Introduction
Bevacizumab (Avastin®; Genentech, San
Francisco, CA, USA), a recombinant humanized monoclonal antibody,
binds vascular endothelial growth factor (VEGF) and inhibits its
interaction with receptors found on endothelial cells (1,2).
Bevacizumab has been increasingly used as an off-label treatment
for exudative age-related macular degeneration (AMD) (2). Retinal angiomatous proliferation, a
variant of exudative neovascular AMD, has been described to occur
in 3 stages: i) intraretinal neovascularization, ii) subretinal
neovascularization with (stage IIB) or without (stage IIA) serous
pigment epithelial detachment (PED) and iii) a vascularized PED
with retinal-choroidal anastomosis (RCA) (3). The ocular complications of
bevacizumab intravitreal injection included corneal abrasion,
chemosis, lens injury, ocular inflammation, retinal pigment
epithelial tear and acute vision loss. The systemic complications
included cerebral infarction, elevation of systolic blood pressure,
facial skin redness, itchy diffuse rash and menstrual
irregularities (4). Another study
demonstrated that retinal pigment epithelial (RPE) tears occur
after intravitreal bevacizumab injections for exudative AMD in
approximately 1.6% of the eyes and may cause severe vision loss.
Intravitreal bevacizumab injections also cause RPE tears in
patients with predominantly classic choroidal neovascularization
(5). The serial intravitreal
injection of bevacizumab caused a large retinal pigment epithelium
rip resulting in a large fibrovascular pigment epithelial
detachment (6). It has been
hypothesized that intravitreal bevacizumab induces contraction of
choroidal neovascular membranes, thus accelerating RPE tear
development (2).
An in vitro study demonstrated that
bevacizumab at a concentration of 2.5 mg/ml caused a moderate
decrease in ARPE-19 cell numbers and cell viability after 2 days of
treatment (7). Bevacizumab caused
a dose-dependent suppression of DNA synthesis in pig choroidal
endothelial (CEC) cells, as a result of a moderate
antiproliferative activity (maximum reduction 36.8%). At higher
doses (2.5 mg/ml) bevacizumab may be harmful to the retinal pigment
epithelium (7). However, after 2
days at a bevacizumab concentration of 2.5 mg/ml, a moderate
decrease in ARPE-19 cell numbers and cell viability was
observed.
However, the mechanisms by which bevacizumab induced
the cell cycle arrest in ARPE-19 cells has yet to be determined.
The present study aimed to clarify the role of bevacizumab in the
cell cycle and the effects of DNA synthesis on human ARPE-19 cells,
as well as provide understanding of the molecular pathway of the
function of bevacizumab in cell cycle regulation.
Materials and methods
Evaluation of the bevacizumab-induced
cell cycle and DNA synthesis inhibition in human ARPE-19 cells
Cells were cultured in 60-mm tissue-culture dishes
(8×105 cells/dish). The culture medium was replaced by a
new medium after 24 h and subsequently exposed to 2.5 mg/ml
bevacizumab for 48 h. After treatment, adherent and floating cells
were pooled, washed with PBS, fixed in PBS-methanol (1:2
volume/volume) solution, and maintained at 4°C for at least 18 h.
Following 2 more washes with PBS, the cell pellets were stained
with the propidium iodide (PI) fluorescent probe solution
containing PBS, 40 μg/ml PI, and 40 μg/ml DNase-free RNase A for 30
min at room temperature in the dark. DNA fluorescence of PI-stained
cells was evaluated by excitation at 488 nm and monitored through a
630/22-nm band pass filter using flow cytometry. A minimum of
10,000 cells was analyzed per sample, and the DNA histograms were
gated and further analyzed using Modfit software on a Mac
workstation to estimate the percentages of cells in various phases
of the cell cycle.
Evaluation of the bevacizumab-induced
expression of cell cycle-regulated proteins in human ARPE-19
cells
ARPE-19 cells (1×106) were cultured in
60-mm tissue-culture dishes for 24 h. The culture medium was
replaced with a new medium and exposed to bevacizumab for the
indicated time points. After treatment, the cells were washed with
PBS, re-suspended in a protein extraction buffer for 10 min and
then centrifuged at 12,000 × g for 10 min at 40°C to obtain total
extracted proteins (supernatant). The protein concentrations were
measured usinga Bio-Rad protein assay reagent (Bio-Rad, Richmond,
CA, USA). The expression of cell cycle-regulated proteins was
evaluated by western blotting. Briefly, the total extracted
proteins were boiled in loading buffer, and an aliquot
corresponding to 50 μg of protein was separated by 12%
SDS-polyacrylamide gel. After electrophoresis, the proteins were
electrotransferred onto a polyvinylidene fluoride transfer
membrane. The membranes were then incubated with the primary
antibody of various cell cycle-regulated proteins overnight, and
washed with PBST solution (0.05% Tween-20 in PBS). Following
washing, the secondary antibody labeled with horseradish-peroxidase
was adjacently incubated for 1 h, and washed with PBST solution
(0.05% Tween-20 in PBS). The antigen-antibody complexes were
detected by enhanced chemiluminescence (Amersham Pharmacia Biotech,
Piscataway, NJ, USA) with a chemiluminescence analyzer.
Evaluation of the bevacizumab-induced
cell viability in human ARPE-19 cells
ARPE-19 cells (8×105) were cultured in
60-mm tissue-culture dishes for 24 h. The culture medium was
replaced with a new medium and then exposed to 0, 2.5 and 5.0 mg/ml
of bevacizumab for 24 h. After treatment, the ARPE-19 cells were
incubated for 2 h with 0.5 mg/ml of MTT reagent and lysed with
DMSO. The absorbance was measured at 570 nm in a microplate
reader.
Results
Bevacizumab induced G1/S phase
arrest in ARPE-19 cells
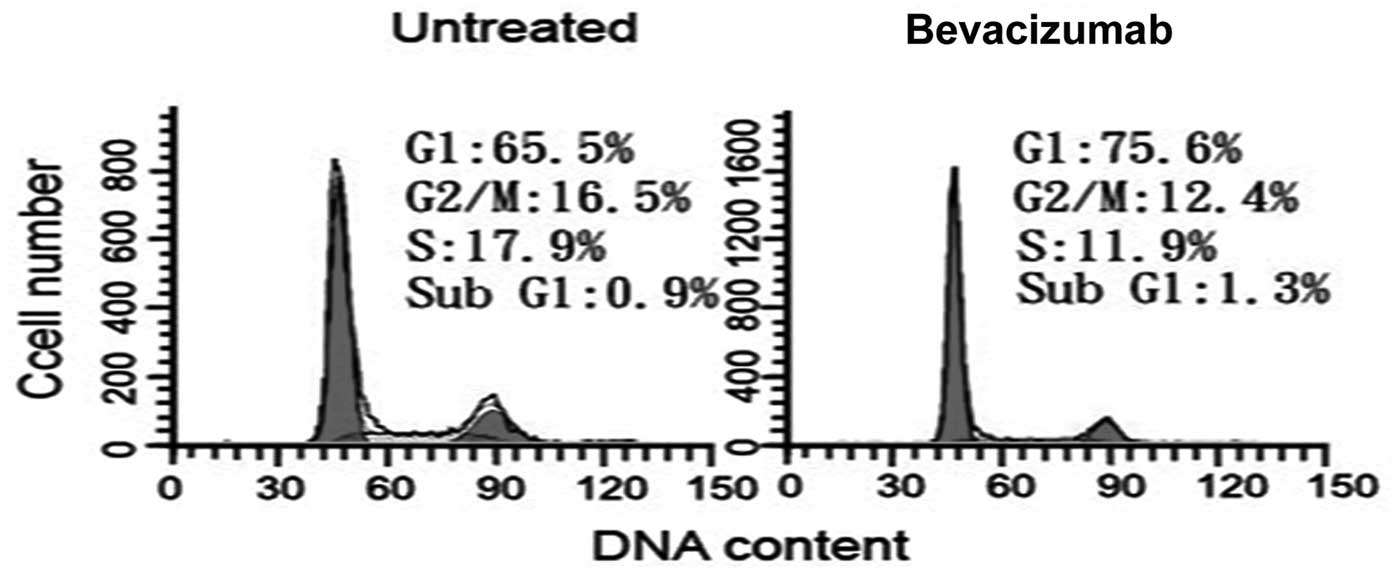
We initially used PI staining and flow cytometry to
evaluate whether bevacizumab is capable of disrupting the cell
cycle progression. The G1 and S phases were 65.5 and
17.9% in untreated cells, respectively (Fig. 1). Treatment with 2.5 mg/ml
bevacizumab resulted in a marked decrease in the S phase (11.9%) at
48 h, suggesting that bevacizumab inhibits DNA synthesis in ARPE-19
cells. The G1 phase increased slightly to 75.6%
following treatment with bevacizumab, indicating a G1/S
arrest during bevacizumab treatment in ARPE-19 cells.
Effect of bevacizumab on cell cycle
regulatory proteins in ARPE-19 cells
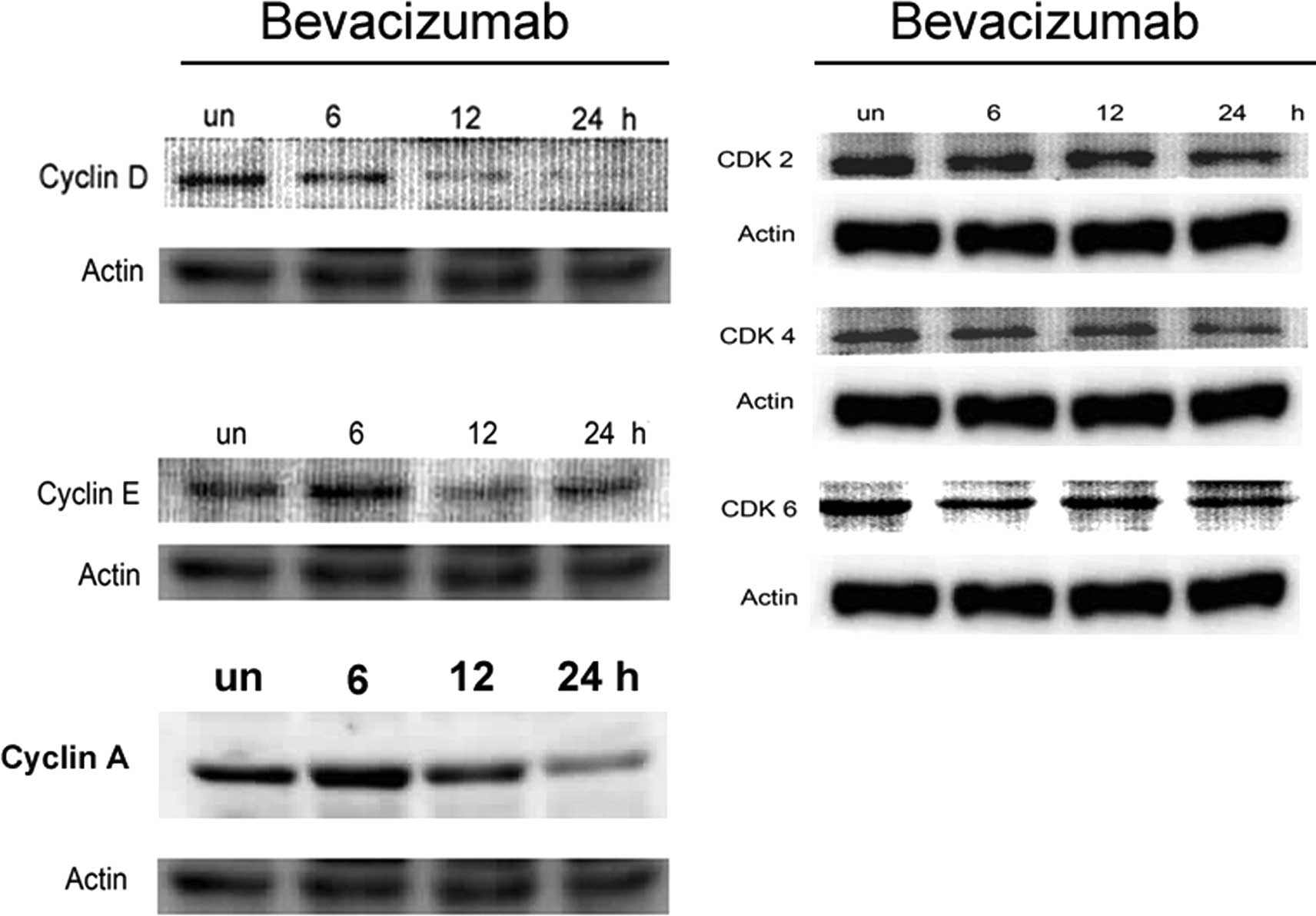
Based on the observation that bevacizumab induced a
G1/S arrest in ARPE-19 cells, we evaluated the effect of
bevacizumab on cell cycle regulatory proteins that are important in
G1/S cell cycle progression, using western blot
analysis. As shown in Fig. 2A,
bevacizumab treatment induced a decrease in the protein level of
cyclin D in ARPE-19 cells that were clearly visible after 6, 12 and
48 h of treatment. The expression of cyclin E increased slightly
after 6 h of treatment, followed by a marked upregulation after 12
and 24 h, compared with the untreated cells. Thus, the
bevacizumab-mediated G1/S phase cell cycle arrest of
ARPE-19 cells correlated with the inhibition of cyclin D and E. The
bevacizumab-treated ARPE-19 cells also demonstrated a decrease in
the protein levels of CDK2, 4 and 6 that was visible after 6, 12
and 24 h of treatment (Fig. 2B).
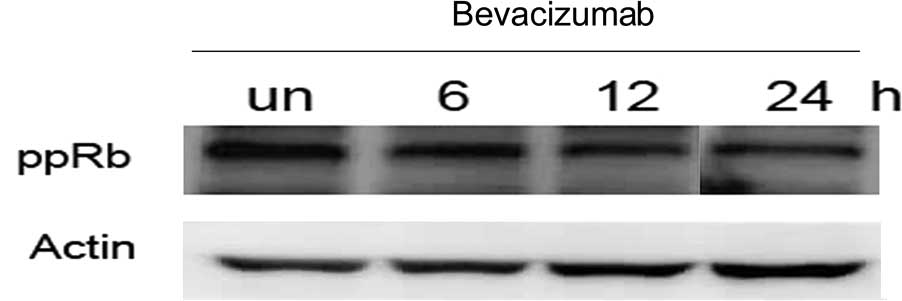
Consistent with cyclin D and CDK4 downregulation, bevacizumab
markedly inhibited the phosphorylation of pRb (ppRb) at 24 h
(Fig. 3). Moreover, to gain
further insights into the mechanism of bevacizumab-mediated
G1/S phase arrest, we determined its effect on p53, p21,
p16 and p27 protein expressions by immunoblotting. Bevacizumab
treatment resulted in a time-dependent increase in the protein
expressions of p53, p21, p16 and p27 in ARPE-19 cells (Fig. 4).
Bevacizumab inhibited cell viability in
ARPE-19 cells
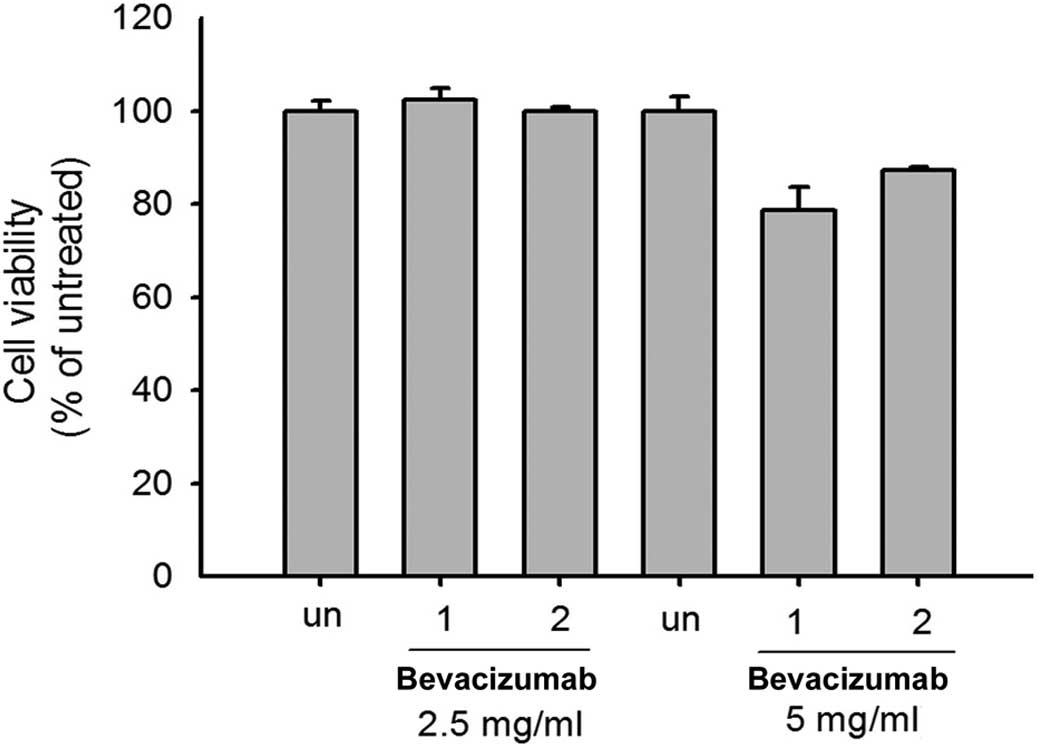
Our study demonstrated that bevacizumab at a
concentration of 2.5 mg/ml in 24 h caused a moderate decrease of
the G1/S phase in ARPE-19 cells (Fig. 1). The MTT assay was subsequently
used to prove that bevacizumab (5.0 mg/ml) treatment inhibited the
cell viability to 80% in ARPE-19 cells after 48 h (Fig. 5).
Discussion
Bevacizumab inhibited monkey choroidal endothelial
cells at 2.0 mg/ml on cell viability, however, no effect was noted
on the ARPE-19 and RGC-5 cell lines at 2.0 mg/ml (8). Results of that study are similar to
our findings demonstrating that bevacizumab treatment did not
reduce the cell viability at 2.5 mg/ml in ARPE-19 cells at 24 h. In
the higher concentration of bevacizumab (5 mg/ml), cell viability
was reduced to 80% suggesting that bevacizumab might affect the
cell cycle of ARPE-19 cells. A recent study reported that
bevacizumab has a cytostatic effect of VEGF inhibition on multiple
myeloma. This malignancy of plasma cells was determined through the
attenuation of critical signaling effectors: VEGF receptor 1, mTOR,
c-Myc, Akt, STAT3 and eIF4E (9).
In addition, bevacizumab has a direct effect on major pathways
critically activated in multiple myeloma that is independent from
its established effect on angiogenesis (9). These effects of bevacizumab were
achieved using high concentrations (2 mg/ml). These results
indicate that bevacizumab had a higher cytostatic effect on
multiple myeloma, compared with the ARPE-19 and RGC-5 cell lines at
2 mg/ml.
CDK2, 4 and 6, as well as cyclins D and E, are
various driving forces for the G1/S phase of the cell
cycle (10). Therefore,
significant decreases in the protein expression of cyclin D, E and
A, as well as CDK2, 4 and 6 due to bevacizumab suggest a potential
inhibition on ARPE-19 cell growth through cell cycle regulation
(Fig. 2A and B). The upregulation
of p53 at 0 and 48 h alone, with a marked induction of the
expression of p16, p21 and p27 at 48 h was also observed (Fig. 2D). Moreover, the accumulation of
p53 induces the upregulation of cell cycle-regulatory proteins,
such as p21 and p27, whereas the induction of these genes leads to
G0/G1 arrest (11).
Cyclins, such as type D and E regulate
G1/S phase cell cycle progression through the activation
of specific CDK that phosphorylates the pRb protein, thereby
decreasing the repression of E2F-DP transactivation of S-phase
genes (12). Loss of cyclin D
causes G1 arrest in some cells, but in other cell lines,
the downstream cyclin E protein replaces cyclin D and aids
G1/S progression (13).
In bevacizumab treatment, the expression of cyclin E increased
slightly at 24 h in ARPE-19 cells (Fig. 2A). However, the downregulation of
cyclin D1 started at 6 h and sustained to 24 h in bevacizumab
treatment (Fig. 2A). This seems to
explain the downregulation of cyclin D1, but not of cyclin E, and
is a main factor for bevacizumab-induced G1/S arrest.
Findings of another study also demonstrated that cyclin D1
degradation is sufficient to induce G1 cell cycle arrest
despite the constitutive expression of cyclin E2 in ovarian cancer
cells (12). CDKs are regulated by
distinct protein sequences, including the cyclins required for the
CDK activity and inhibitor (CKI) proteins (14). Results of the western blotting
demonstrated that bevacizumab decreased cyclin D1 levels (Fig. 2A). The protein levels of CDK2, 4
and 6 were also inhibited by bevacizumab at 24 h (Fig. 2B). In addition, CKIs, such as p16,
p21 and p27 were increased by bevacizumab, at least within a 6- to
24-h treatment period (Fig. 2D).
Therefore, the inhibitory effect of bevacizumab on cell
cycle-regulated proteins, not only inhibited the expression of
cyclins/CDKs, but also promoted the expression of CKIs.
Factors associated with the Rb family proteins are
recognized as determining downstream targets of
G1-specific cyclin/CDK complexes (15). In hypophosphorylation, the Rb
proteins correlate with and suppress the activity of E2F family
transcription factors, which are involved in the transcription of
key cell cycle-regulatory proteins (16). Upon growth stimulation, the
G1-specific cyclins/CDKs phosphorylate Rb, resulting in
the release of E2F factors and progression into the S phase
(16). The phosphorylation of Rb
provides a crucial role in the progression of the G1
phase and the transition of G1 to S phase (17). Ser795 in pRb is a specific site for
phosphorylation by the cyclin D/CDK4 complex in the
G0/G1 phase (18). The treatment with bevacizumab
resulted in the inhibition of cyclin D and CDK4 expression in
ARPE-19 cells, as well as the prevention of CDK4 from
phosphorylating pRb at Ser795, thereby arresting cell growth
(Fig. 2). Another study
demonstrated that the p21 protein binds to cyclin/CDK complexes
resulting in inhibition of the G1/S phase transition by
inhibiting the phosphorylation of the Rb protein (19). The decrease in phosphorylated Rb
proteins, therefore, might result from bevacizumab-triggered p21
expression and the decreased expression of cyclin-CDK. Generally,
we observed that bevacizumab inhibited cell growth in ARPE-19
cells. We, therefore, suggest a novel pathway by which bevacizumab
regulates cell cycle progression in ARPE-19 cells. Bevacizumab was
able to induce p53 production, and then upregulate the expression
of p16, p21 and p27. These events decrease the expression of cyclin
D and E, and CDK2, 4 and CDK 6, followed by the reduction of ppRb
as well as the triggering of the G1/S arrest of the cell
cycle.
Acknowledgements
This study was supported by a grant from the Chang
Gung Memorial Hospital, ROC: CMRPG6B0041.
References
|
1
|
Luthra S, Narayanan R, Marques LE, Chwa M,
Kim DW, Dong J, Seigel GM, Neekhra A, Gramajo AL, Brown DJ, et al:
Evaluation of in vitro effects of bevacizumab (Avastin) on
retinal pigment epithelial, neurosensory retinal, and microvascular
endothelial cells. Retina. 26:512–518. 2006.PubMed/NCBI
|
|
2
|
Garg S, Brod R, Kim D, Lane RG, Maguire J
and Fischer D: Retinal pigment epithelial tears after intravitreal
bevacizumab injection for exudative age-related macular
degeneration. Clin Experiment Ophthalmol. 36:252–256. 2008.
View Article : Google Scholar
|
|
3
|
Forooghian F, Cukras C and Chew EY:
Retinal angiomatous proliferation complicated by pigment epithelial
tear following intravitreal bevacizumab treatment. Can J
Ophthalmol. 43:246–248. 2008. View
Article : Google Scholar
|
|
4
|
Shima C, Sakaguchi H, Gomi F, Kamei M,
Ikuno Y, Oshima Y, Sawa M, Tsujikawa M, Kusaka S and Tano Y:
Complications in patients after intravitreal injection of
bevacizumab. Acta Ophthalmol. 86:372–376. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arias L, Caminal JM, Rubio M, Pujol O and
Arruga J: Retinal pigment epithelial tears after intravitreal
bevacizumab injection for predominantly classic choroidal
neovascularization. Eur J Ophthalmol. 17:992–995. 2007.
|
|
6
|
Subramanyam A, Phatak S and Chudgar D:
Large retinal pigment epithelium rip following serial intravitreal
injection of avastin in a large fibrovascular pigment epithelial
detachment. Indian J Ophthalmol. 55:483–486. 2007. View Article : Google Scholar
|
|
7
|
Spitzer MS, Wallenfels-Thilo B, Sierra A,
Yoeruek E, Peters S, Henke-Fahle S, Bartz-Schmidt KU and Szurman P;
Tuebingen Bevacizumab Study Group. Antiproliferative and cytotoxic
properties of bevacizumab on different ocular cells. Br J
Ophthalmol. 90:1316–1321. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brar VS, Sharma RK, Murthy RK and Chalam
KV: Evaluation of differential toxicity of varying doses of
bevacizumab on retinal ganglion cells, retinal pigment epithelial
cells, and vascular endothelial growth factor-enriched choroidal
endothelial cells. J Ocul Pharmacol Ther. 25:507–511. 2009.
View Article : Google Scholar
|
|
9
|
Attar-Schneider O, Drucker L, Zismanov V,
Tartakover-Matalon S, Rashid G and Lishner M: Bevacizumab
attenuates major signaling cascades and eIF4E translation
initiation factor in multiple myeloma cells. Lab Invest.
92:178–190. 2012. View Article : Google Scholar
|
|
10
|
Mueller A, Odze R, Jenkins TD, Shahsesfaei
A, Nakagawa H, Inomoto T and Rustgi AK: A transgenic mouse model
with cyclin D1 overexpression results in cell cycle, epidermal
growth factor receptor, and p53 abnormalities. Cancer Res.
57:5542–5549. 1997.PubMed/NCBI
|
|
11
|
Yoo YA, Kim MJ, Park JK, Chung Young Min,
Jong HL, Sung-Gil C, Jun SK and Young DY: Mitochondrial ribosomal
protein L41 suppresses cell growth in association with p53 and
p27Kip1. Mol Cell Biol. 25:6603–6616. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Masamha CP and Benbrook DM: Cyclin D1
degradation is sufficient to induce G1 cell cycle arrest despite
constitutive expression of cyclin E2 in ovarian cancer cells.
Cancer Res. 69:6565–6572. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu Q, Geng Y and Sicinski P: Specific
protection against breast cancers by cyclin D1 ablation. Nature.
411:1017–1021. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sherr CJ and Roberts JM: Inhibitors of
mammalian G1 cyclin-dependent kinases. Genes Dev. 9:1149–1163.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sherr CJ: The Pezcoller lecture: cancer
cell cycles revisited. Cancer Res. 60:3689–3695. 2000.PubMed/NCBI
|
|
16
|
Vivar OI, Lin C, Firestone GL and
Bjeldanes LF: 3,3′-Diindolylmethane induces a G1 arrest in human
prostate cancer cells irrespective of androgen receptor and p53
status. Biochem Pharmacol. 78:469–476. 2009.
|
|
17
|
Jinno S, Hung SC and Okayama H: Cell cycle
start from quiescence controlled by tyrosine phosphorylation of
Cdk4. Oncogene. 18:565–571. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Das SK, Hashimoto T and Kanazawa K: Growth
inhibition of human hepatic carcinoma HepG2 cells by fucoxanthin is
associated with down-regulation of cyclin D. Biochim Biophys Acta.
1780:743–749. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ukomadu C and Dutta A: p21-dependent
inhibition of colon cancer cell growth by mevastatin is independent
of inhibition of G1 cyclin-dependent kinases. J Biol Chem.
278:43586–43594. 2003. View Article : Google Scholar : PubMed/NCBI
|