Introduction
Individual cardiomyocytes are connected to each
other via three cell-cell junctions, desmosomes (macula adherens),
adherens junctions (fascia adherens) and gap junctions which
collectively make up intercalated disks. These membrane junctions
ensure mechanical coupling between cells and enable the propagation
of electrical impulses through cardiac muscles to propel muscle
contraction (1).
Desmosomes are of particular interest as they bear a
high mechanical load and are particularly abundant in the heart.
Desmoplakin (DSP), a member of the plakin family, is an extremely
large desmosomal protein that serves as an intracellular link
between desmosomes and intermediate filaments. A wide array of
mutations in the DSP protein have been observed in individuals with
arrhythmogenic right ventricular cardiomyopathy (ARVC) (2–4). A
mutation in the DSP protein has been suggested to disrupt various
cell targets, resulting in abnormal protein interactions at
intercellular junctions. This process may ultimately lead to
electrical dysfunction, with the possibility of life-threatening
arrhythmias and sudden cardiac death (5).
While the mutated proteins in ARVC may lead to the
loss of desmosomal integrity, this observation does not account as
to why life-threatening arrhythmias occur early in the course of
the disease before cardiomyocytes are replaced by fibro-fatty
tissue. The mechanical functions of desmosomes in heart tissue
depend on complex binding interactions between multiple proteins.
Disruption of cell junctions associated with desmosomal protein
mutations is considered to affect the structure and function of
electrical connections at gap junctions, which may contribute to
impairment in cardiac electrical conduction and associated
arrhythmias. Structurally, the gap junction consists of two
hemichannels or connexons and the principal subtype in the human
heart is connexin43 (Cx43) (6). Of
note, the correlation between DSP and gap junctions in cardiac
arrhythmias has not been investigated.
Apart from cell-cell junctions, other molecules that
reside in the intercalated disk include voltage-gated sodium
channels. A previous study indicated that Nav1.5, the
major α-subunit of the cardiac sodium channel, is situated in the
intercalated disk (7).
Nav1.5 is responsible for rapid depolarization of
intercalated disks and is important for action potential
propagation. In addition, while Nav1.5 may be involved
in the occurrence of arrhythmias in patients with ARVC (8–10),
no studies have currently shown Nav1.5 to be involved in
ventricular arrhythmias due to desmosomal protein mutations.
In the present study, the potential link between DSP
desmosomal protein expression and the impact it may have on Cx43
and Nav1.5 was investigated. To test this hypothesis,
the study was designed to explore the distribution and function of
gap junctions and Nav1.5 while inhibiting the expression
of DSP in HL-1 cardiomyocytes.
Materials and methods
Preparation of HL-1 cardiomyocytes
HL-1 cells are a cardiac muscle cell line derived
from AT-1 mice that have the ability to be continuously passaged.
These cells accurately represent the electrophysiological
functioning of adult cardiomyocytes in vitro, making them
ideal for the current study (11,12).
HL-1 cells were obtained from the laboratory of William Claycomb
(Louisiana State University Health Science Center, New Orleans, LA,
USA) and were maintained as previously described (11). HL-1 cells were grown in Claycomb
medium (JRH Biosciences, Lenexa, KS, USA), supplemented with 10%
v/v fetal bovine serum (JRH Biosciences), 100 mM norepinephrine
(Sigma-Aldrich, St. Louis, MO, USA), 4 mM L-glutamine and 1%
penicillin/streptomycin (Invitrogen Life Technologies, Carlsbad,
CA, USA) in a humidified 5% CO2 incubator at 37ºC.
Culture flasks, dishes and plates were precoated with 1
μg/cm2 fibronectin/0.02% w/v gelatin solution
(Sigma-Aldrich). The medium was changed every 24–48 h.
Small interfering RNA (siRNA)
transfection
siRNA specific for mouse and control desmoplakin
(NM_0238422) were purchased from GenePharma (Shanghai, China). The
sequence for targeting and negative control siRNA contained the
following four polynucleotides (sense sequences only):
5′-GCACCAGCAGGACGUUCUATT-3′; 5′-CGAUCAGAU GGAAAUCAUATT-3′;
5′-GGAGCGACAAGAACACCA ATT-3′ and 5′-UUCUCCGAACGUGUCUCGUTT-3′
(negative control). The transfection procedure was performed
according to the manufacturer’s instructions with Lipofectamine
2000® reagent (Invitrogen Life Technologies).
Transfection was performed on 1–2×105 cells/well in
6-well plates in serum- and antibiotic-free cell culture medium.
Cells were washed in Opti-MEM (Invitrogen Life Technologies) twice
prior to the addition of 2 ml serum-free culture medium. Then, 500
μl mixed Opti-MEM, siRNA of negative control or desmoplakin and
Lipofectamine 2000 (5 μl/well) were added into each well to a final
siRNA concentration of 50 nM. The medium was changed the following
day and cells were used experimentally 48–120 h following
transfection.
Western blotting
HL-1 cells in 6-well plates were rinsed twice with
PBS and then added to 100 μl RIPA lysis buffer and complete
protease inhibitor (Roche Diagnostics, Mannheim, Germany). The
plates were agitated for 15 min at 4ºC and centrifuged at 14,800 ×
g for 20 min. Protein concentrations were determined and 30 μg
samples were diluted with 4X loading buffer (Invitrogen Life
Technologies) and heated at 95ºC for 5 min. Next, 10 μl 4X SDB was
added to 30 μl sample, run in a 4–12% Bis-Tris precast gel
(Invitrogen Life Technologies) and transferred to nitrocellulose
membranes (Amersham Pharmacia Biotech, Piscataway, NJ, USA)
according to the manufacturer’s instructions. Membranes were
blocked in dried skimmed milk powder in Tween 20 Tris-buffered
saline for 2 h at room temperature prior to overnight incubation at
4ºC with the following primary antibodies: mouse anti-desmoplakin
I/II (1:200; Abcam, Cambridge, UK), rabbit anti-desmoplakin I/II
(1:200; Santa Cruz Biotechnology Inc., CA, USA), rabbit anti-Cx43
(1:500; Cell Signaling Technology, Inc, Danvers, MA, USA) and
rabbit polyclonal anti-Nav1.5 (1:100; Alomone Labs,
Jerusalem, Israel). Anti-GAPDH (1:1,000; Zymed Laboratories,
Carlsbad CA, USA) was used as the loading control. The secondary
antibody against mouse primary antibodies was goat anti-mouse
IgG-HRP (1:5,000), while the secondary antibody used against rabbit
primary antibodies was goat anti-rabbit IgG-HRP (1:5,000; Santa
Cruz Biotechnology Inc.). Protein bands were visualized using
electrochemiluminescence reagents (Invitrogen Life Technologies).
The images were evaluated densitometrically using Gel-Pro Analyzer
4.0 software (Media Cybernetics, Inc., Rockville, MD, USA).
Flow cytometry
HL-1 cells were routinely dissociated with
trypsin/EDTA which was pre-warmed to 37ºC. For intracellular
protein labeling, the cells were fixed and permeabilized according
to the manufacturer’s instructions by Fix and Perm Cell Reagent
(MultiSciences Biotech Co., Ltd., Shanghai, China). The cells were
fixed in 100 μl fixation reagent for 15 min at room temperature,
washed in PBS and then resuspended in 100 μl permeabilization
reagent for 20 min at room temperature together with the primary
antibodies: mouse anti-desmoplakin I/II, rabbit anti-connexin 43
and rabbit polyclonal to Nav1.5. After washing the cells
the following secondary antibodies were added: Alexa Fluor 488 goat
anti-rabbit IgG (Invitrogen Life Technologies) and goat anti-mouse
IgG-PE (MultiSciences Biotech). The mixtures were incubated for 20
min at room temperature. Following a final wash, the cells were
resuspended in PBS and then analyzed using flow cytometry (Beckman
Coulter Inc., Brea, CA, USA). Alexa Fluor 488 IgG or IgG-PE
staining alone was used as the negative control.
Immunofluorescent staining and laser
scanning confocal microscopy
siRNA-transfected cells, cells treated with
transfection reagent only and untreated cells were all grown on
collagen-coated coverslips, rinsed twice in PBS and fixed with
methanol at −20ºC for 20 min. Following fixation, the coverslips
were directly washed in PBS for 5 min, followed by incubation with
PBS, 0.2% Triton X-100 and 5% bovine serum albumin for 20 min at
room temperature. Following rinsing with PBS, the cells were
incubated with primary antibody at 4ºC in a humidity box. Cx43,
desmoplakin and Nav1.5 proteins were examined following
incubation with mouse anti-desmoplakin I/II (1:200; Abcam), rabbit
anti-Cx43 (1:200; CST) and rabbit polyclonal to Nav1.5
(1:200; Alomone). Coverslips were subsequently washed 3 times with
PBS and incubated with the corresponding secondary antibodies for 2
h at room temperature in a dark, humid box. Secondary antibodies
included Alexa Fluor 647 goat anti-mouse (Invitrogen Life
Technologies) and Alexa fluor 488 goat anti-rabbit (Invitrogen Life
Technologies) diluted to 1:500 in blocking buffer. DAPI staining
was then performed to identify nuclei. Following a final rinsing
step, coverslips were mounted using antifade mounting media
(Applygen Technologies Inc, Beijing, China). Cardiomyocytes were
observed at 48, 72, 96 and 120 h following incubation with siRNAs
targeting desmoplakin, with non-targeting siRNAs or with
transfection reagent only and were compared with untreated control
cells at each time point.
Scrape loading dye transfer
The functions of gap junctional intercellular
communication in adjacent cells were assessed by scrape loading dye
transfer (SLDT) with fluorescent dye Lucifer Yellow (LY;
Sigma-Aldrich). At 90% confluence, the cultured cells were rinsed
three times with PBS. A cross scrape through the monolayer was made
using a surgical scalpel, followed by incubation with 100 g/l LY in
0.33 mol/l LiCl for 3 min in a dark room at room temperature and
several PBS washes to remove any background fluorescence. Cells
were then fixed with 4% paraformaldehyde and the optical density
was detected using confocal laser scanning microscopy (Leica,
Wetzlar, Germany).
Electrophysiology
HL-1 cells were isolated from culture dishes
following infection with siRNA targeting desmoplakin or with
non-targeting siRNA for 5 days. Whole cell cardiac sodium current
(INa) was recorded from HL-1 cells. Recorded
cells were rod-shaped and appeared striated. Cells were perfused
with extracellular solution containing: 136 mM NaCl, 1 mM
MgCl2, 1 mM CaCl2, 0.5 mM CoCl, 10
mM HEPES, 0.33 mM NaH2PO4, 5.4 mM CsCl and 10
mM glucose (pH 7.3). The pipette solution contained: 20 mM CsCl,
110 mM CsF, 5 mM NaCl, 10 mM HEPES, 5 mM Na2ATP, 1mM
MgCl and 10 mM EGTA (pH 7.2). Pipette resistances were 2–3 MΩ.
Access resistance was compensated to 1–2 MΩ. Recordings were
performed using an Axopatch 200B amplifier and data acquisition and
analysis were performed using the Digidata 1200B-pClamp 7.0
data-acquisition system (Axon Instruments, Jakarta, Indonesia).
Signals were filtered at 5 kHz and stored on a computer. All
experiments were performed at room temperature. To characterize the
voltage dependence of the peak INa, single cells
were held at −120 mV and 200 msec voltage steps were applied from
−90 to +30 mV in 5 mV increments. The interval between voltage
steps was 3 sec. Voltage-dependence of inactivation was assessed by
holding cells at various potentials between −130 and −40 mV
followed by a 30 msec test pulse to −40 mV to elicit
INa. Recovery from inactivation was studied by
holding cells at −120 mV and applying two 20 msec test pulses (S1,
S2) to −40 mV separated by increasing increments of 2 msec to a
maximum S1–S2 interval of 80 msec. The S1–S1 interval was
maintained at 3 sec. Data are presented as mean ± SEM.
Results
HL-1 cells treated with DSP siRNA exhibit
downregulation of DSP
DSP siRNA was introduced into HL-1 cells in
vitro to investigate the effect of loss of DSP expression on
the function and distribution of Cx43/Nav1.5. Initially,
DSP mRNA expression levels in HL-1 cells were assessed by real-time
PCR analysis following transfection with DSP siRNA. GAPDH was used
as the internal control. Three distinct siRNA sequences decreased
levels of DSP mRNA expression; DSP mRNA levels were decreased by
>70% from the control using a pair of DSP mRNA sequences
(5′GGAGCGACAAGAACACCAATT3′, sense sequence). Subsequent experiments
were conducted using this DSP siRNA sequence.
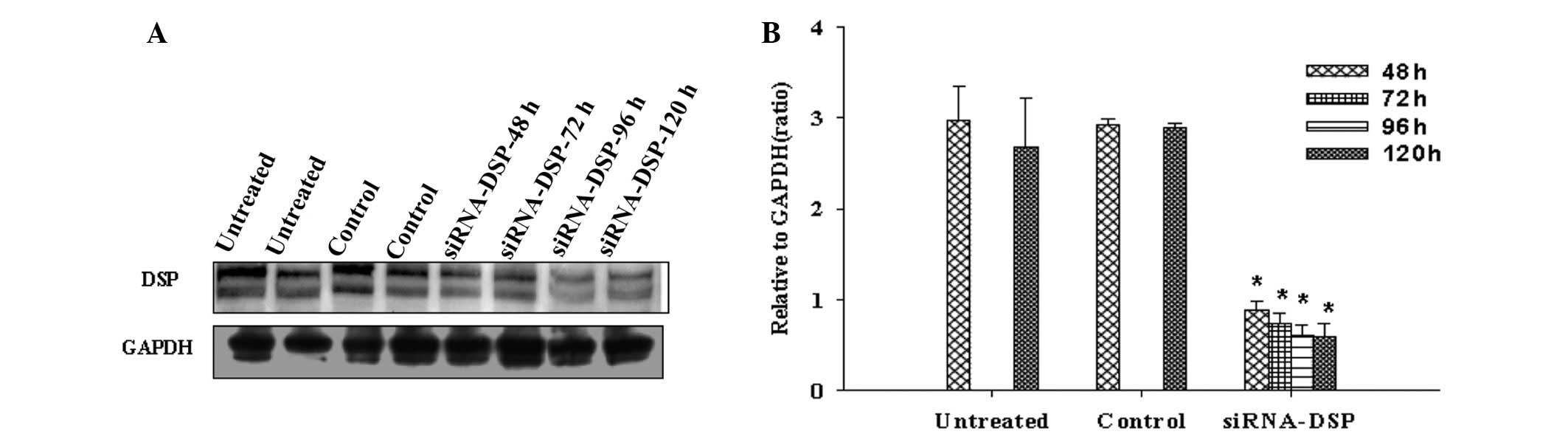
Western blot analysis following treatment with DSP
siRNA consistently revealed a significantly reduced expression of
DSP in HL-1 cells when compared with untreated and non-targeting
siRNA control treatments (GAPDH was used as a loading control;
n=3). Fig. 1 shows that DSP
reduction persisted between 48 and 120 h. For quantification, each
band density was measured relative to its corresponding GAPDH
control.
Quantification of immunoblot signals revealed that
the expression of DSP protein was markedly reduced following 48–120
h DSP siRNA treatment compared with control siRNA or untreated
cells.
DSP siRNA alters Cx43 expression and
function in HL-1 cells
Effect of DSP silencing on content and
distribution of Cx43
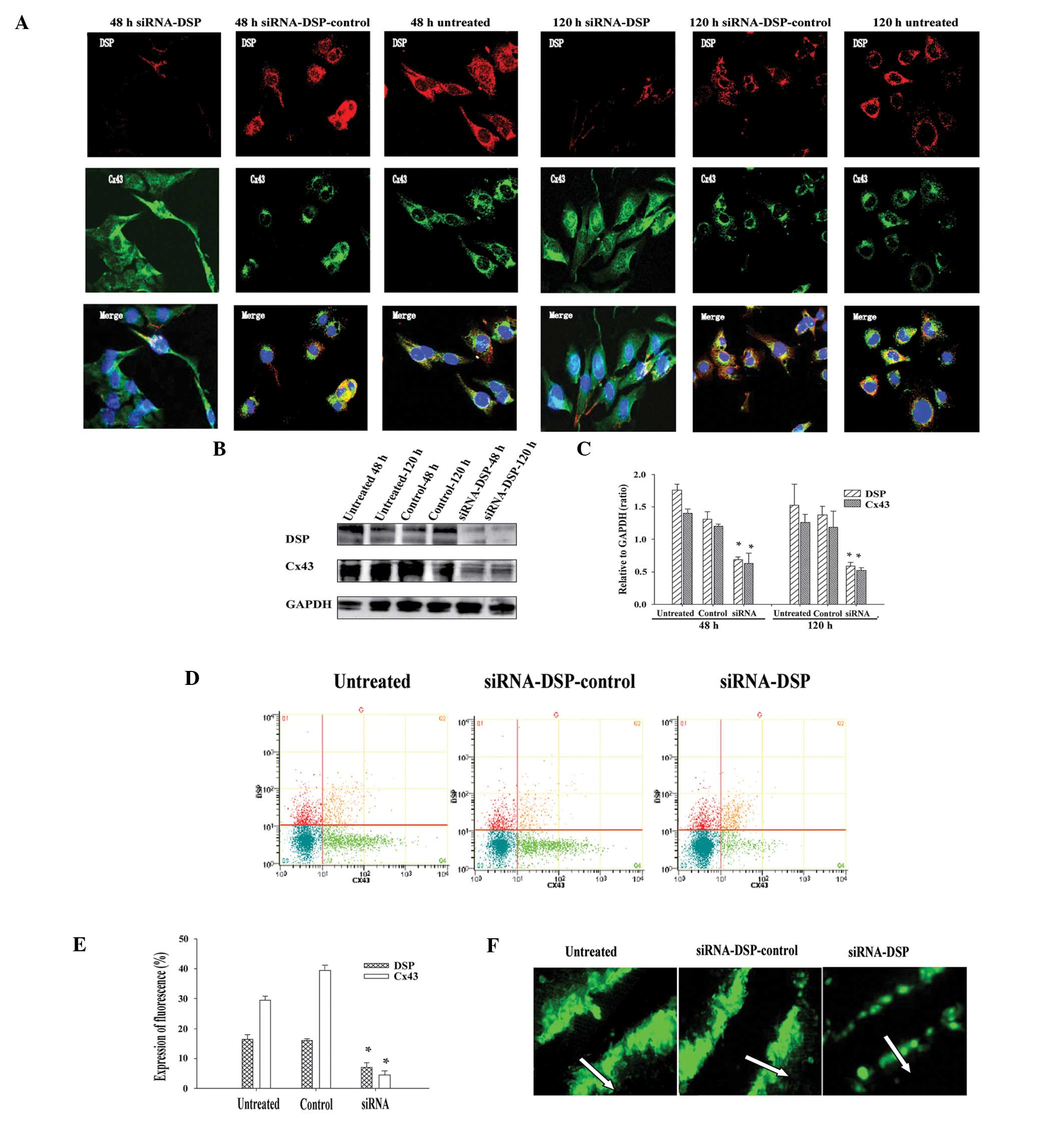
Results of immunolocalization of Cx43 and DSP in
HL-1 cells indicated that Cx43 undergoes progressive and marked
reorganization following DSP siRNA silencing. These effects were
not detected with non-targeting control siRNA (control siRNA) and
untreated cells. Fig. 2A shows
typical results observed at the first (48 h) and last (120 h)
time-points following transfection with DSP siRNA, compared with
control siRNA or untreated cells. Colocalization to the site of
cell-cell contact was apparent in untreated conditions and when
cells were infected with non-targeting control siRNA. The content
of Cx43 at the cell-cell borders was significantly distributed in
transfected cells, as Cx43 was identified within the intracellular
space. In addition, Cx43 protein expression levels were
investigated using western blot analysis. Cx43 protein content was
reduced following DSP knockdown by DSP siRNA (Fig. 2B). Quantitative evaluations of the
density of the Cx43 signal also revealed that the Cx43 level was
markedly reduced following 48–120 h DSP siRNA treatment compared
with control siRNA or untreated cells. To clarify Cx43 expression
in HL-1 cells treated with DSP siRNA, levels of Cx43 fluorescence
were measured by flow cytometry. It was observed that the rate of
Cx43 fluorescence expression detected in HL-1 cells treated with
DSP mRNA was decreased when compared with control siRNA or
untreated cells (Fig. 2C and D).
Overall, these results suggest that a loss of DSP expression leads
to a significant decrease and redistribution of Cx43.
Changes in the function of gap
junctions in DSP-silenced cells
To determine whether the loss of DSP expression
affected the function of gap junctions between cell pairs, the
transfer of LY across gap junctions in adjacent cells was assessed
by SLDT. The distance over which LY spread in the cells treated
with DSP siRNA was significantly decreased compared with the
control siRNA or untreated cells (Fig.
2E).
DSP siRNA affects Nav1.5
expression and sodium currents in HL-1 cells
DSP siRNA alters Nav1.5
expression in HL-1 cells
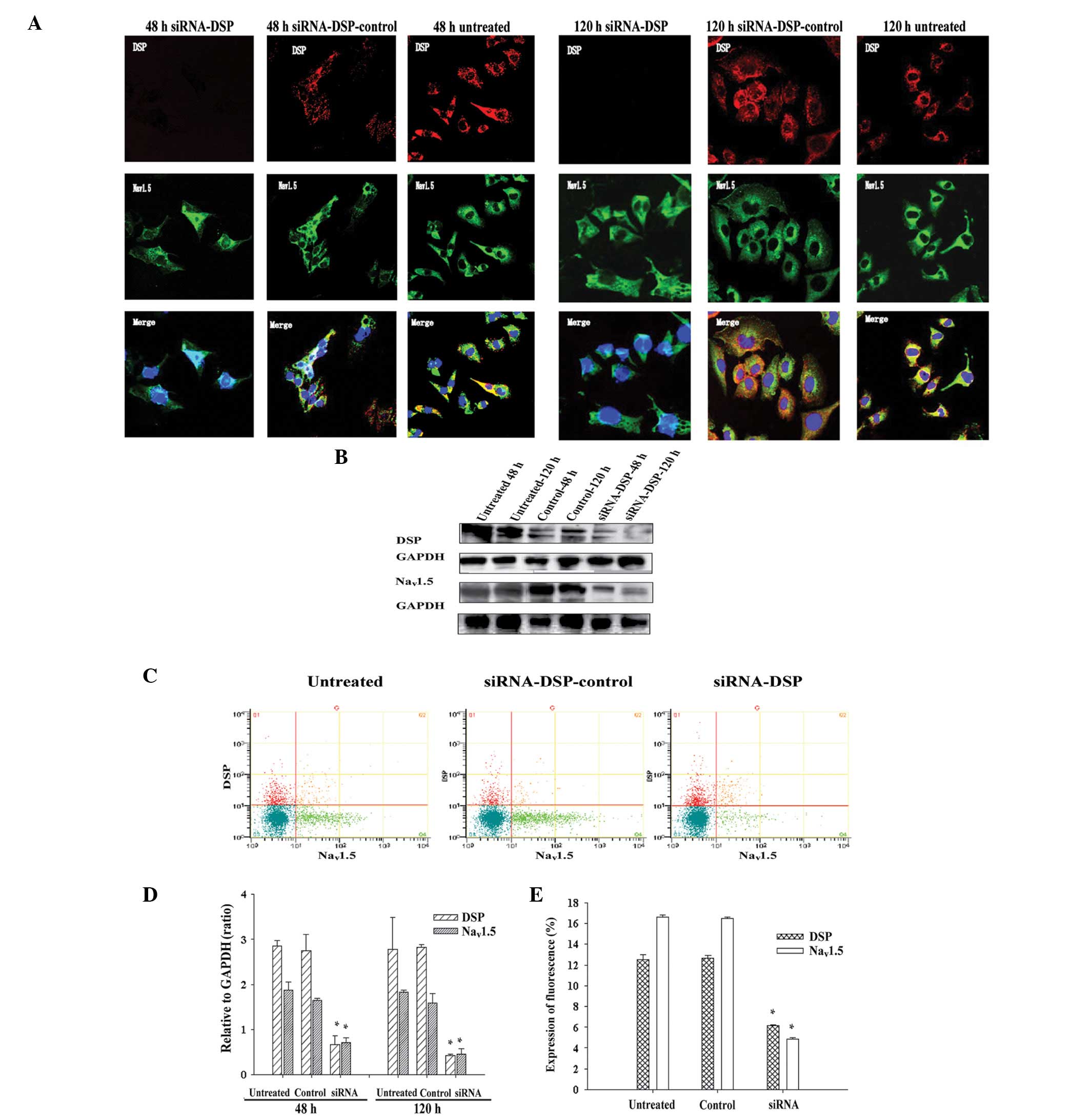
To characterize the effects of DSP siRNA on
Nav1.5, immunofluorescent experiments were performed in
transfected HL-1 cells. Results of the immunofluorescence
experiments suggested that Nav1.5 undergoes significant
alterations following treatment with DSP siRNA, which were not
observed with non-targeting control siRNA and untreated cells
(Fig. 3A). The expression of
Nav1.5 at the cell-cell borders was markedly reduced in
transfected cells (Fig. 3A, left
panel), while residual Nav1.5 protein tended to relocate
in the cytoplasmic space. Colocalization of Nav1.5 and
DSP proteins at cell borders was observed in HL-1 cells infected
with non-targeting control siRNA (Fig.
2A, middle panel) and untreated conditions (Fig. 3A, left panel). Western blotting and
flow cytometry measuring the expression of DSP siRNA and
Nav1.5 protein revealed that the Nav1.5
protein content was reduced following DSP siRNA silencing. The rate
of Cx43 fluorescence expression in HL-1 cells treated with DSP mRNA
was decreased compared with control siRNA or untreated cells. The
results of immunofluorescence, western blotting and flow cytometry
suggested that DSP siRNA silencing has a deleterious effect on
Nav1.5 localization and expression.
Effect of DSP siRNA on sodium currents
in HL-1 cells
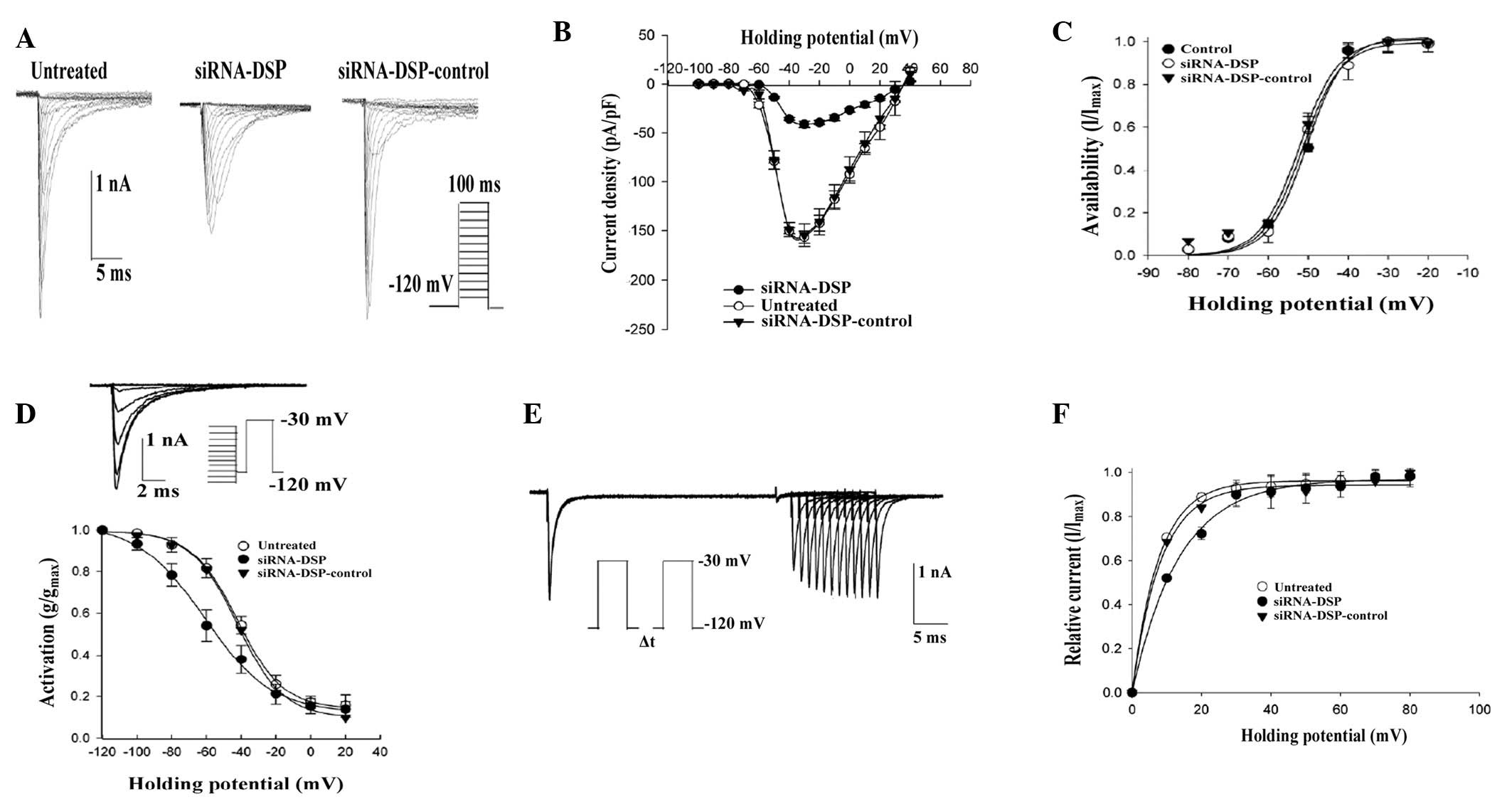
The properties of INa as a
function of DSP expression were assessed and sodium currents were
recorded using a voltage clamp. Fig.
4A shows an example of currents obtained from cells untreated
or treated with an oligonucleotide mixture. Composite data are
presented in Fig. 4B-F. Treatment
with control siRNA did not affect the current parameters. However,
a decrease in peak current density (Fig. 4B), a shift in voltage dependent
steady-state inactivation (Fig.
4D) and a prolongation of time-dependent recovery from
inactivation (Fig. 4F) were
observed in DSP-silenced cells.
Discussion
It has been well documented that DSP is a major
component of desmosomes and plays a significant role in the
maintenance of cardiac tissue integrity. Previous studies have
proposed that DSP and plakophilin 2 (PKP2) interact with each other
at their N-terminal domains (13).
Previously, Oxford et al demonstrated that Cx43 and PKP2 may
coexist in the same macromolecular complex (6). The present study expands this concept
by demonstrating that a loss of DSP leads to a decreased expression
and redistribution of Cx43 protein. This phenomenon has a
significant impact on gap junction function and reveals a reduction
in the distance of LY spread from cell to cell. The current
observations are the first to clearly demonstrate an association
between DSP downregulation and abnormal cell-to-cell communication
mediated by Cx43. The observations indicate that DSP may have a
greater impact on regulating the formation and interaction of
mechanical and electrical junctional complexes than previously
hypothesized.
Gap junctions are intercellular channels that
connect adjacent cells via the cytoplasm. Previous studies have
indicated that the disruption of desmosomal proteins results in a
strong effect on the integrity or stability of the gap junction
(1). Carvajal syndrome is a
cardiomyopathy caused by a mutation in DSP that is characterized by
distinct ultrastructural abnormalities of the intercalated disk
(5). Thus, the mutation in DSP may
not only interfere with molecular interactions among the desmosomal
proteins, but also cause contractile- and electrical- related
dysfunctions.
A previous study identified other proteins localized
at the intercalated disk that interconnected with Cx43 (14). While the functional role of these
protein-protein interactions has not been clearly established, it
has been hypothesized that they are important in coupling between
inter- and intracellular signaling in the heart. The current study
reveals that loss of DSP expression leads to Cx43 remodeling.
Notably, loss of the DSP signal may be associated with the change
in the integrity of DSP-Cx43 interactions and the heterozygous
deletion of DSP in mice ultimately led to fibro-fatty infiltration.
While gap junction plaques do not define the location of all gap
junctions, a decrease in cell-cell dye coupling was observed. Based
on these observations, it was suggested that DSP and Cx43 represent
a molecular network that may be disrupted by an ARVC relevant
mutation. In human DSP mutation carriers, there was a significant
conduction delay at short coupling intervals with no evidence of
fibro-fatty replacement, despite a downregulation in cardiac Cx43
protein expression. Therefore, future studies must determine the
manner in which Cx43 relocalization prior to major structural and
histological changes affects development in animal models as well
as patients.
Little is known concerning the signaling
correlations between cell-cell junctions and the voltage-gated
sodium channel in cardiac cells. Voltage-dependent sodium channels
are responsible for generating action potentials and the rapid
conduction of electrical signals in excitable cells (15). While it has been observed that a
mutation in Nav1.5 may cause cardiac arrhythmias and
sudden cardiac death, it is not yet clear how mutations in
Nav1.5 are responsible for such a large spectrum of
diseases along with the overlapping syndromes (16). Gomes et al(17) previously demonstrated that sodium
channels localize to the intercalated disk in the biopsy samples of
an ARVC patient, potentially contributing to a reduced conduction
velocity. It was suggested that a loss of DSP expression would
significantly affect propagation properties in cardiomyocytes due
to the change in sodium current function. The current study
effectively demonstrates that loss of DSP leads to a decreased
expression and redistribution of the Nav1.5 protein and
knockdown of DSP expression decreases the sodium current while also
slowing the conduction velocity in cultured HL-1 cells. However, it
is important to emphasize that the present observations do not
disprove a possible effect of DSP silencing on the membrane and
other unrelated factors may have affected the slow conduction
velocity in the current experiments. Future investigations are
likely to be directed towards addressing this possibility.
Mutations in DSP have been observed to be the main
cause of ARVC that presents with clinical manifestations, including
ventricular arrhythmias and sudden death under strenuous
circumstances (2,18). The underlying mechanisms by which
mutations in cell-cell junction proteins affect electric synchrony
remains undefined.
Results of the current study indicate a correlation
between three components of the intercalated disk, desmosomes, gap
junctions and the voltage-gated sodium channel Nav1.5
complex. This is the first association identified between DSP
expression and function of the sodium channel complex,
demonstrating that the molecular integrity of mechanical
connections may be associated with sodium channel function.
Additional studies are required to understand the pathogenesis of
ventricular arrhythmias in ARVC. However, current observations are
likely to be a stepping stone towards identification of a treatment
for ARVC patients.
Acknowledgements
The study was supported by grants from Guangdong
Nature Science Foundation to Shulin Wu (no. 10151008002000011).
References
|
1
|
Noorman M, van der Heyden MA, van Veen TA,
et al: Cardiac cell-cell junctions in health and disease:
Electrical versus mechanical coupling. J Mol Cell Cardiol.
47:23–31. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bauce B, Basso C, Rampazzo A, et al:
Clinical profile of four families with arrhythmogenic right
ventricular cardiomyopathy caused by dominant desmoplakin
mutations. Eur Heart J. 26:1666–1675. 2005. View Article : Google Scholar
|
|
3
|
Rampazzo A, Nava A, Malacrida S, et al:
Mutation in human desmoplakin domain binding to plakoglobin causes
a dominant form of arrhythmogenic right ventricular cardiomyopathy.
Am J Hum Genet. 71:1200–1206. 2002. View
Article : Google Scholar
|
|
4
|
Norman M, Simpson M, Mogensen J, et al:
Novel mutation in desmoplakin causes arrhythmogenic left
ventricular cardiomyopathy. Circulation. 112:636–642. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaplan SR, Gard JJ, Carvajal-Huerta L, et
al: Structural and molecular pathology of the heart in Carvajal
syndrome. Cardiovasc Pathol. 13:26–32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oxford EM, Musa H, Maass K, et al:
Connexin43 remodeling caused by inhibition of plakophilin-2
expression in cardiac cells. Circ Res. 101:703–711. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roden DM, Balser JR, George AL JR and
Anderson ME: Cardiac ion channels. Annu Rev Physiol. 64:431–475.
2002. View Article : Google Scholar
|
|
8
|
Abriel H: Cardiac sodium channel Na(v)1.5
and interacting proteins: Physiology and pathophysiology. J Mol
Cell Cardiol. 48:2–11. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peters S, Trümmel M, Denecke S and Koehler
B: Results of ajmaline testing in patients with arrhythmogenic
right ventricular dysplasia-cardiomyopathy. Int J Cardiol.
95:207–210. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peters S: Arrhythmogenic right ventricular
dysplasia-cardiomyopathy and provocable coved-type ST-segmen
elevation in right precordial leads: clues from long-term
follow-up. Europace. 10:816–820. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chaudary N, Shuralyova I, Liron T, et al:
Transport characteristics of HL-1 cells: A new model for the study
of adenosine physiology in cardiomyocytes. Biochem Cell Biol.
80:655–665. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Claycom WC, Lanson NA Jr, Stallworth BS,
et al: Hl-1 cells: A cardiac muscle cell line that contracts and
retains phenotypic characteristics of the adult cardiomyocyte. Proc
Natl Acad Sci USA. 95:2979–2984. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stokes DL: Desmosomes from a structural
perspective. Curr Opin Cell Biol. 19:565–571. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shaw RM, Fay AJ, Puthenveedu MA, et al:
Microtubule plus-end-tracking proteins target gap junctions
directly from the cell interior to adherens junctions. Cell.
128:547–560. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Plaisier AS, Burgmans MC, Vonken EP, et
al: Image quality assessment of the right ventricle with three
different delayed enhancement sequences in patients suspected of
ARVC/D. Int J Cardiovasc Imaging. 28:595–601. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Derangeon M, Montnach J, Baró I and
Charpentier F: Mouse models of SCN5A-related cardiac arrhythmias.
Frontiers Physiol. 3:2102003.PubMed/NCBI
|
|
17
|
Gomes J, Finlay M, Ahmed AK, et al:
Electrophysiological abnormalities precede overt structural changes
in arrhythmogenic right ventricular cardiomyopathy due to mutations
in desmoplakin-a combined murine and human study. Eur Heart J.
33:1942–1953. 2012. View Article : Google Scholar
|
|
18
|
Delmar M and McKenna WJ: The cardiac
desmosome and arrhythmogenic cardiomyopathies: from gene to
disease. Circ Res. 107:700–714. 2010. View Article : Google Scholar : PubMed/NCBI
|