Introduction
Heat shock treatment protects against a variety of
cell and tissue injuries, including sepsis, acute lung injury,
ischemia reperfusion injury and endotoxin-mediated apoptosis
(1). One of the mechanisms by
which heat shock treatment induces tolerance is via the inhibition
of key signaling pathways, including nuclear factor κB (NF-κB),
which transcriptionally regulate stress-response genes. Heat shock
treatment has been demonstrated to inhibit activation of the NF-κB
pathway by mechanisms involving inhibition of the inhibitor of
NF-κB (IκB) kinase activation (2–4).
Heat shock has also been demonstrated to increase the mRNA
stability of a number of genes (5,6).
NF-κB is a critical transcription factor involved in
a broad range of biological processes, including immune responses,
cell survival, stress responses and maturation of various cell
types. NF-κB activation is required to protect organisms from
environmental factors, however, deregulated NF-κB activity has been
observed in various diseases, including chronic inflammation and
cancer. Thus, understanding the regulation of NF-κB signaling is
important for maintaining human health.
The IκB proteins are important for the regulation of
NF-κB signaling. The classical inhibitor proteins in the NF-κB
signaling system consist of the single polypeptide IκBs, IκB-α,
IκB-β and IκB-ɛ. In resting cells, IκB binds and sequesters the
NF-κB dimer and prevents DNA binding and transcriptional
activation. Stimulus-responsive activation of the IκB kinase
results in the degradation of the IκBs to release and activate
NF-κB (4).
From these observations, the mechanism by which heat
shock treatment modulates NF-κB signaling cascades was
investigated, in particular the Iκ-Bα gene expression with a focus
on post-transcriptional regulation.
Materials and methods
Cell culture and heat shock
NIH 3T3 and RAW264.7 murine macrophages (American
Type Culture Collection, Manassas, VA, USA) were cultured in DMEM
(Gibco-BRL, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (Gibco-BRL) in a 95% room air-5% CO2 incubator at
37°C. Heat shock treatment was performed by incubating cells at
43°C for 1 h in a 95% room air-5% CO2 incubator with
subsequent return to 37°C.
Plasmid construction
Mouse (flag)-tagged HuR and the pRL-TK plasmid were
used. Full-length HuR mRNA 3′UTR was amplified using specific
primers and cloned into the pcDNA3-flag vector (Promega, Madison,
WI, USA) using the KpnI and XbaI sites. The following
primers were used: sense, 5′-GTGGTACCA TGTCTAATGGTTATGAAGACCAC-3′
and anti-sense, 5′-GCTCTAGAGCTGTCTGTAAAAATCT GTTTAAT-3′.
Full-length IκB-α mRNA 3′UTR was amplified using specific primers
and cloned into the pGL3 vector (Promega) using the XbaI and
NcoI sites and named pLuNB. The following primers were used:
sense, 5′-ATCGCCGTGTAATGGAAAGTGGCAAAAAG AATG-3′ and anti-sense,
5′-GCTCTAGAGCTGTCTGTAAA AATCTGTTTAAT-3′.
Transient transfection
pLuNB and control plasmids were transfected using
Lipofectamine 2000™ (Invitrogen Life Technologies, Carlsbad, CA,
USA). Transfected cells were used for analysis 48 h following
transfection.
Luciferase assays
Cells were transfected and cultured in six-well
plates at a density of 3×105 cells/well. Following 1 h
heat shock, cells were washed with PBS once and cellular proteins
were extracted and analyzed for luciferase activity according to
the manufacturer’s instructions (Promega GmbH) with the use of a
luminometer (SpectarMax M5, Molecular Devices, Sunnyvale, CA, USA).
Luciferase activity was corrected for total cellular protein and
reported as relative induction over respective control cells (cells
that were transfected with pGL3 control plasmid).
RNA interference
A mixture of small interfering RNAs (siRNAs; 25 nM;
GenePharma, Shanghai, China) specifically targeting HuR was
transfected into cells using Lipofectamine 2000. The following sets
of primers were used: Elavl1-mus-360 sense,
5′-GCGAGGUUGAAUCUGCAAATT-3′ and antisense,
5′-UUUGCAGAUUCAACCUCGCTT-3′; Elavl1-mus-1099 sense,
5′-GACCAUGACAAACUAUGAATT-3′ and antisense,
5′-UUCAUAGUUUGUCAUGGUCTT-3′; Elavl1-mus-522 sense,
5′-CAAGCUCAGAGGUCAUCAATT-3′ and antisense
5′-UUGAUGACCUCUGAGCUUGTT-3′.
RNP immunoprecipitation (IP) assays
For the IP assay of endogenous RNA-protein complexes
from whole-cell extracts, cell lysates were incubated with protein
A-Sepharose beads (Sigma-Aldrich, St. Louis, MO, USA) that had been
precoated with 30 μg mouse immunoglobulin G1 (IgG1; BD Biosciences
San Jose, CA, USA) or antibodies recognizing HuR (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) for 3 h at 4°C. Beads
were washed with NT2 buffer [50 mM Tris-HCl (pH 7.4), 150 mM
NaCl2, 1 mM MgCl2 and 0.05% Nonidet P-40],
incubated with 20 units RNase-free DNase I (15 min; 30°C) and
further incubated in 100 μl NT2 buffer containing 0.1% SDS and 0.5
mg/ml proteinase K (30 min; 55°C).
RNA isolated from the IP material was reverse
transcribed using random hexamers and SSII reverse transcriptase
(Invitrogen Life Technologies). Transcription abundance was
measured by amplification of cDNA, using quantitative PCR (qPCR)
employing SYBR-Green PCR master mix (Applied Biosystems China,
Beijing, China). PCR primers for the detection of IκB-α and β-actin
were as follows: IκB-α sense, 5′-GAAGAGAAGCCGCTGACCAT-3′ and
antisense, 5′-CAGAAGTGCCTCAGCAATTCC-3′; β-actin sense,
5′-ACGGCCAGGTCATCACTATTG-3′ and antisense,
5′-AGAGGTCTTTACGGATGTCAACGT-3′.
Biotin pull-down analysis
For in vitro synthesis of biotinylated
transcripts, cDNA from RAW264.7 cells was used as a template for
PCR, whereby the T7 RNA polymerase promoter sequence
[CCAAGCTTCTAATACGACTCACTATAGGGAGA(T7)] was added to the 5′ end of
all fragments. The primers used for the preparation of biotinylated
transcripts spanning the 3′UTR of IκB-α (GenBank accession, no.
NM_004417) and GAPDH were as follows: primers (T7),
5′-ATCGCCGTGTAATGGAAAGTGGCAAAAAGAATG-3′ and
5′-GCTCTAGAGCTGTCTGTAAAAATCTGTTT AAT-3′, were used for preparing
the IκB-α 3′UTR and primers (T7), 5′-CAGCAAGAGCACAAGAGGAA-3′ and
5′-AGGGGTCTACATGGCAACTG-3′ were used to prepare the GAPDH
transcripts. Biotinylated RNAs were synthesized using a MaxiScript
T7 kit (Ambion, Carlsbad, CA, USA). Whole-cell lysates were
incubated with 2 μg purified biotinylated transcripts for 1 h at
room temperature. Complexes were isolated with paramagnetic
streptavidin-conjugated Dynabeads (Dynal®, Invitrogen,
Carlsbad, CA, USA) and bound proteins in the pulldown material were
assayed by western blotting using antibodies against HuR (Santa
Cruz Biotechnology Inc.).
mRNA stability assays
Cells were subjected to heat shock treatment for 1
h. Following heat shock, the cells were treated with actinomycin D
(5 μg/ml; Sigma-Aldrich) to induce transcriptional arrest. Total
RNA was harvested at 0.5 h intervals. Control cells were maintained
at 37°C, treated with actinomycin D and harvested for total RNA
isolation at 0.5 h intervals. Total RNA samples were subjected to
qPCR as previously described (3).
Nuclear protein extraction
Nuclear extraction procedures were performed on ice.
Cells were washed twice with PBS, harvested in 1 ml PBS and
pelleted at 3,300 × g for 5 min. The pellet was washed twice with
PBS, resuspended in one packed cell volume of lysis buffer [10 mM
HEPES, (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 1.5 mM MgCl2,
0.2% v/v Nonidet P-40, 1 mM DTT and 0.1 mM PMSF] and incubated for
5 min. Following centrifugation at 6,000 rpm, one cell pellet
volume extraction buffer [20 mM HEPES, (pH 7.9), 420 mM NaCl, 0.1 M
EDTA, 1.5 mM MgCl2, 25% v/v glycerol, 1 mM DTT and 0.5
mM PMSF] was added to the nuclear pellet and incubated on ice for
15 min with occasional vortexing. Nuclear proteins were isolated by
centrifugation at 14,000 rpm for 15 min. Protein concentrations
were determined by the Bradford assay (Bio-Rad, Hercules, CA, USA)
and stored at −80°C for western blot analysis.
Protein stability assays
CHX (10 μg/ml; Sigma-Aldrich) was added to the
culture medium 1 h following heat shock treatment and at the
indicated time points (0.5, 1, 2, 3 and 4 h), cells were collected
and protein stability was analyzed by immunoblotting.
Western blot analysis
Cells were harvested and washed twice with PBS.
Harvested cells were lysed and 80 μg total protein was separated by
SDS-PAGE on 10% polyacrylamide gels and transferred onto
nitrocellulose membranes. Following 1 h blocking with 5% skimmed
milk in TBS buffer (10 mM Tris and 150 mM NaCl), membranes were
incubated with primary antibodies at 4°C overnight. Membranes were
washed four times for 15 min with TBST buffer (10 mM Tris, 150 mM
NaCl and 0.1% Tween 20) and incubated with the appropriate
HRP-conjugated secondary antibody for 1 h at room temperature.
Protein bands were detected using an enhanced chemiluminescene
western blotting detection kit (Amersham, Buckinghamshire, UK). All
antibodies were purchased from Santa Cruz Biotechnology.
Immunofluorescence
Cells were washed with ice-cold PBS and fixed for 10
min using 4% formaldehyde in PBS. The cells were permeabilized
using 0.1% Triton X-100 in PBS for 5 min, washed with ice-cold PBS
and blocked with 5% bovine serum albumin in PBS for 1 h at 37°C.
Subsequently, cell preparations were incubated with anti-HuR or
anti-G3BP1 antibodies diluted in 5% bovine serum albumin at 4°C
(Santa Cruz Biotechnology) and underwent additional washes with
ice-cold PBS, cells were incubated with a secondary antibody (Santa
Cruz Biotechnology), washed with PBS and incubated with a solution
of DAPI for 10 min. Following preparation, cells were washed
thoroughly and were embedded using ProLong Gold antifade reagent
(Invitrogen Life Technologies). Images were captured using a
fluorescence microscope (Zeiss Axiovert 35).
Statistical analysis
Data were presented as mean ± SD of three
independent assays. The statistical analysis was conducted by
one-way ANOVA. P<0.05 was considered to indicate a statistically
significant difference.
Results
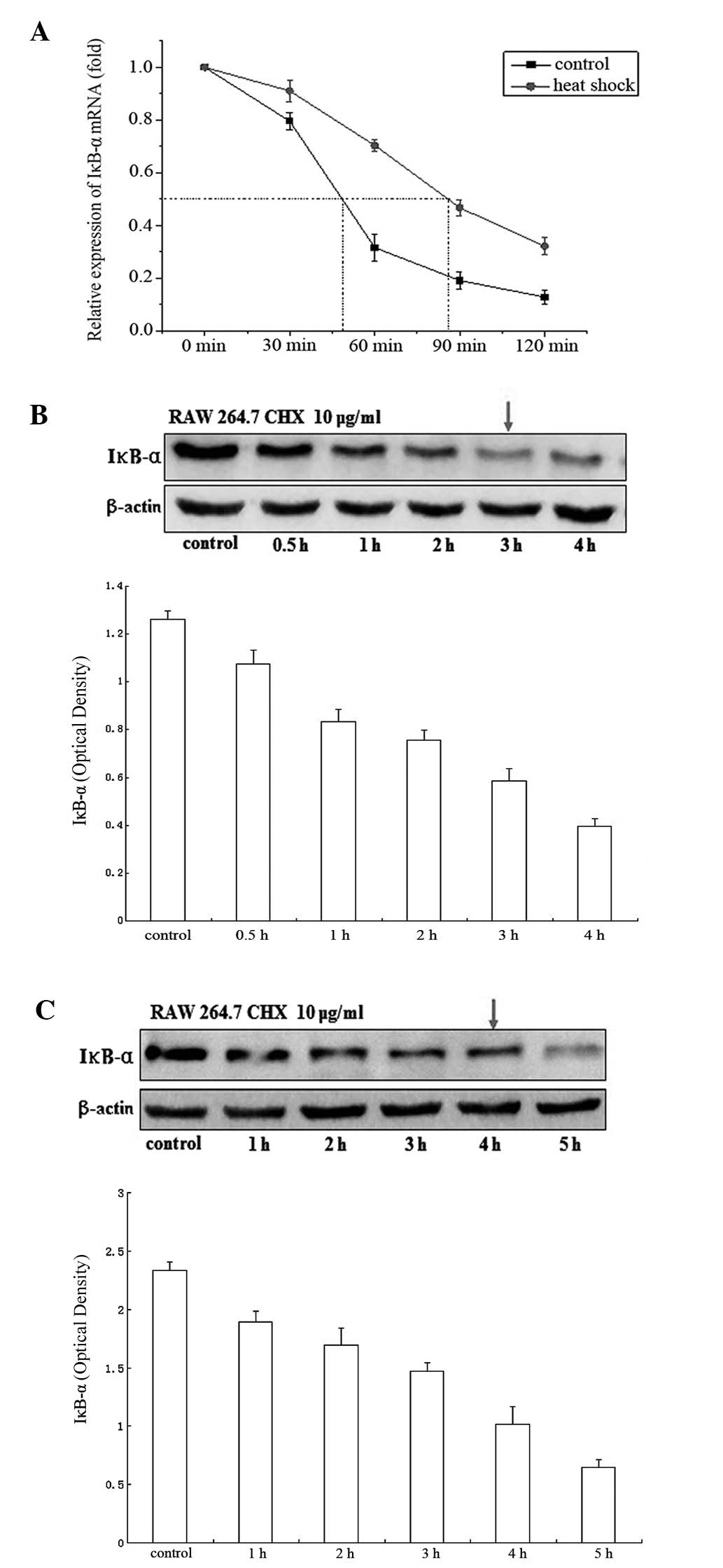
Heat shock increases IκB-α mRNA and
protein stability
To explore the effect of heat shock on IκB-α,
RAW264.7 murine macrophages were subjected to heat shock treatment.
RNA and protein were extracted and examined for IκB-α mRNA and
protein stability by qPCR and western blotting, respectively. The
results showed that the IκB-α mRNA had a half-life of ~48 min in
untreated cells and this increased to ~80 min following heat shock
treatment (Fig. 1A). IκB-α protein
had a half-life of ~3 h (Fig. 1B)
which increased to ~4 h (Fig. 1C)
following heat shock treatment. The results indicate that treatment
with heat shock potently stabilizes IκB-α mRNA and proteins.
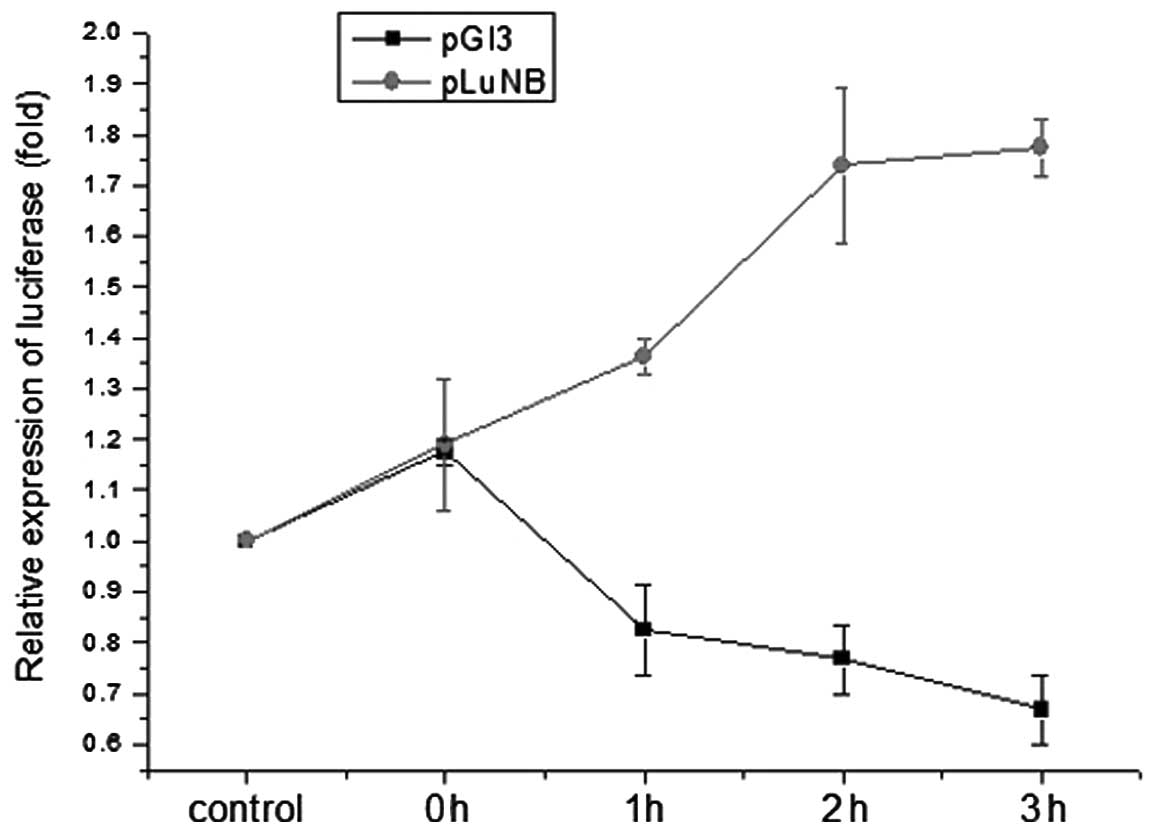
3′UTR IκB-α mRNA is important for
regulating the function of IκB-α upon heat shock treatment
3′-UTR mRNA may regulate the mRNA stability and
translation efficiency, thus, the mechanism by which the 3′UTR of
IκB-α mRNA regulates the function of IκB-α upon heat shock
treatment was investigated. RAW264.7 cells were transiently
transfected with a 3′UTR IκB-α luciferase reporter plasmid and
luciferase activity was measured in cells subjected to heat shock
treatment. Exposure to heat shock significantly increased
luciferase activity in transfected cells compared with cells
transfected with the control plasmid (Fig. 2). These observations demonstrate
that the 3′UTR of IκB-α mRNA exerts an important role in regulating
the function of IκB-α upon heat shock treatment.
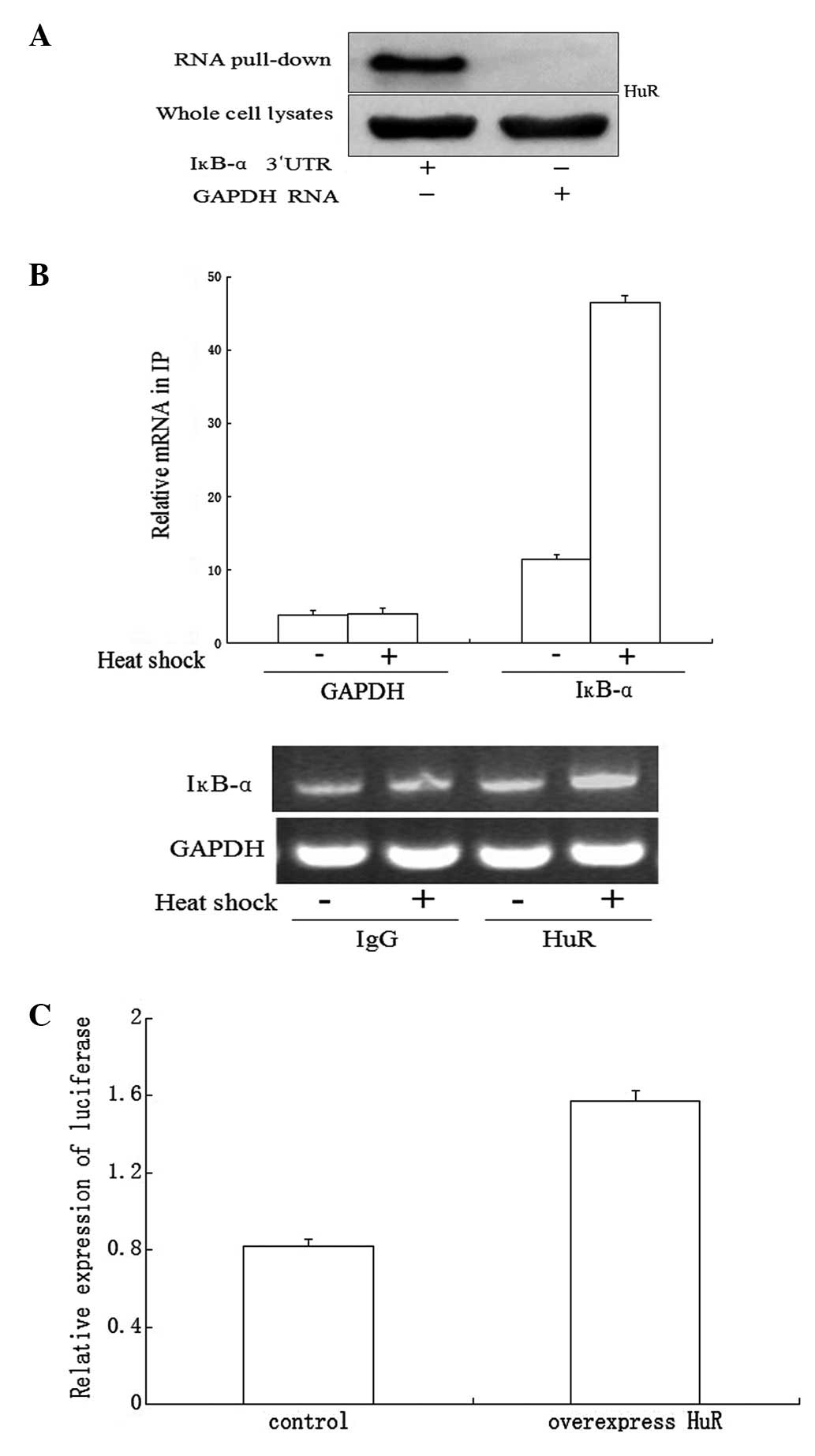
HuR binds to endogenous and recombinant
IκB-α mRNA
The IκB-α mRNA has a long AU-rich 3′UTR containing a
number of predicted hits of a previously identified HuR motif.
Thus, we determined whether IκB-α mRNA is the target of HuR in mRNA
stabilization and/or translation and if such RNA protein
associations changed in a heat shock-dependent manner. First, the
existence of such RNA binding protein was analyzed by employing a
biotin pull-down assay. Following the pull-down assay using
streptavidin-coated beads, western blotting analysis revealed that
HuR associates specifically with IκB-α 3′UTR transcripts but not
with GAPDH 3′UTR transcripts (Fig.
3A).
RNP IP analysis was then performed to investigate
the association of endogenous HuR with endogenous mRNAs. Using
lysates from untreated RAW264.7 cells, RNP IP assays revealed an
~11-fold enrichment in IκB-α mRNA associated with HuR in anti-HuR
antibody IP reactions compared with the control IgG IP reactions.
Using lysates from heat shock-treated cells, the association of
MKP-1 mRNA with HuR was >45-fold higher than that observed in
control cells (Fig. 3B). As a
negative control, the abundance of housekeeping GAPDH mRNA in HuR
IP samples was comparable with that observed following IgG IP and
remained unchanged by heat shock treatment (Fig. 3B). The relative enrichment of these
mRNAs in each RNP IP reaction was also investigated by qPCR,
followed by conventional PCR amplification and visualization on
agarose gels (Fig. 3B).
HuR affects the function of the IκB-α
mRNA 3′UTR
To determine the role of HuR in heat shock-mediated
regulation of the IκB-α mRNA 3′UTR, NIH 3T3 cells were transiently
cotransfected with the 3′UTR of IκB-α luciferase reporter plasmid
and flag-HuR plasmid as described and luciferase activity was
measured following heat shock treatment. Overexpression of HuR
increased luciferase activity compared with the control group
cotransfected with luciferase reporter control plasmid and flag-HuR
plasmid (Fig. 3C).
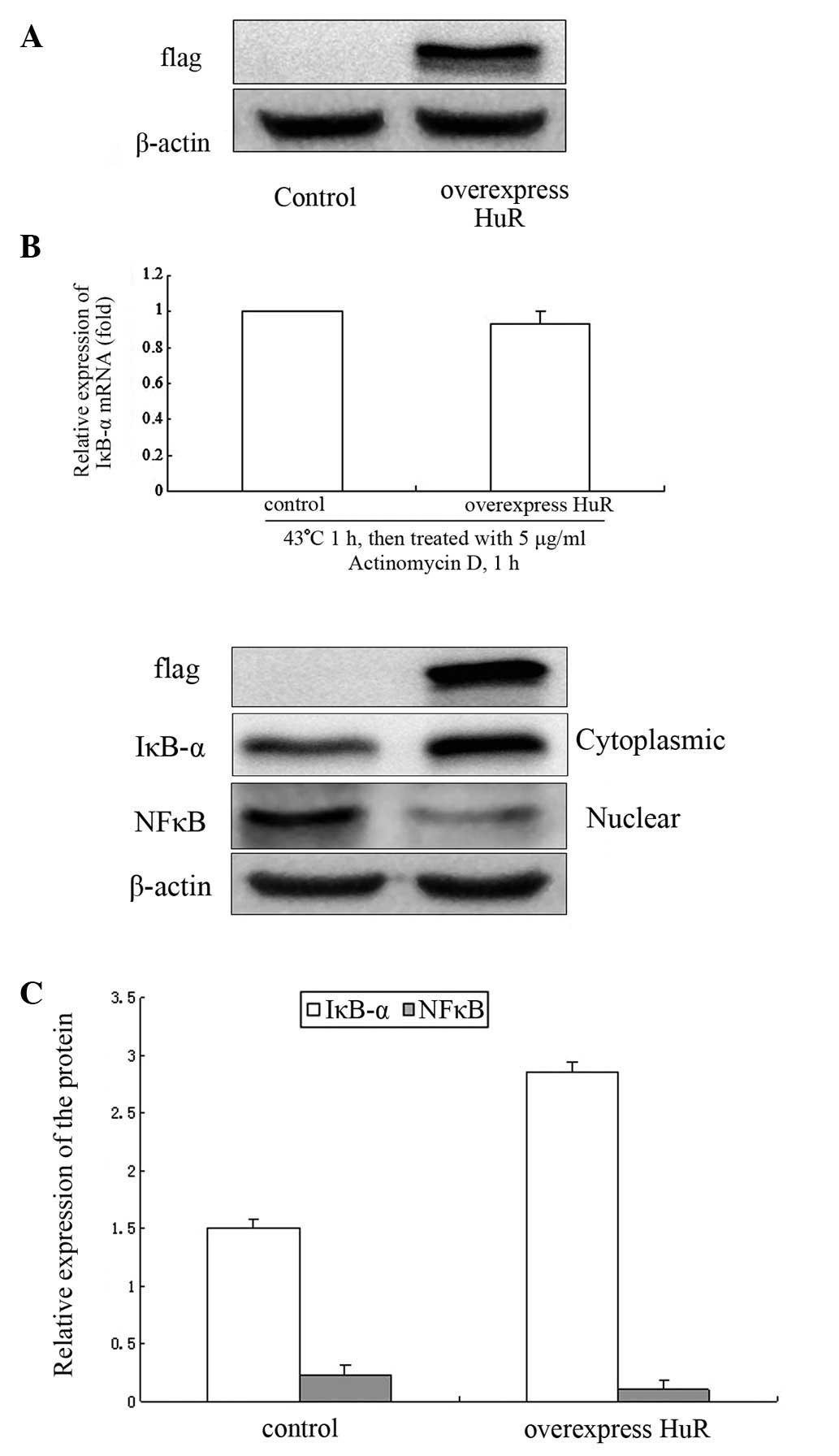
Effect of overexpression of HuR on IκB-α
mRNA stability and protein expression
To explore the effect of HuR on the regulation of
IκB-α at the post-transcriptional level, NIH 3T3 cells were
transfected with flag-HuR plasmid for 48 h and IκB-α mRNA stability
was detected by qPCR and IκB-α and NF-κB protein by western
blotting. The flag-HuR plasmid was expressed at high levels in NIH
3T3 cells (Fig. 4A). There was
little change in IκB-α mRNA stability following heat shock
treatment (Fig. 4B).
Overexpression of HuR may increase the expression of IκB-α protein.
IκB-α protein increased by ~50% compared with the control group.
NF-κB protein levels in the nucleus were decreased by ~55%
(Fig. 4C).
Effect of HuR on IκB-α mRNA stability and
protein expression
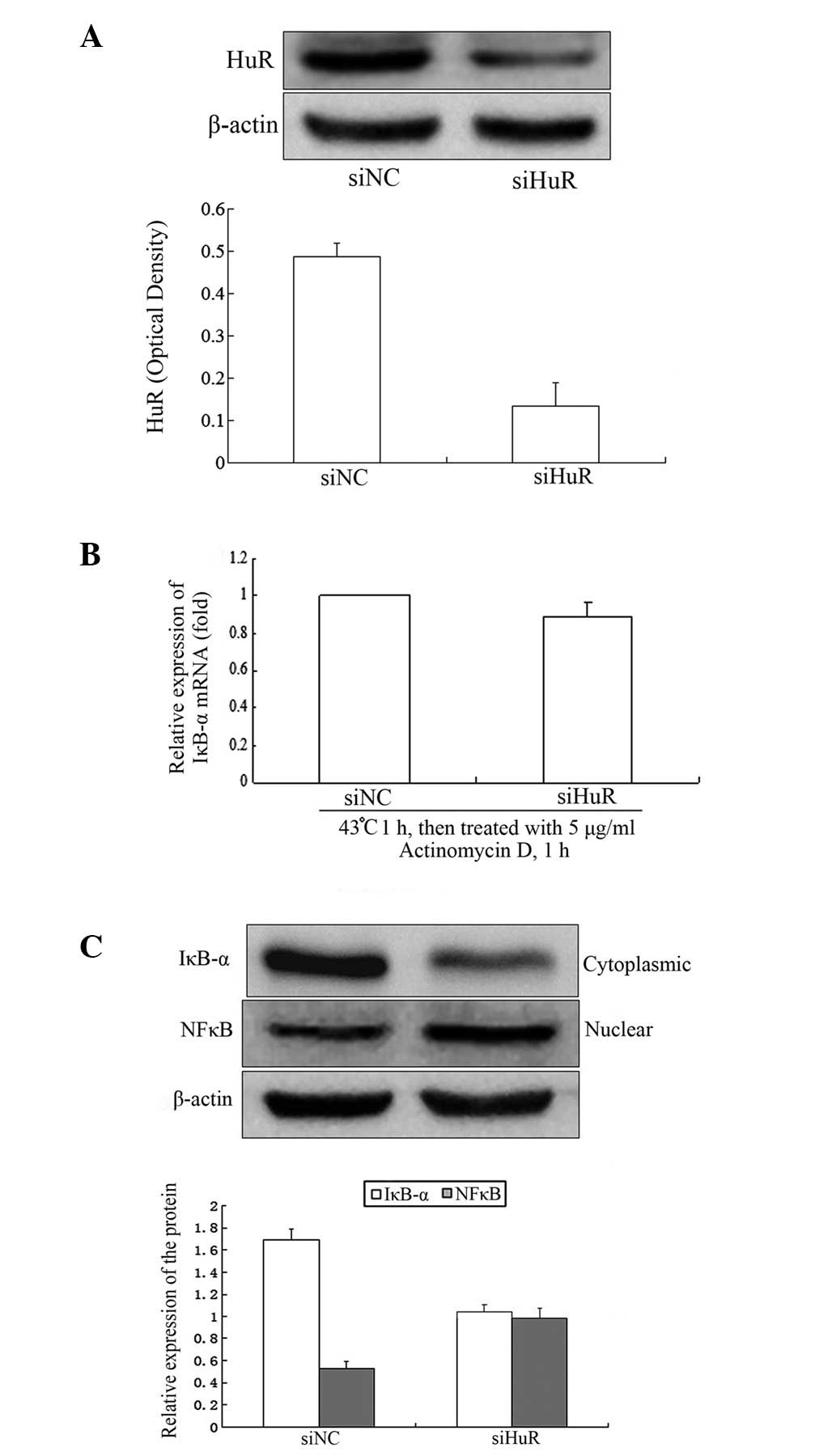
NIH 3T3 cells were transfected with siRNA
specifically targeting HuR for 48 h. The results revealed that HuR
protein decreased by ~75% (Fig.
5A). There was little effect on IκB-α mRNA stability following
heat shock treatment (Fig. 5B).
IκB-α protein levels decreased by ~40% compared with the control
group and the nuclear NF-κB protein levels increased by ~50%
(Fig. 5C).
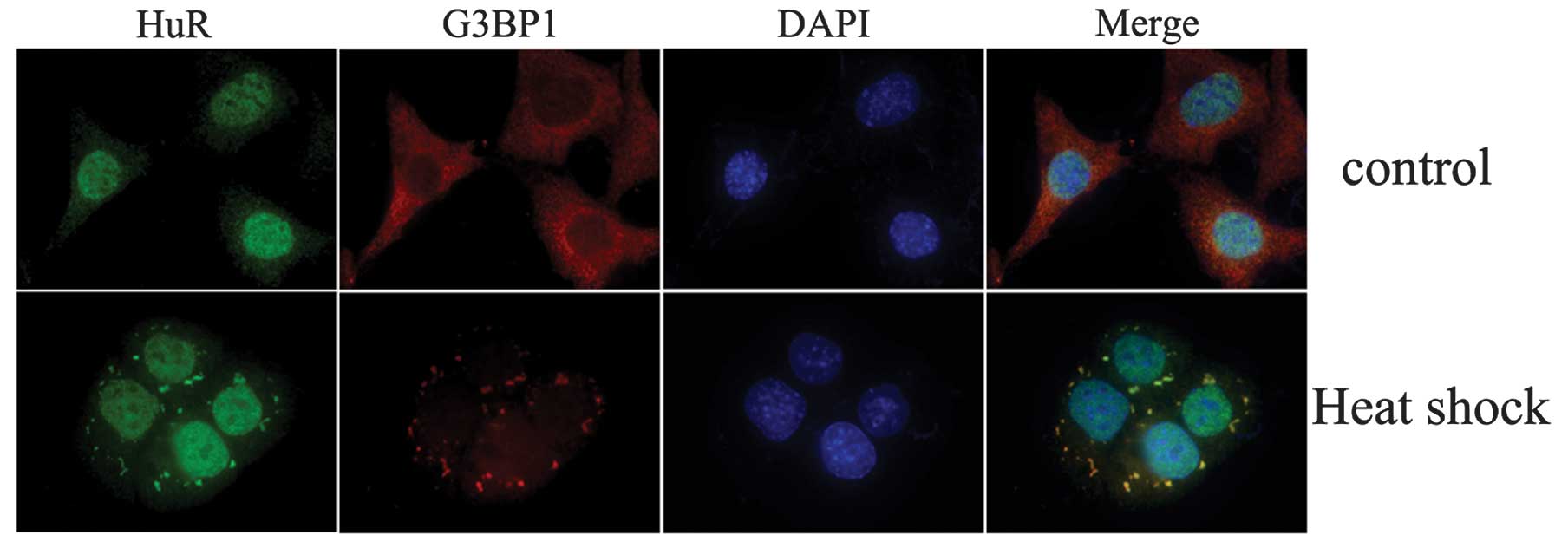
Subcellular distribution changes of
HuR
To investigate whether HuR undergoes
nucleus-to-cytoplasmic shuttling upon heat shock treatment, NIH 3T3
cells were treated by heat shock for 1 h. It was observed that HuR
was primarily localized in the nucleus and G3BP1 was primarily
localized in the cytoplasm of resting cells, whereas relocalization
of HuR from the nucleus to the cytoplasm was observed following 1 h
of heat shock treatment. HuR was observed to colocalize with G3BP1
protein to form stress granules (SGs; Fig. 6).
Discussion
It has been established that preconditioning with
heat shock treatment confers protection against
inflammation-associated tissue and organ injury (7,8).
Accordingly, the mechanisms by which heat shock modulates
inflammation-associated signal transduction were investigated,
particularly in cells of the monocyte/macrophage line. The present
study focused on the IκB/NF-κB pathway, clearly demonstrating that
heat shock inhibits NF-κB activation in vitro and in
vivo, in agreement with Malhotra and Wong (9). The mechanisms which govern this
modulation have been of particular interest.
Activation of the NF-κB pathway results in the
expression of inflammatory and immune response genes. These signals
must be transient and well regulated as prolonged NF-κB activation
may lead to abnormal gene expression and the deregulation of NF-κB
activation has been involved in various pathologies, including
chronic inflammation and cancer. The biological importance of the
IκBs as negative feedback regulators in NF-κB signaling has been
established. The expression of IκB-α and IκB-ɛ may be induced and
function as negative feedback regulators of NF-κB. IκB-α knockouts
cause mortality 7–10 days following birth due to hyperinflammation
(10). In addition, mice with
mutated κB enhancers of the IκB-α promoter exhibit a shortened life
span (13–15 months), hypersensitivity to septic shock and abnormal
T-cell development/activation (11). Negative feedback limits the
duration of stimulus-induced NF-κB activity and the magnitude of
the gene expression response, but may also mediate the transduction
of stimulus-specific information via a ‘temporal signaling code’
that determines which of the many possible target genes are
activated in response to a specific stimulus. IκB-α deficiency
results in inappropriate gene expression (12). The distinct degradation pathways
between the free and bound IκBs result in highly dynamic
homeostatic control of the NF-κB signaling module, which imparts
functional robustness to this signaling system.
Eukaryotes employ multiple post-transcriptional
mechanisms to adjust their gene expression programs in response to
external cues and changes in the physiological status of the cell
(13,14). A major role in post-transcriptional
hierarchy is played by RNA-binding proteins (RBPs) that regulate
virtually every aspect of mRNA metabolism, from nuclear processing
reactions and export to the cytoplasm to translation and stability
(15,16). HuR is one of the most popular RBPs.
HuR associates with mRNAs bearing U/AU-rich sequences, typically
present in 3′UTRs (17). The IκB-α
mRNA possesses a long, AU-rich 3′UTR, containing predicted binding
sites of an HuR motif, thus, the RNA binding protein HuR was
selected as a target. RNA pull-down and RNP IP was employed to
investigate the association between HuR and IκB-α mRNA. HuR was
observed to be associated specifically with IκB-α 3′UTR transcripts
upon heat shock treatment and RNP IP assays revealed a 45-fold
enrichment in IκB-α mRNA associates with HuR in anti-HuR antibody
IP reactions relative to that of control IgG IP reactions. These
observations indicate that HuR associates prominently with IκB-α
mRNA following heat shock treatment. In addition, the effect of HuR
on the regulation of IκB-α at the post-transcriptional level was
investigated. The results showed that overexpression of HuR
increases the expression of IκB-α protein which, in turn, decreases
NF-κB in the nucleus. Knockdown of HuR decreased IκB-α protein
levels, increasing NF-κB in the nucleus, but had little effect on
the stability of IκB-α mRNA.
Although HuR is located predominantly in the
nucleus, its post-transcriptional effect is linked to its
translocation to the cytoplasm, where it stabilizes and/or
modulates the translation of numerous target mRNAs. Changes in the
subcellular distribution of HuR upon heat shock treatment was
observed. The results indicate that HuR is primarily located in the
nucleus without heat shock treatment and translocates to the
cytoplasm with heat shock where HuR and G3BP1 protein colocalize.
G3BP1 proteins are the main content of the SG (18). SGs typically contain stalled mRNAs,
40S ribosomal subunits and a number of translational initiation
factors, including eIF2 and eIF3 and RNA binding proteins.
In the present study, HuR was demonstrated to
translocate between the nucleus and the cytoplasm, binding to the
IκB-α mRNA upon heat shock treatment. HuR was observed to modulate
the translation of IκB-α mRNA without affecting the half-lives. The
exact mechanism by which HuR modulates the translation of IκB-α
mRNA and the effect on the NF-κB pathway remain to be
elucidated.
Acknowledgements
Financial support was provided by grants from the
National Science Foundation of China (no. 81171476), Natural
Science Funds for Distinguished Youth Team of Zhejiang Province
(no. R2101166), the Scientific Innovation Team Project of Ningbo,
China (no. 2011B82014) China and the Scientific Project of Ningbo,
China (no. XKL11D2116). Sponsored by K.C. Wong Magna Fund of Ningbo
University, Ningbo, China.
References
|
1
|
Shanley TP, Ryan MA, Eaves-Pyles T and
Wong HR: Heat shock inhibits phosphorylation of IkappaBalpha.
Shock. 14:447–450. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grossman BJ, Shanley TP, Odoms K, Dunsmore
KE, Denenberg AG and Wong HR: Temporal and mechanistic effects of
heat shock on LPS-mediated degradation of IkappaBalpha in
macrophages. Inflammation. 26:129–137. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kohn G, Wong HR, Bshesh K, Zhao B, Vasi N,
Denenberg A, Morris C, Stark J and Shanley TP: Heat shock inhibits
tnf-induced ICAM-1 expression in human endothelial cells via
Ikappakinase inhibition. Shock. 17:91–97. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pittet JF, Lee H, Pespeni M, O’Mahony A,
Roux J and Welch WJ: Stress-induced inhibition of the NF-kappaB
signaling pathway results from the insolubilization of the IkappaB
kinase complex following its dissociation from heat shock protein
90. J Immunol. 174:384–394. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Deng L, Wang C, Spencer E, Yang L, Braun
A, Slaughter C, Pickart C and Chen ZJ: Activation of the IkappaB
kinase complex by TRAF6 requires a dimeric ubiquitin conjugating
enzyme complex and a unique polyubiquitin chain. Cell. 103:351–361.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ouaaz F, Arron J, Zheng Y, Choi Y and Beg
AA: Dendritic cell development and survival require distinct
NF-kappaB subunits. Immunity. 16:257–270. 2002. View Article : Google Scholar
|
|
7
|
Wong HR: Potential protective role of the
heat shock response in sepsis. New Horiz. 6:194–200.
1998.PubMed/NCBI
|
|
8
|
Wong HR and Wispé JR: The stress response
and the lung. Am J Physiol Lung. 273:L1–L9. 1997.PubMed/NCBI
|
|
9
|
Malhotra V and Wong HR: Interactions
between the heat shock response and the nuclear factor-kappaB
signaling pathway. Crit Care Med. 30(1 Supp): S89–S95. 2002.
View Article : Google Scholar
|
|
10
|
Beg AA, Sha WC, Bronson RT and Baltimore
D: Constitutive NF-kappaB activation, enhanced granulopoiesis and
neonatal lethality in IkappaBalpha-deficient mice. Genes Dev.
9:2736–2746. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peng B, Ling J, Lee AJ, Wang Z, Chang Z,
Jin W, Kang Y, Zhang R, Shim D, Wang H, Fleming JB, Zheng H, Sun SC
and Chiao PJ: Defective feedback regulation of NF-kappaB underlies
Sjogren’s syndrome in mice with mutated kappaB enhancers of the
IkappaBalpha promoter. Proc Natl Acad USA. 107:15193–15198.
2010.PubMed/NCBI
|
|
12
|
Hoffmann A, Levchenko A, Scott ML and
Baltimore D: The IkappaB-NF-kappaB signaling module: temporal
control and selective gene activation. Science. 298:1241–1245.
2002. View Article : Google Scholar
|
|
13
|
Moore MJ: From birth to death: the complex
lives of eukaryotic mRNAs. Science. 309:1514–1518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moore MJ and Proudfoot NJ: Pre-mRNA
processing reaches back to transcription and ahead to translation.
Cell. 136:688–700. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Glisovic T, Bachorik JL, Yong J and
Dreyfuss G: RNA-binding proteins and post-transcriptional gene
regulation. FEBS Lett. 582:1977–1986. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sanchez-Diaz P and Penalva LO:
Post-transcription meets post-genomic: the saga of RNA binding
proteins in a new era. RNA Biol. 3:101–109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
López de Silanes I, Zhan M, Lal A, Yang X
and Gorospe M: Identification of a target RNA motif for RNA-binding
protein HuR. Proc Natl Acad USA. 101:2987–2992. 2004.PubMed/NCBI
|
|
18
|
Anderson P and Kedersha N: Stressful
initiations. Cell. 115:3227–3234. 2002.PubMed/NCBI
|