J: Role
of the acidic N’ region of cardiac troponin I in regulating
myocardial function. FASEB J. 22:1246–1257. 2008.
|
|
10
|
Yu ZB, Zhang LF and Jin JP: A proteolytic
NH2-terminal truncation of cardiac troponin I that is
up-regulated in simulated microgravity. J Biol Chem.
276:15753–15760. 2001.PubMed/NCBI
|
|
11
|
Zhang L, Wang YY and Yu ZB: Depressed
responsiveness of cardiomyocytes to isoproterenol in simulated
weightlessness rats. Sheng Li Xue Bao. 59:845–850. 2007.(In
Chinese).
|
|
12
|
Yin W, Liu JC, Fan R, Sun XQ, Ma J, Feng
N, Zhang QY, Yin Z, Zhang SM, Guo HT, Bi H, Wang YM, Sun X, Cheng
L, Cui Q, Yu SQ, Yi DH and Pei JM: Modulation of
{beta}-adrenoceptor signaling in the hearts of 4-wk simulated
weightlessness rats. J Appl Physiol. 105:569–574. 2008.
|
|
13
|
Morey-Holton ER and Globus RK: Hindlimb
unloading rodent model: technical aspects. J Appl Physiol.
92:1367–1377. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nagata K, Liao R, Eberli FR, Satoh N,
Chevalier B, Apstein CS and Suter TM: Early changes in
excitation-contraction coupling: transition from compensated
hypertrophy to failure in Dahl salt-sensitive rat myocytes.
Cardiovasc Res. 37:467–477. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fabiato A and Fabiato F: Effects of
magnesium on contractile activation of skinned cardiac cells. J
Physiol. 249:497–517. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
MacDonnell SM, García-Rivas G, Scherman
JA, Kubo H, Chen X, Valdivia H and Houser SR: Adrenergic regulation
of cardiac contractility does not involve phosphorylation of the
cardiac ryanodine receptor at serine 2808. Circ Res. 102:e65–e72.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stelzer JE, Patel JR, Walker JW and Moss
RL: Differential roles of cardiac myosin-binding protein C and
cardiac troponin I in the myofibrillar force responses to protein
kinase A phosphorylation. Circ Res. 101:503–511. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sadayappan S, Gulick J, Klevitsky R,
Lorenz JN, Sargent M, Molkentin JD and Robbins J: Cardiac myosin
binding protein-C phosphorylation in a {beta}-myosin heavy chain
background. Circulation. 119:1253–1262. 2009.PubMed/NCBI
|
|
19
|
Sadayappan S, Osinska H, Klevitsky R,
Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG and
Robbins J: Cardiac myosin binding protein C phosphorylation is
cardioprotective. Proc Natl Acad Sci USA. 103:16918–16923. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li L, Desantiago J, Chu G, Kranias EG and
Bers DM: Phosphorylation of phospholamban and troponin I in
beta-adrenergic-induced acceleration of cardiac relaxation. Am J
Physiol Heart Circ Physiol. 278:H769–H779. 2000.PubMed/NCBI
|
|
21
|
Saucerman JJ and McCulloch AD: Mechanistic
systems models of cell signaling networks: a case study of myocyte
adrenergic regulation. Prog Biophys Mol Biol. 85:261–278. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang R, Zhao J, Mandveno A and Potter JD:
Cardiac troponin I phosphorylation increases the rate of cardiac
muscle relaxation. Circ Res. 76:1028–1035. 1995. View Article : Google Scholar : PubMed/NCBI
|