Introduction
Cataracts are opacities of the crystalline lens that
can be categorized into early onset (congenital or infantile) and
age-related conditions. Congenital cataracts exhibit a prevalence
of ~1–6 cases per 10,000 live births and are a significant cause of
blindness in childhood (1). Among
the cases of congenital cataracts, approximately one-third are
hereditary, with most occurring in a nonsyndromic autosomal
dominant fashion (2). Congenital
cataracts exhibit high clinical and genetic heterogeneity. A total
of 40 genetic loci have been associated with congenital cataracts,
with ≥26 susceptibility genes cloned and sequenced (3). Among the disease-causing mutations
identified, ~50% are found in crystallins, 25% in connexins and the
remainder within genes such as heat shock transcription factor-4,
aquaporin-0 and beaded filament structural protein-2 (3). Crystallins are abundant, soluble
proteins located in the eye lens. The major human crystallins
comprise 90% of protein in the mature lens and contain two
different superfamilies: the small heat-shock proteins
(α-crystallins) and the βγ-crystallins.
In this study a functional candidate approach was
used to investigate the known crystallin genes, including
CRYAA, CRYAB, CRYBA1/A3, CRYBB1,
CRYBB2, CRYGC, CRYGD and CRYGS, in
which a major proportion of the mutations identified in a large
family with congenital cataracts were found.
Subjects and methods
Subjects
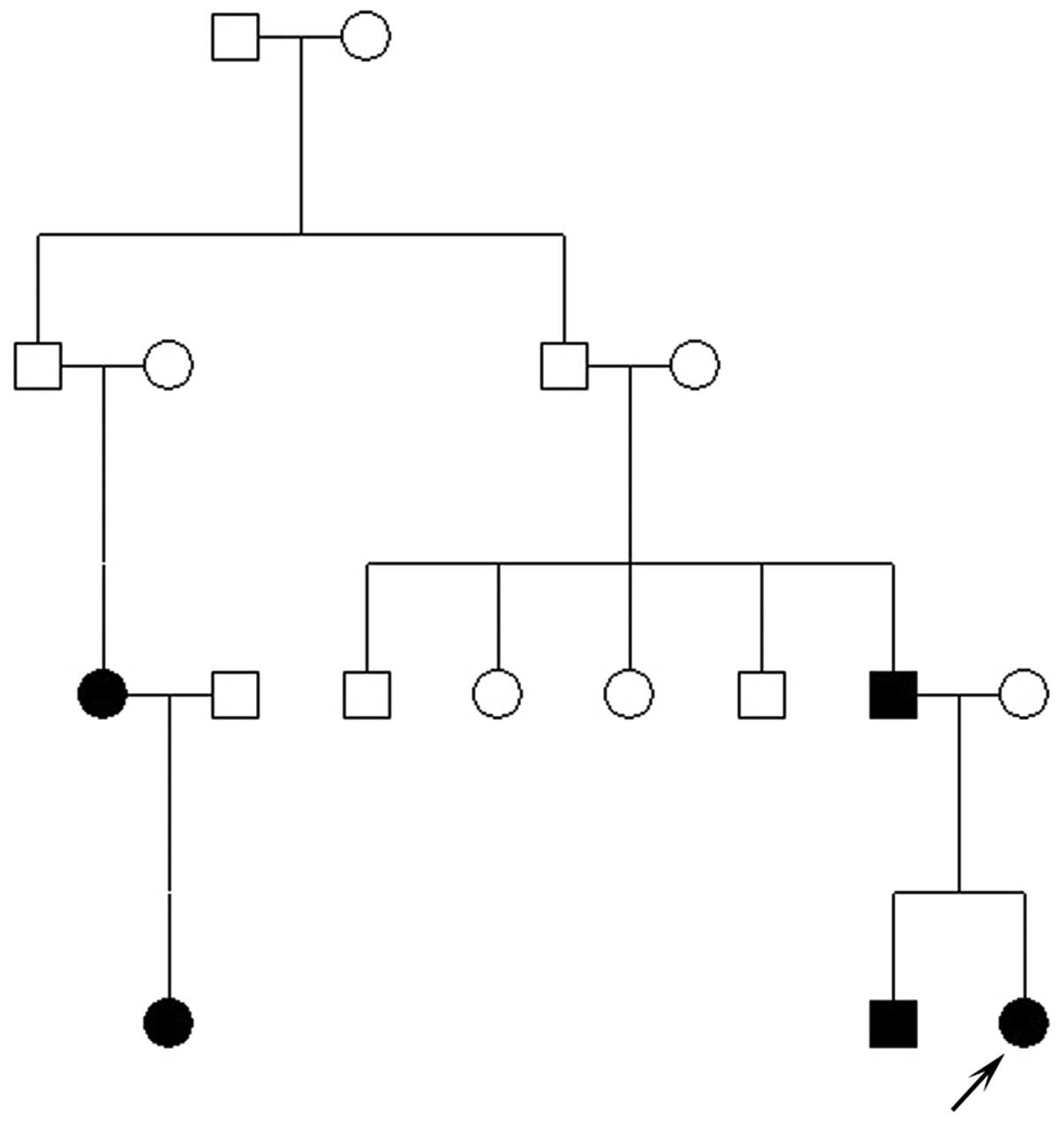
A four-generation Chinese family with autosomal
dominant congenital cataracts was identified from the Shijiazhuang
Obstetrics and Gynecology Hospital (Shijiazhuang, China) (Fig. 1). The study was approved by the
Ethics Committee of the Shijiazhuang Obstetrics and Gynecology
Hospital, and the study protocol followed the principles of the
Declaration of Helsinki. Informed consent was obtained from all
family participants. One hundred unrelated subjects without eye
disease were recruited from the Shijiazhuang Obstetrics and
Gynecology Hospital as unaffected controls. A history of cataract
extraction or ophthalmologic examination was used to determine
affected family members. In total, eight family members (III:1,
III:3, III:6, III:7, III:8, IV:1, IV:2 and IV:3) participated in
the study. All participating family members and controls underwent
ophthalmic examination, including visual acuity, slit-lamp and
fundus examination. Phenotypes were documented by slit-lamp
photography and 5 ml venous blood was collected in BD Vacutainer™
tubes (BD Biosciences, San Jose, CA, USA) containing EDTA for
further analysis. Genomic DNA was extracted by QIAamp DNA Blood
Mini kits (Qiagen Sciences, Inc., Germantown, MD, USA).
Mutation detection
All coding exons and intron-exon junctions of
candidate genes known to be associated with congenital cataracts
were amplified by polymerase chain reaction (PCR) using the primers
listed in Table I. Each reaction
mix (25 μl) contained 20 ng genomic DNA, 1X PCR buffer, 1.5 mM
MgCl2, 0.2 mM deoxynucleotide triphosphates, 0.5 μM each
of forward and reverse primer and 2.5 units Taq DNA polymerase
(Tiangen Biotech, Beijing, China). The PCR conditions for DNA
amplification were: 95°C for 5 min, followed by 35 cycles at 95°C
for 30 sec, 57–63°C for 30 sec (annealing temperature dependent on
the primer), 72°C for 30 sec, with a final extension at 72°C for 10
min. The PCR products were sequenced using an ABI 3730 Automated
Sequencer (Applied Biosystems, Foster City, CA, USA). Sequencing
results were analyzed using Chromas 2.33 (http://www.technelysium.com.au/chromas.html) and
compared with the National Center for Biotechnology Information
(NCBI) reference sequences (http://www.ncbi.nlm.nih.gov/).
 | Table IPrimers used for the polymerase chain
reaction. |
Table I
Primers used for the polymerase chain
reaction.
| Name | Forward (5′-3′) | Reverse (5′-3′) |
|---|
| CRYAA-1 |
AGCAGCCTTCTTCATGAGC |
CAAGACCAGAGTCCATCG |
| CRYAA-2 |
GGCAGGTGACCGAAGCATC |
GAAGGCATGGTGCAGGTG |
| CRYAA-3 |
GCAGCTTCTCTGGCATGG |
GGGAAGCAAAGGAAGACAGA |
| CRYAB-1 |
AACCCCTGACATCACCATTC |
AAGGACTCTCCCGTCCTAGC |
| CRYAB-2 |
CCATCCCATTCCCTTACCTT |
GCCTCCAAAGCTGATAGCAC |
| CRYAB-3 |
TCTCTCTGCCTCTTTCCTCA |
CCTTGGAGCCCTCTAAATCA |
| CRYBA1-1 |
GGCAGAGGGAGAGCAGAGTG |
CACTAGGCAGGAGAACTGGG |
| CRYBA1-2 |
AGTGAGCAGCAGAGCCAGAA |
GGTCAGTCACTGCCTTATGG |
| CRYBA1-3 |
AAGCACAGAGTCAGACTGAAGT |
CCCCTGTCTGAAGGGACCTG |
| CRYBA1-4 |
GTACAGCTCTACTGGGATTG |
ACTGATGATAAATAGCATGAACG |
| CRYBA1-5 |
GAATGATAGCCATAGCACTAG |
TACCGATACGTATGAAATCTGA |
| CRYBA1-6 |
CATCTCATACCATTGTGTTGAG |
GCAAGGTCTCATGCTTGAGG |
| CRYBB1-1 |
CCCTGGCTGGGGTTGTTGA |
TGCCTATCTGCCTGTCTGTTTCTC |
| CRYBB1-2 |
TAGCGGGGTAATGGAGGGTG |
AGGATAAGAGTCTGGGGAGGTGG |
| CRYBB1-3 |
CCTGCACTGCTGGCTTTTATTTA |
TCTCCAGAGCCCAGAACCATG |
| CRYBB1-4 |
CCAACTCCAAGGAAACAGGCATA |
CCTCCCTACCCACCATCATCTC |
| CRYBB1-5 |
TAGACAGCAGTGGTCCCTGGAGA |
AGCACTGGGAGACTGTGGAAGG |
| CRYBB1-6 |
CCTAGAAAAGGAAACCGAGGCC |
AGCGAGGAAGTCACATCCCAGTA |
| CRYBB2-1 |
GTTTGGGGCCAGAGGGGAGTGGT |
TGGGCTGGGGAGGGACTTTCAGTA |
| CRYBB2-2 |
CCTTCAGCATCCTTTGGGTTCTCT |
GCAGTTCTAAAAGCTTCATCAGTC |
| CRYBB2-3 |
GTAGCCAGGATTCTGCCATAGGAA |
GTGCCCTCTGGAGCATTTCATAGT |
| CRYBB2-4 |
GGCCCCCTCACCCATACTCA |
CTTCCCTCCTGCCTCAACCTAATC |
| CRYBB2-5 |
CTTACCCTTGGGAAGTGGCAATGG |
TCAAAGACCCACAGCAGACAAGTT |
| CRYGC-1 |
TGCATAAAATCCCCTTACCG |
CCTCCCTGTAACCCACATTG |
| CRYGC-2 |
TGGTTGGACAAATTCTGGAAG |
CCCACCCCATTCACTTCTTA |
| CRYGD-1 |
CAGCAGCCCTCCTGCTAT |
GGGTCCTGACTTGAGGATGT |
| CRYGD-2 |
GCTTTTCTTCTCTTTTTATTTCTGG |
AAGAAAGACACAAGCAAATCAGT |
| CRYGS-2 |
GAAACCATCAATAGCGTCTAAATG |
TGAAAAGCGGGTAGGCTAAA |
| CRYGS-3 |
AATTAAGCCACCCAGCTCCT |
GGGAGTACACAGTCCCCAGA |
| CRYGS-4 |
GACCTGCTGGTGATTTCCAT |
CACTGTGGCGAGCACTGTAT |
Bioinformatics analysis
The amino acid sequences of CRYBA1/A3 protein from
four species (human, mouse, rat and cow) were obtained from the
NCBI GenBank (https://www.ncbi.nih.gov/genbank/). Conservation
analysis was performed using CLC Main Workbench Software (CLC bio,
Aarhus, Denmark). Hydrophobicity changes were predicted using
ProtScale (http://web.expasy.org/protscale/).
Results
Clinical evaluation
Using medical records and ophthalmic examinations,
five family members (III:1, III:7, IV:1, IV:2 and IV:3) were
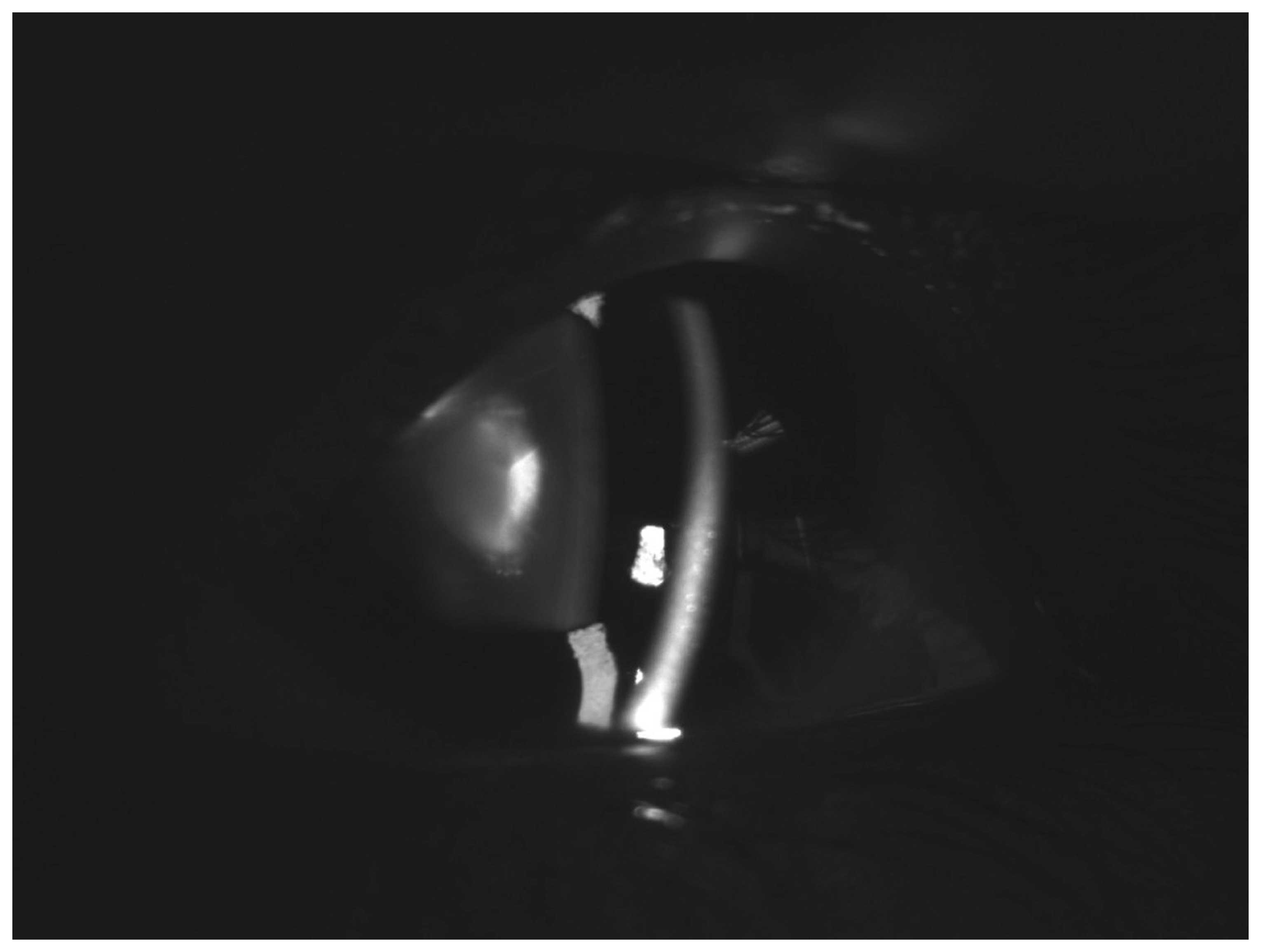
diagnosed with bilateral congenital cataracts. Slit-lamp
examination of the left eye of the proband (IV:3) showed nuclear
lens opacities (Fig. 2). No other
ocular or systemic abnormalities were identified.
Mutation analysis
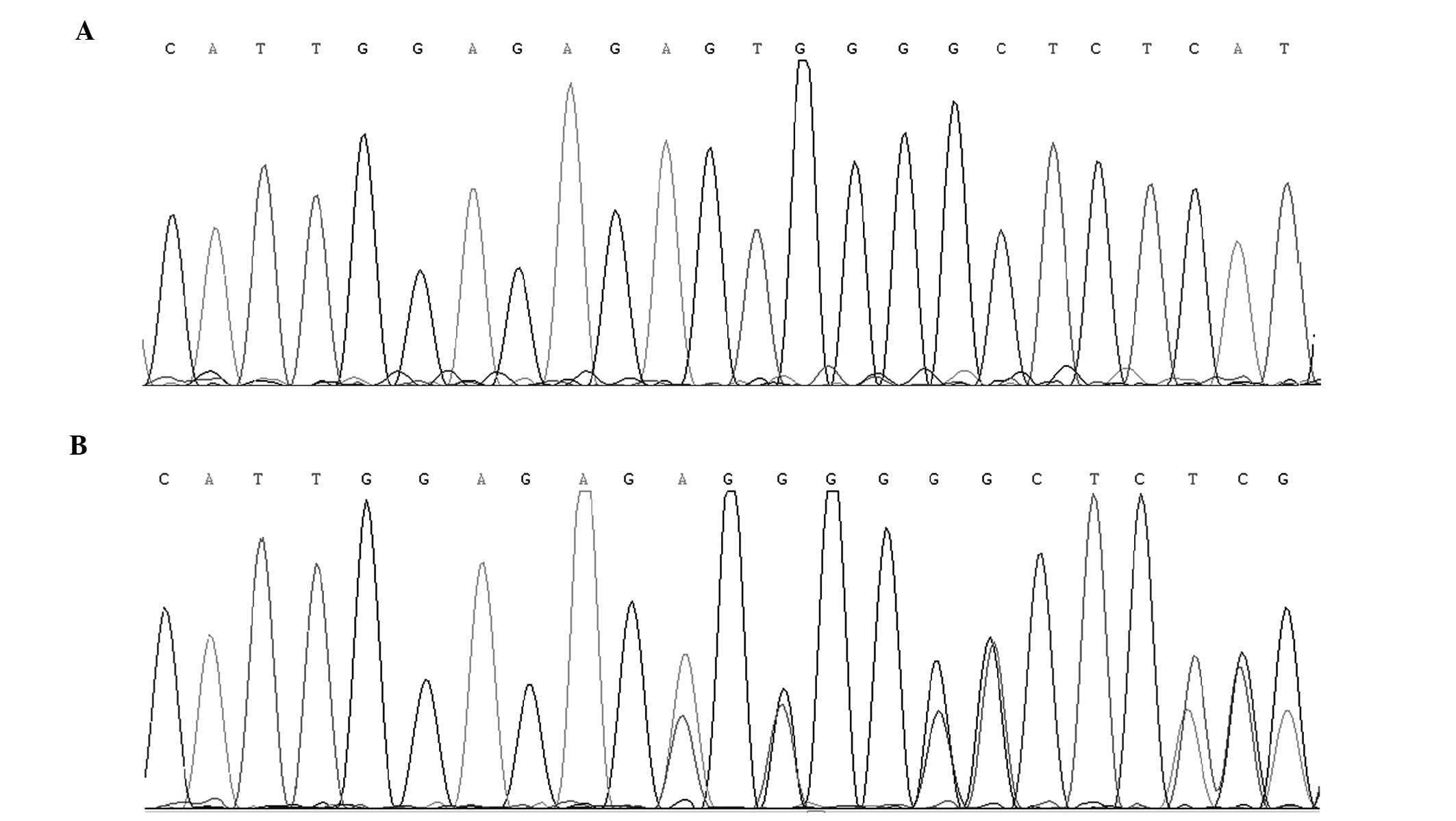
By direct sequencing of the coding regions of
candidate genes, a novel heterozygous 2-bp deletion mutation
(c.590-591delAG) in exon 6 of CRYBA1/A3 was
identified (Fig. 3). This deletion
led to a frameshift starting at amino acid residue 197, with a
substitution of glutamic acid to valine, followed by an altered
amino acid sequence (wild type, -EWGSHAQTSQIQSIRRIQQ; mutant,
-VGLSCPDFADPIDSPNPTV). This altered sequence was identified in all
affected family members, but was not detected in any unaffected
family members or the 100 unrelated control subjects.
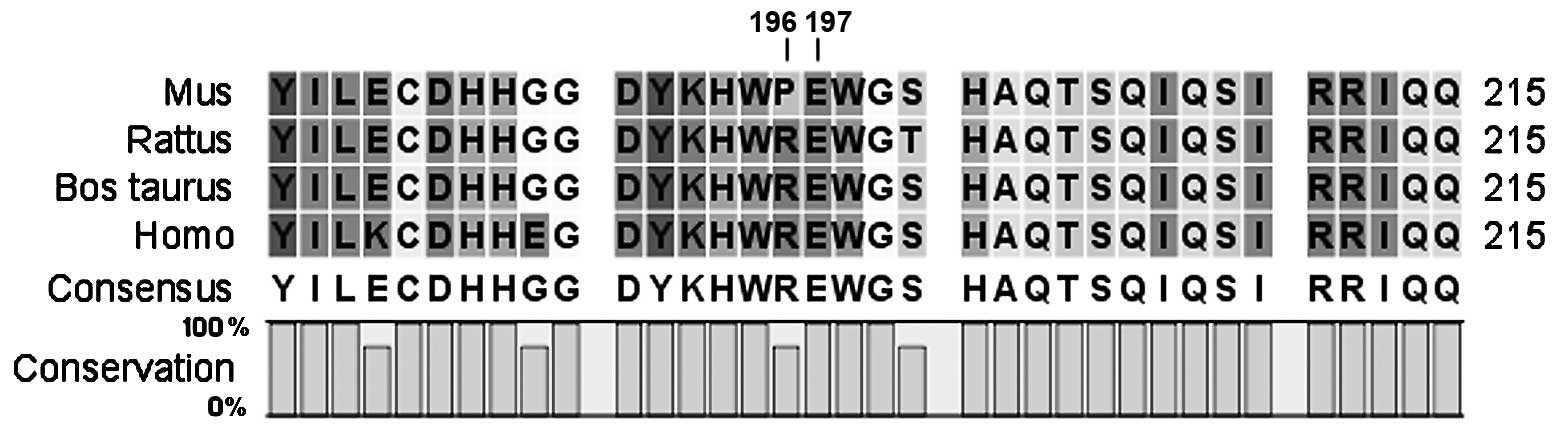
Bioinformatic analysis
Conservation analysis revealed that the sequence
from amino acid residue 197 to the COOH-terminal end is highly
conserved among species (Fig. 4).
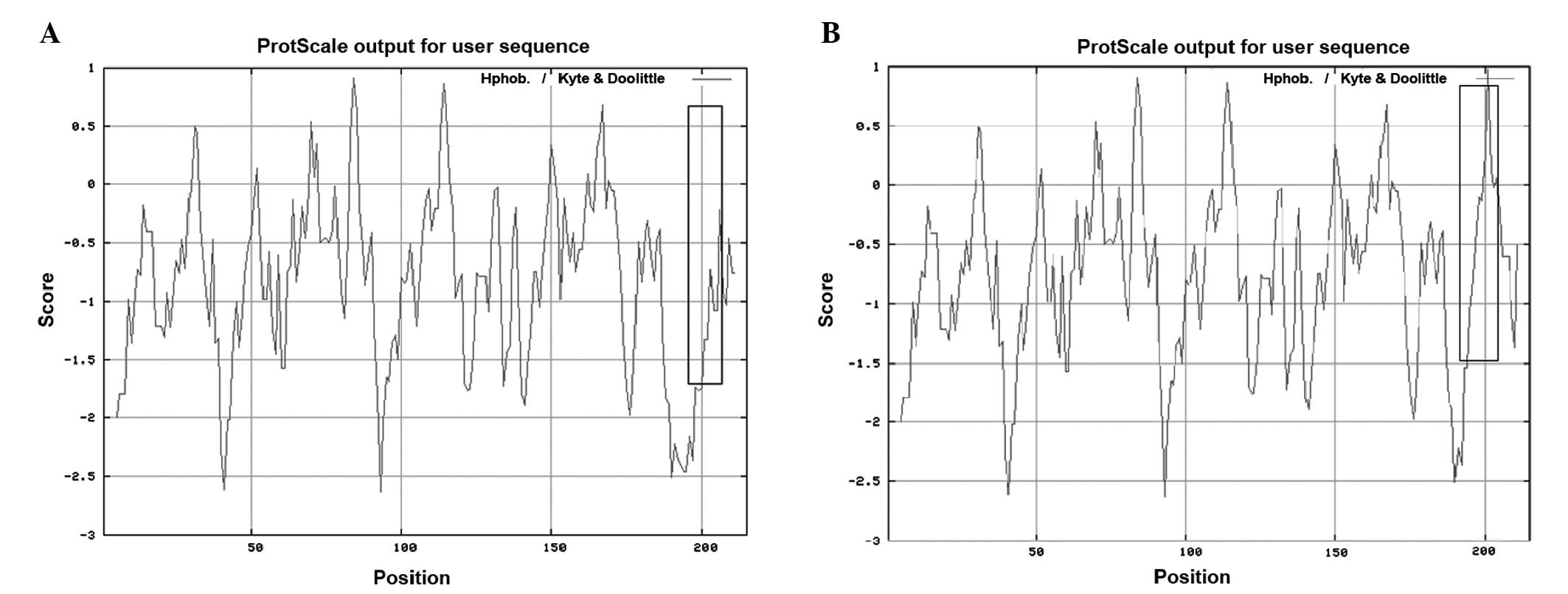
Furthermore, an increase in local hydrophobicity around the
mutation site was predicted by ProtScale (Fig. 5).
Discussion
Lens crystallin is fundamental for the establishment
and maintenance of lens transparency (4). Previous studies in affected patients
and transgenic animals indicate that mutations in crystallin genes
cause cataracts (14–15). The human lens contains α-, β- and
γ-crystallins, of which, β-crystallins comprise the greatest
proportion (5). CRYBA3/A1
is located at 17q11.2 and is a member of the β-crystallin family.
The gene utilizes an alternate translation initiation site to
encode two proteins (βA3- and βA1-crystallin) from a single mRNA,
and consists of six exons. β-crystallin comprises seven protein
domains: Four homologous Greek key motifs, a connecting peptide and
NH2- and COOH-terminal extensions. The first two exons
of CRYBA3/A1 encode the N-terminal arm and the Greek key
motifs are encoded by exons 3–6 (6). βA3- and βA1-crystallin are identical,
with the exception of 17 additional amino acid residues found on
the NH2-terminal arm of βA3-crystallin (7).
To date, several CRYBA3/A1 gene mutations
have been associated with autosomal dominant congenital cataracts
(Table II). All known
CRYBA3/A1 gene mutations can be divided into two clusters,
one located in the exon 3 splice site (8–9), and
the other in an exon 4 in-frame deletion (10). In the present study, a novel 2-bp
deletion mutation (590-591delAG) in exon 6 of
CRYBA1/A3 was identified. This mutation caused a
frameshift and resulted in an alternative βA1/βA3-crystallin
COOH-terminal. Typically, β- or γ-crystallin mutations cause major
abnormalities in protein structure, stability, solubility or the
ability to oligomerize, and are predicted to precipitate from
solution to cause lens opacity formation (11–12).
The COOH-terminal of βA1/βA3-crystallin is highly conserved, and it
has been reported that the C-terminal domain plays a role in
structural stability (11). The
ProtScale protein analysis in the present study showed a notable
increase in local hydrophobicity around the deletion site in
CRYBA3/A1. As demonstrated in other lens proteins,
hydrophobicity is associated with crystallin activities, and an
increased hydrophobic interaction can reduce protein solubility or
lead to abnormal protein folding (12–13).
In the present study, it was speculated that the mutant
COOH-terminal produces an abnormal protein structure with altered
stability and/or solubility.
| Table IISummary of mutations in
CRYBA1/A3 responsible for congenital cataracts. |
Table II
Summary of mutations in
CRYBA1/A3 responsible for congenital cataracts.
| Locus | Nucleotide | Amino acid |
|---|
| IVS3 (14) | IVS3+1 G.>C | Splice site
mutation |
| IVS3 (5) | IVS3+1 G.>A | Splice site
mutation |
| IVS3 (8) | IVS3+1 G.>T | Splice site
mutation |
| IVS3 (9) | IVS3+2 T.>G | Splice site
mutation |
| Exon 4 (10,13) | 276-281delGGAGGA | p.90Glydel
p.91Glydel |
| Exon 4 | 279-281delGGAGGA | p.91Glydel |
In conclusion, this is the first study, to the best
of our knowledge, to associate a frameshift mutation in exon 6 of
CRYBA1/A3 with the development of congenital
cataracts, and highlights the physiological importance of
βA1/A3-crystallin. The possible effect of the mutation on
βA1/A3-crystallin structure and function requires further
investigation.
Acknowledgements
The authors would like to thank the family members
for their participation in this study. The study was supported by a
grant from the Plan Issue of Hebei Province Health Department,
(project no.20110186).
References
|
1
|
Francis PJ, Berry V, Bhattacharya SS and
Moore AT: The genetics of childhood cataract. J Med Genet.
37:481–488. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reddy MA, Francis PJ, Berry V,
Bhattacharya SS and Moore AT: Molecular genetic basis of inherited
cataract and associated phenotypes. Surv Ophthalmol. 49:300–315.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hejtmancik JF: Congenital cataracts and
their molecular genetics. Semin Cell Dev Biol. 19:134–149. 2008.
View Article : Google Scholar
|
|
4
|
Jester JV: Corneal crystallins and the
development of cellular transparency. Semin Cell Dev Biol.
19:82–93. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu Y, Shentu X, Wang W, Li J, Jin C and
Yao K: A Chinese family with progressive childhood cataracts and
IVS3+1G>A CRYBA3/A1 mutations. Mol Vis. 16:2347–2353.
2010.PubMed/NCBI
|
|
6
|
Wistow G, Turnell B, Summers L, Slingsby
C, Moss D, Miller L, Lindley P and Blundell T: X-ray analysis of
the eye lens protein gamma-II crystallin at 1.9 A resolution. J Mol
Biol. 170:175–202. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Werten PJ, Carver JA, Jaenicke R and de
Jong WW: The elusive role of the N-terminal extension of beta A3-
and beta A1-crystallin. Protein Eng. 9:1021–1028. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang Z, Li Q, Ma Z, Guo Y, Zhu S and Ma X:
A G→T splice site mutation of CRYBA1/A3 associated with autosomal
dominant suture cataracts in a Chinese family. Mol Vis.
17:2065–2071. 2011.
|
|
9
|
Yang Z, Su D, Li Q, Yang F, Ma Z, Zhu S
and Ma X: A novel T→G splice site mutation of CRYBA1/A3 associated
with autosomal dominant nuclear cataracts in a Chinese family. Mol
Vis. 18:1283–1288. 2012.
|
|
10
|
Lu S, Zhao C, Jiao H, Kere J, Tang X, Zhao
F, Zhang X, Zhao K and Larsson C: Two Chinese families with
pulverulent congenital cataracts and deltaG91 CRYBA1 mutations. Mol
Vis. 13:1154–1160. 2007.PubMed/NCBI
|
|
11
|
Gupta R, Srivastava K and Srivastava OP:
Truncation of motifs III and IV in human lens betaA3-crystallin
destabilizes the structure. Biochemistry. 45:9964–9978. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Srivastava K, Gupta R, Chaves JM and
Srivastava OP: Truncated human betaB1-crystallin shows altered
structural properties and interaction with human betaA3-crystallin.
Biochemistry. 48:7179–7189. 2009. View Article : Google Scholar
|
|
13
|
Reddy MA, Bateman OA, Chakarova C, Ferris
J, Berry V, Lomas E, Sarra R, Smith MA, Moore AT, Bhattacharya SS
and Slingsby C: Characterization of the G91del CRYBA1/3-crystallin
protein: a cause of human inherited cataract. Hum Mol Genet.
13:945–953. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bateman JB, Geyer DD, Flodman P, Johannes
M, Sikela J, Walter N, Moreira AT, Clancy K and Spence MA: A new
betaA1-crystallin splice junction mutation in autosomal dominant
cataract. Invest Ophthalmol Vis Sci. 41:3278–3285. 2000.PubMed/NCBI
|
|
15
|
Andley UP, Hamilton PD, Ravi N and Weihl
CC: A knock-in mouse model for the R120G mutation of αB-crystallin
recapitulates human hereditary myopathy and cataracts. PLoS One.
6:e176712011.
|