Introduction
Non-small cell lung cancer (NSCLC) accounts for
~80–85% of all lung cancer cases, and is the most common cause of
worldwide cancer mortality (1).
Platinum-based combination regimens offer a modest, but
significant, survival advantage to NSCLC patients with advanced or
metastatic disease, although the majority of patients eventually
experience disease progression (2). Lung cancer is a heterogeneous disease
with multiple signaling pathways influencing tumor cell survival
and proliferation, and it is likely that blocking only one of these
pathways allows others to act as salvage or escape mechanisms for
cancer cells (3–5). Whether combined inhibition therapy
has greater antitumor activity compared with single inhibition
therapy is a matter of debate.
Sorafenib is a multi-kinase inhibitor that was
originally developed as an inhibitor of RAF-1, a component of the
extracellular signal-regulated kinase 1/2 (ERK1/2) pathway, but
which was subsequently shown to inhibit multiple other kinases,
including class III tyrosine kinase receptors, such as
platelet-derived growth factor, vascular endothelial growth factor
receptors 1 and 2, c-Kit and FLT3. Sorafenib is currently approved
for the treatment of metastatic renal cell carcinoma and for
advanced hepatocellular carcinoma (6,7).
Molecularly-targeted therapies, including sorafenib,
which disrupts molecular defects in signaling pathways, may provide
clinical benefits in the treatment of NSCLC (8). The present study aimed to
systematically characterize the in vitro effects of
sorafenib on proliferation, apoptosis and intracellular signaling,
using two NSCLC cell lines.
Materials and methods
Cell culture and reagents
The A549 and NCI-H1975 human lung cancer cell lines
were purchased from the American Type Culture Collection (Manassas,
VA, USA). Cells were maintained in Dulbecco’s modified Eagle’s
medium supplemented with 10% fetal calf serum, L-glutamine (5
mmol/l), non-essential amino acids (5 mmol/l), penicillin (100
U/ml), and streptomycin (100 U/ml) (Invitrogen Life Technologies,
Carlsbad, CA, USA) at 37°C in a humidified 5% CO2
atmosphere. The mitogen-activated or extracellular signal-regulated
protein kinases 1 and 2 (MEK1/2) inhibitor, U0126, was obtained
from Cell Signaling Technology (Beverly, MA, USA). Sorafenib was
purchased from Bayer Corporation (West Haven, CT, USA) and
dissolved in dimethyl sulfoxide (DMSO) with cell medium to the
given concentration, with a final DMSO concentration of 0.1%.
Inhibitor treatment
Stock solutions of sorafenib and U0126 were prepared
at appropriate concentrations in DMSO and stored at −20°C. For
treatment, inhibitor solutions were diluted 1:1,000 to appropriate
working concentrations (5 or 20 μmol/l cyclopamine and 3 μmol/l
U0126) in serum-free medium. Control cultures received medium
containing the solvent DMSO at a concentration of 0.1%.
Cell viability assay
Cell proliferation was determined using an MTT
viability assay, the most commonly used assay for determining cell
growth and death. The MTT survival assay has been described in
detail in previous studies (9).
Exponentially growing cells were recultured (5,000 cells/well)
overnight in 96-well tissue culture plates. Up to 20 μl MTT
(Sigma-Aldrich, St. Louis, MO, USA) was directly added to the media
in each well, with a final concentration of 2 mg/ml. After 4 h
incubation, the medium containing MTT was discarded, and 120 μl
DMSO was added for 10 min. The absorbance was measured using an
enzyme-linked immunosorbent assay reader (Bio-Tek, Inc., Winooskie,
VT, USA) at 570 nm, with the absorbance at 630 nm as the background
correction. The cell viability is expressed as the percentage of
untreated controls. All experiments were performed at least three
times.
Apoptosis assay
Apoptosis analysis by a flow cytometer Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) kit (BD
Biosciences, Sparks, MD, USA) was used to measure the percentage of
apoptosis induced by sorafenib. Cells were removed with trypsin and
collected into centrifuge tubes together with the culture medium. A
total of 5 μl Annexin V-FITC solution and 10 μl PI (1 μg/ml) were
added to these cells for 30 min in the dark. Flow cytometry and
Annexin V-FITC apoptosis analysis were performed as previously
described (10). Apoptotic rates
were calculated from 10,000 cells using ModFit LT software with
FACSCalibur (both Becton Dickinson, San Jose, CA, USA).
Cell cycle assays
The cells were removed with trypsin and collected in
centrifuge tubes, together with the culture medium. The contents
were centrifuged for 5 min at 1,800 × g. The supernatant was poured
out, washed once with 1X phosphate-buffered saline (PBS), and
centrifuged for a subsequent 5 min. The cells were then fixed with
5 ml pre-cooled 70% ethanol for at least 4 h. The fixed cells were
centrifuged and washed with 1X PBS. Following centrifugation, the
cell pellets were resuspended in 500 μl PI (10 μg/ml) containing
300 μg/ml RNase (Sigma-Aldrich). The cells were incubated on ice
for 30 min, and filtered with a 53 μm nylon mesh. The cell cycle
distribution was calculated from 10,000 cells using ModFit LT
software (Becton Dickinson) using FACSCalibur.
Western blot assay
Cell lysates were prepared and western blot analysis
was performed as previously described (10). Equal aliquots of total cell protein
(50 μg/lane) were electrophoresed on sodium dodecyl
sulfate-polyacrylamide gels, transferred onto polyvinylidene
fluoride membranes, and blotted using the primary antibodies:
β-actin (C-4), cyclin D1 (A-12), cyclin-dependent kinase (CDK)2
(M2), nuclear factor (NF)-κB (P65A), matrix metallopeptidase
(MMP)-2 (2C1), MMP-9 (6-6B), BAX (P-19) and Bcl-2 (C-2; all 1:1,000
dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
followed by the secondary antibodies horseradish peroxidase-labeled
goat anti-mouse (GAM-007) and goat anti-rabbit (SC-2004) IgG, and
phospho-ERK1/2 (T202/Y204) and ERK1/2 (1:1,000 dilution; Cell
Signaling Technology, Beverly, MA, USA). The protein bands were
visualized using an enhanced chemiluminescence system (Union
Bioscience Corporation, Hangzhou, China) with prestained markers as
molecular size standards. At least three independent experiments
were performed for each cell type studied.
Statistical analysis
Data are presented as the mean ± standard deviation.
Experimental results of the treated and control groups were
compared using two-tailed Student’s t-test. All statistical tests
were performed using SPSS version 17.0 (SPSS Inc., Chicago, IL,
USA). P≤0.05 was considered to indicate a statistically significant
difference.
Results
Sorafenib decreases breast cancer cell
proliferation
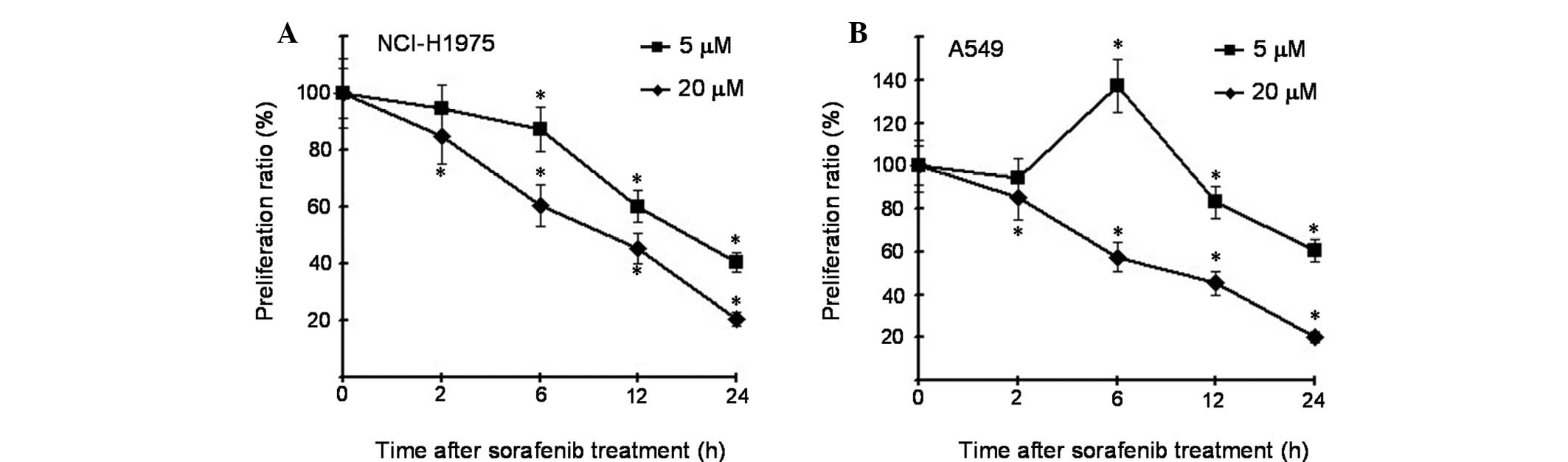
MTT viability assays were conducted to elucidate the
potential biological effects of sorafenib in different lung cancer
cells. As shown in Fig. 1A, the
NCI-H1975 cells treated with cyclopamine exhibited significant
reduction in proliferation rates compared with the control cells at
the two concentrations (5 and 20 μM), and sorafenib also markedly
inhibited A549 cells at a concentration of 20 μM. However, at a
concentration of 5 μM and 6 h time point, the proliferation rate
was increased compared with the control cells (Fig. 1B).
Effect of sorafenib on cell cycle
distribution and apoptosis in different cells
To investigate the different proliferation results
in sorafenib-treated lung cancer cells, cell cycle proliferation
and apoptosis was examined by flow cytometry. The results showed
that sorafenib decreased the proportion of A549 cells in the G1
phase of the cell cycle and increased that of cells in the S phase
(Fig. 2A), and the cell cycle
redistribution was not observed in NCI-H1975 cells at this time
point (Fig. 2B). The apoptosis
assay revealed that sorafenib decreased the proportion of apoptotic
cells in A549 cells at the 6 h time point, which was not observed
in NCI-H1975 cells (Fig. 2C and
D). These data indicate that cell cycle distribution and
apoptosis were involved in the proliferation of sorafenib-treated
lung cancer cells.
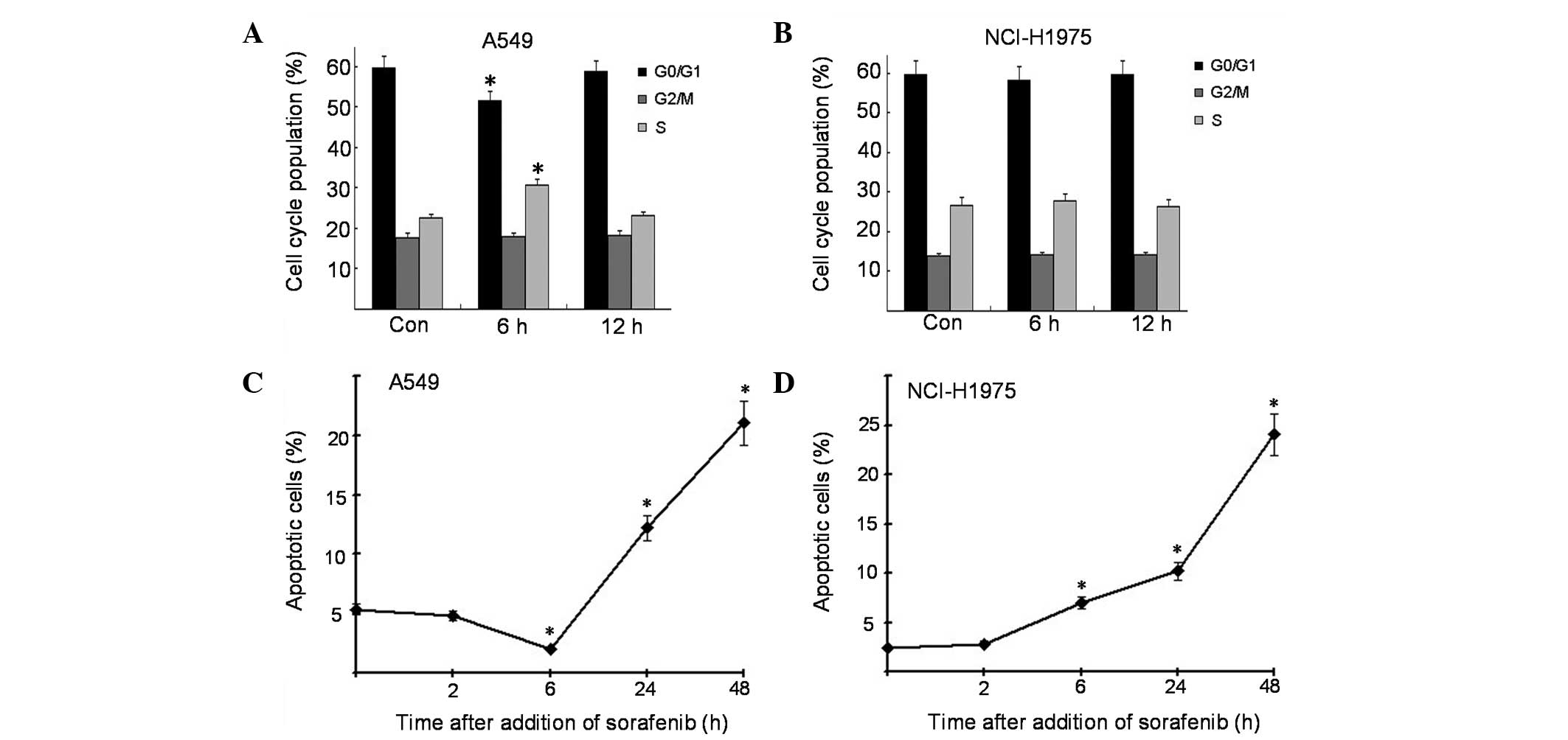 | Figure 2Effects of sorafenib on cell cycle
distribution and apoptosis in NSCLC cells. (A and B) Cells were
incubated for 0, 6, or 12 h in complete culture medium containing 5
μM sorafenib and were fixed, stained with PI, and analyzed for cell
cycle distribution by flow cytometry. (C and D) Cells were
incubated for 0, 2, 6, 24 or 48 h in complete culture medium
containing 5 μM sorafenib and were fixed, stained with Annexin
V-FITC/PI and analyzed for apoptosis by flow cytometry. All samples
were prepared in triplicate. *P<0.05, vs. the control
group. NSCLC, non-small-cell lung cancer; PI, propidium iodide;
FITC, fluorescein isothiocyanate. |
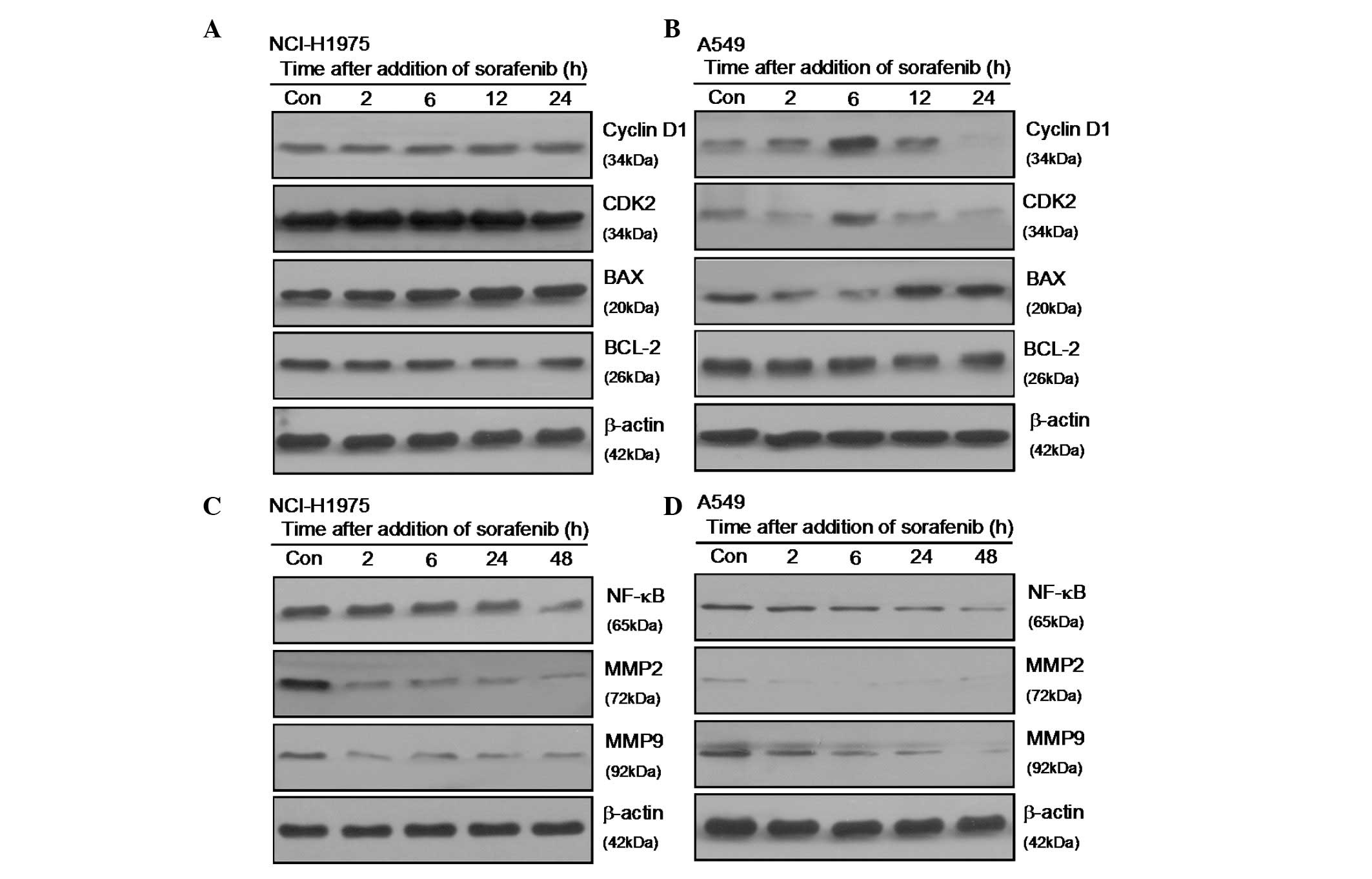
Sorafenib affects the expression of cell
cycle, invasion and apoptosis-associated proteins
The alterations in the cell cycle, invasion- and
apoptosis-associated proteins in sorafenib-treated cells were
evaluated and compared with the control cells using western blot
analysis. As shown in Fig. 3A, the
expression levels of cyclin D1, CDK2, NF-κB, MMP2, MMP9 and Bcl 2
decreased significantly in sorafenib-treated NCI H1975 cells. In
A549 cells, sorafenib significantly enhanced the expression of
cyclinD1 and CDK2, and suppressed BAX expression at the 6 h time
point (Fig. 3B). With regard to
invasion-associated proteins, there was no difference between the
two cell lines (Fig. 3C and D).
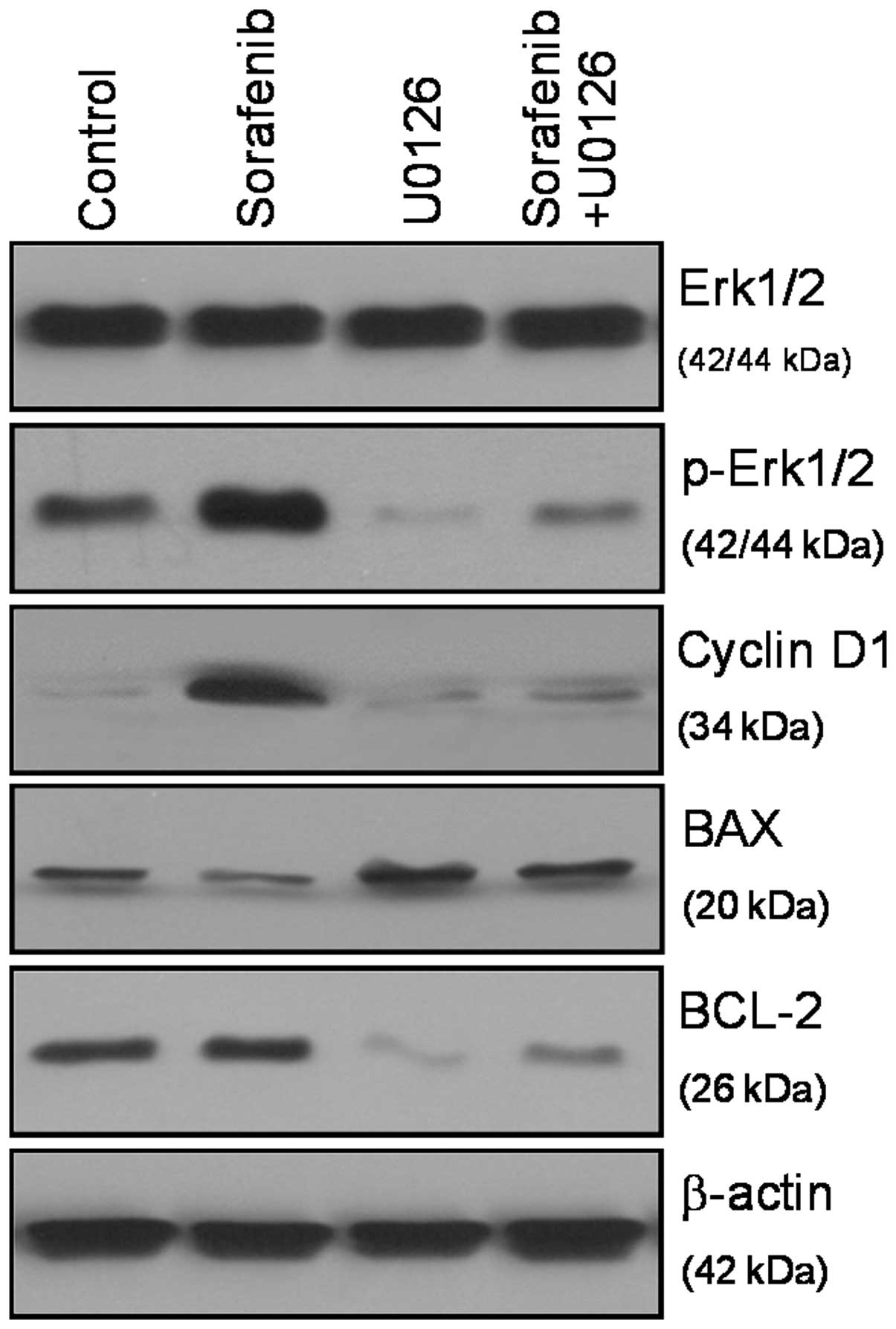
Sorafenib regulates the proliferation and apoptosis of A549 cells
by activating the ERK1/2 signaling pathway. To examine the effects
of sorafenib on the ERK1/2 signaling pathway in A549 cells,
immunoblot analysis was performed with antibodies specific for
phosphorylated ERK1/2. The results of this study reveal that
sorafenib markedly enhanced ERK phosphorylation in A549 cells at
the 5 μM concentration and 6 h time point, but had no marked effect
on the expression of this protein (Fig. 4). Phosphorylation of ERK1/2 is a
key initial response to cell cycle distribution and apoptosis. With
regard to downstream proteins, sorafenib markedly upregulated the
expression of cyclin D1, and inhibited the expression of BAX at the
same concentration and time point. The specific signaling cascade
involved in this response was investigated using the MEK1/2
inhibitor (U0126) specific to the MAPK/ERK pathway. The increased
phospho-ERK1/2 following sorafenib-treatment was inhibited by
U0126, as well as the downstream protein cyclin D1, Bcl-2 and BAX
(Fig. 4), which suggests that
sorafenib mediates cell cycle distribution and apoptosis by
modulating the ERK signaling pathway.
Discussion
NSCLC remains a leading cause of mortality worldwide
among patients diagnosed with malignancies (11). The standard therapeutic concept of
lung cancer is based on cisplatin chemotherapy. However, during the
last few decades, no significant improvement in the prognosis of
patients with lung cancer has been achieved (12).
Sorafenib, an oral multi-kinase inhibitor,
inhibiting serine/threonine Raf kinases and RTKs, including
vascular endothelial growth factor receptor and platelet-derived
growth factor receptor-b, is currently used for the therapy of
advanced renal carcinoma. Furthermore, antitumor activity and an
improved outcome have also been observed for patients with other
carcinomas, including hepatocellular carcinoma and NSCLC (6,7,13–16).
In the present study, it was observed that at a low
concentration and early time point, sorafenib is capable of
significantly stimulating the proliferation of A549 cells, which
was not observed in NCI-H1975 cells. The cell cycle assay showed
that sorafenib decreased the proportion of A549 cells in the G1
phase of the cell cycle, and increased that of cells in the S
phase, which was different with NCI-H1975 cells. The apoptosis
assay also revealed that sorafenib decreased the proportion of
apoptotic cells in A549 cells at a specific time point.
Phosphorylated ERK is a key downstream target of the
Ras/Raf/MEK/ERK signaling pathway, and dysregulation of this
pathway occurs in approximately one-third of all human cancers
(17). ERK is the terminal kinase
in this pathway. During the phosphorylation cascade, ERK1/2 is
activated by mitogen-activated protein kinases. Mitogen-stimulated
cell growth, through survival/apoptosis and cell cycle control, is
frequently linked to phosphorylated ERK translocation from the
cytoplasm to the nucleus and to the initiation of transcriptional
activation of cell regulatory genes (18,19).
Therefore, the current study focused on the effects of sorafenib on
the phosphorylation status of ERK1/2. As described in the results,
high concentrations of sorafenib decreased the phosphorylation
levels of ERK-1/2. Notably, a significant stimulatory effect of
sorafenib was observed at low concentrations on ERK-1/2
phosphorylation. To further elucidate the underlying signaling
pathways, the MEK inhibitor U0126 was used. The results showed that
the increased phospho-ERK1/2 following sorafenib-treatment was
inhibited by U0126, as well as the downstream protein cyclin D1,
Bcl-2 and BAX, which suggests that sorafenib mediates cell cycle
distribution and apoptosis by modulating the ERK signaling
pathway.
The current observations demonstrate the
antimigratory and antiproliferatory effects of sorafenib in
different human NSCLC cells. However, the stimulatory effects at
low concentrations of sorafenib in certain cancer cells may have
marked consequences for this antitumor drug. This may be
particularly relevant for selecting dosing regimens that avoid low
plasma concentrations of sorafenib in order to optimize the
treatment with this promising antitumor drug.
References
|
1
|
Kyle F and Spicer J: Targeted therapies in
non-small cell lung cancer. Cancer Imaging. 8:199–205. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ray MR, Jablons D and He B: Lung cancer
therapeutics that target signaling pathways: an update. Expert Rev
Respir Med. 4:631–645. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang X, Li Y, Li H, et al: Combined EGFR
and VEGFR versus single EGFR signaling pathways inhibition therapy
for NSCLC: a systematic review and meta-analysis. PLoS One.
7:e401782012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Larsen JE, Cascone T, Gerber DE, et al:
Targeted therapies for lung cancer: clinical experience and novel
agents. Cancer J. 17:512–527. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pal SK, Figlin RA and Reckamp K: Targeted
therapies for non-small cell lung cancer: an evolving landscape.
Mol Cancer Ther. 9:1931–1944. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ramakrishnan V, Timm M, Haug JL, et al:
Sorafenib, a multikinase inhibitor, is effective in vitro against
non-Hodgkin lymphoma and synergizes with the mTOR inhibitor
rapamycin. Am J Hematol. 87:277–283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilhelm S, Carter C, Lynch M, et al:
Discovery and development of sorafenib: a multikinase inhibitor for
treating cancer. Nat Rev Drug Discov. 5:835–844. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wilhelm SM, Adnane L, Newell P, et al:
Preclinical overview of sorafenib, a multikinase inhibitor that
targets both Raf and VEGF and PDGF receptor tyrosine kinase
signaling. Mol Cancer Ther. 7:3129–3140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiao Y, Sun KK, Zhao L, et al: Suppression
of human lung cancer cell proliferation and metastasis in
vitro by the transducer of ErbB-2.1 (TOB1). Acta Pharmacol Sin.
33:250–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun KK, Zhong N, Yang Y, Zhao L and Jiao
Y: Enhanced radiosensitivity of NSCLC cells by transducer of
erbB2.1 (TOB1) through modulation of the MAPK/ERK pathway. Oncol
Rep. 29:2385–2391. 2013.PubMed/NCBI
|
|
11
|
Scagliotti G, Novello S, von Pawel J, et
al: Phase III study of carboplatin and paclitaxel alone or with
sorafenib in advanced non-small-cell lung cancer. J Clin Oncol.
28:1835–1842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bearz A, Berretta M, Lleshi A and Tirelli
U: Target therapies in lung cancer. J Biomed Biotechnol.
2011:9212312011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rose A, Grandoch M, vom Dorp F, et al:
Stimulatory effects of the multi-kinase inhibitor sorafenib on
human bladder cancer cells. Br J Pharmacol. 160:1690–1698. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bareford MD, Park MA, Yacoub A, et al:
Sorafenib enhances pemetrexed cytotoxicity through an
autophagy-dependent mechanism in cancer cells. Cancer Res.
71:4955–4967. 2011. View Article : Google Scholar
|
|
15
|
Takahashi O, Komaki R, Smith PD, et al:
Combined MEK and VEGFR inhibition in orthotopic human lung cancer
models results in enhanced inhibition of tumor angiogenesis,
growth, and metastasis. Clin Cancer Res. 18:1641–1654. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takezawa K, Okamoto I, Yonesaka K, et al:
Sorafenib inhibits non-small cell lung cancer cell growth by
targeting B-RAF in KRAS wild-type cells and C-RAF in KRAS mutant
cells. Cancer Res. 69:6515–6521. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marampon F, Gravina GL, Di Rocco A, et al:
MEK/ERK inhibitor U0126 increases the radiosensitivity of
rhabdomyosarcoma cells in vitro and in vivo by downregulating
growth and DNA repair signals. Mol Cancer Ther. 10:159–168. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Klein PJ, Schmidt CM, Wiesenauer CA, et
al: The effects of a novel MEK inhibitor PD184161 on MEK-ERK
signaling and growth in human liver cancer. Neoplasia. 8:1–8. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shukla A, Hillegass JM, MacPherson MB, et
al: ERK2 is essential for the growth of human epithelioid malignant
mesotheliomas. Int J Cancer. 129:1075–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|