Introduction
Cells respond to DNA damage by activating cell cycle
checkpoints and DNA repair mechanisms or by engaging proapoptotic
pathways (1–3). Genotoxic chemotherapeutic agents
target DNA in order to activate mitochondrial apoptotic pathways in
cancer cells (4,5). Dysregulation of the DNA
damage-induced apoptotic pathway triggers tumorigenesis and may
lead to unwanted chemoresistance (6,7).
Therefore, it is important to identify the mechanisms of resistance
to DNA damage-induced apoptosis and to target these mechanisms in
order to increase the effectiveness of cancer therapy.
Aven has been identified as an intracellular
apoptosis inhibitor by interacting with B-cell lymphoma-extra large
(Bcl-xL) and apoptotic protease activating factor 1 (Apaf-1)
(8). Aven interacts with the
N-terminal region and BH1 domain of Bcl-xL, which enhances the
anti-apoptotic function of Bcl-xL (9). In addition, Aven interrupts Apaf-1
oligomerization, leading to inactivation of caspase-9 and
inhibition of apoptosis (8).
Furthermore, it has been previously reported that Aven may be
cleaved at L144/196 sites by the aspartic protease, cathepsin D
(10). The cleaved C-terminal of
Aven, ΔN-Aven, is essential for its anti-apoptotic function. In
addition to its anti-apoptotic functions, Aven serves as a signal
transducer in the ataxia telangiectasia mutated (ATM) activation
pathway during DNA damage (11,12).
Activated ATM phosphorylates Aven on S135/308 and induces full
activation of Aven. The activated Aven then further enhances ATM
activation, leading to the activation of downstream pathway
components to inhibit Cdc25 activation and enhance Wee1/Myt kinase
activity. This results in the subsequent inhibition of mitotic
entry. Notably, it has been reported that the protein levels of
Aven are increased in acute leukemias (13,14).
Aven overexpression was found to be associated with a poor
prognosis in childhood acute lymphoblastic leukemia and may be
useful as a novel prognostic indicator in this malignancy (13). These studies suggest a potential
oncogenic role for the anti-apoptotic protein Aven. However, the
molecular mechanism underlying the expression of Aven remains to be
elucidated.
The present study described the regulation of Aven
protein levels by the Akt signaling pathway and cathepsin D.
Cellular levels of full-length Aven are maintained by proteolytic
cleavage as a result of cathepsin D activity, which is associated
with the Akt signaling pathway, rather than transcriptional control
and protein stability. Higher expression of cathepsin D decreases
levels of full length Aven, but increases ΔN-Aven levels in cancer
cells, leading to the resistance of cancer cells to anticancer
drugs and vice versa. The present results suggest that Aven level
is regulated through the Akt signaling pathway by cathepsin D
activity, which contributes to the sensitivity of cancer cells to
chemotherapeutic agents.
Materials and methods
Cells and reagents
MCF7 human breast cancer cell line (American Type
Culture Collection, Manassas, VA, USA) was cultured in Dulbecco’s
modified Eagle’s medium (Invitrogen Life Technologies, Carlsbad,
CA, USA) supplemented with 10% fetal bovine serum (HyClone
Laboratories, Logan, UT, USA) and 1% penicillin/streptomycin
(Invitrogen Life Technologies). MCF7 constitutively active (CA)-Akt
cells were established by stable transfection with pcDNA3 Myr HA
Akt1 (#9008; Addgene, Cambridge, MA, USA) into MCF7 cells.
Cycloheximide (CHX), doxorubicin and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma-Aldrich (St. Louis, MO, USA). LY294002
was purchased from Biomol Research Labs (Farmingdale, NY, USA). All
other reagents were obtained from Sigma-Aldrich, unless otherwise
specified.
Plasmids and transfection
Human Aven cDNA was cloned into the pEGFP-C1 vector
to generate GFP-Aven. The cells on 60 mm plates were transfected
using TransIT-2020 (Mirus, Madison, WI, USA) according to the
manufacturer’s instructions. The cells were subcultured for 24 h
prior to transfection on 60 mm plates. For use in transfection, 15
μl TransIT-2020 was mixed with 5 μg of plasmid and incubated for 30
min at room temperature. The TransIT-2020 reagent:DNA complexes
were added dropwise into the wells and then incubated for 72 h.
Following transfection, cell lysates were collected and analyzed
further.
Western blot analysis
Cells were lysed with 50 mM Tris-HCl (pH 7.5), 120
mM NaCl, 20 mM NaF, 1 mM EDTA, 5 mM EGTA, 15 mM sodium
pyrophosphate, 30 mM p-nitrophenyl phosphate, 1 mM benzamidine, 0.1
mM phenylmethylsulfonyl fluoride and 1% Nonidet P-40 for 20 min at
4°C and subsequently centrifuged at 15,000 × g for 15 min at 4°C.
Cell lysates were boiled in Laemmli sample buffer for 3 min, and 30
μg protein was subjected to either 10 or 12% SDS-PAGE (Mini-Protean
Electrophoresis system; Bio-Rad, Hercules, CA, USA) depending on
the molecular weight of the target proteins. Proteins were then
transferred to polyvinylidene difluoride membranes (Millipore
Corporation, Billerica, MA, USA) according to standard procedures.
Membranes were blocked in Tris-buffered saline containing 0.05%
Tween 20 (M. Biotech, Hanam, South Korea) and 5% non-fat dry milk
for 1 h. Following blocking, membranes were probed overnight at 4°C
with primary antibodies in antibody dilution buffer (Tris-buffered
saline containing 0.05% Tween 20 and 1% nonfat dry milk), followed
by 1 h incubation with secondary antibodies at room temperature.
Antibody detection was accomplished with WesternBright™ ECL
(Advansta Inc., Menlo Park, CA, USA) and exposed on Hyperfilm
(Eastman Kodak Co., Rochester, NY, USA). The following primary
antibodies were used: rabbit polyclonal immunoglobulin G (IgG)
anti-green fluorescent protein (GFP; sc-8334; 1:1,000), rabbit
polyclonal IgG anti-poly ADP ribose polymerase (PARP; sc-7150;
1:1,000) and rabbit polyclonal IgG anti-p21 (sc-756; 1:1,000) were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA);
rabbit polyclonal anti-pAkt (#9271s; 1:1,000), rabbit polyclonal
anti-Akt (#9272s; 1:1,000), rabbit polyclonal anti-Aven (#2300s;
1:1,000) and mouse monoclonal IgG2a anti-pS6 (#9206s;
1:1,000) obtained from Cell Signaling Technology, Inc. (Beverly,
MA, USA); mouse monoclonal anti-cathepsin D (#610800; 1:1,000)
purchased from BD Biosciences (San Diego, CA, USA) and mouse
monoclonal IgG1 anti-β-actin (A1978; 1:3,000) purchased
from Sigma-Aldrich. Peroxidase-conjugated secondary antibodies
polyclonal goat anti-rabbit IgG (#111-035-003; 1:1,000) and
polyclonal goat anti-mouse IgG (#115-035-003; 1:1,000) were
obtained from Jackson Immunoresearch Laboratories, Inc. (West
Grove, PA, USA).
Determination of RNA and protein
stability
Single-stranded cDNA was made with reverse
transcriptase (M. Biotech, Hanam, South Korea) from 1 μg of RNA
using an oligo-(dT)18 primer. Subsequently,
reverse-transcription quantitative polymerase chain reaction
(RT-qPCR) analysis was performed to determine the relative
expression levels of the Aven and GAPDH genes. PCR was performed
for 35 cycles with an annealing temperature of 55°C. PCR products
were analyzed by electrophoresis on agarose gels.
Protein stability assay
For the protein stability analysis, the cells were
treated with 10 μg/ml CHX and then whole cell lysates were prepared
at 0, 0.25, 0.5, 1, 3, 6, 12 and 24 h. The lysates (50 μg) were
then subjected to western blot analysis to identify the Aven and
β-actin proteins.
MTT assay
The MTT viability assay was performed with slight
modifications as previously described (15,16).
MTT was first prepared as a stock solution of 5 mg/ml in
phosphate-buffered saline (PBS; pH 7.2) and was filtered. Briefly,
cells (200 μl/well) were seeded at 3×104 cells/ml into
96 well plates in the presence of various concentrations (0.005–1
μM) of doxorubicin (or vehicle control) for 72 h. MTT was added to
the cultures during the last 4 h of incubation and then the media
was removed with a needle and syringe. The blue formazan crystals
trapped in cells were dissolved in sterile dimethyl sulfoxide (100
μl) by incubating at room temperature for 30 min. The well plate
was read using the Epoch Microplate Spectrophotometer™ (BioTek
Instruments, Inc., Winooski, VT, USA) with absorbance at a
wavelength of 570 nm with a reference wavelength of 630 nm.
Results
Expression of Aven is regulated through
the Akt signaling pathway
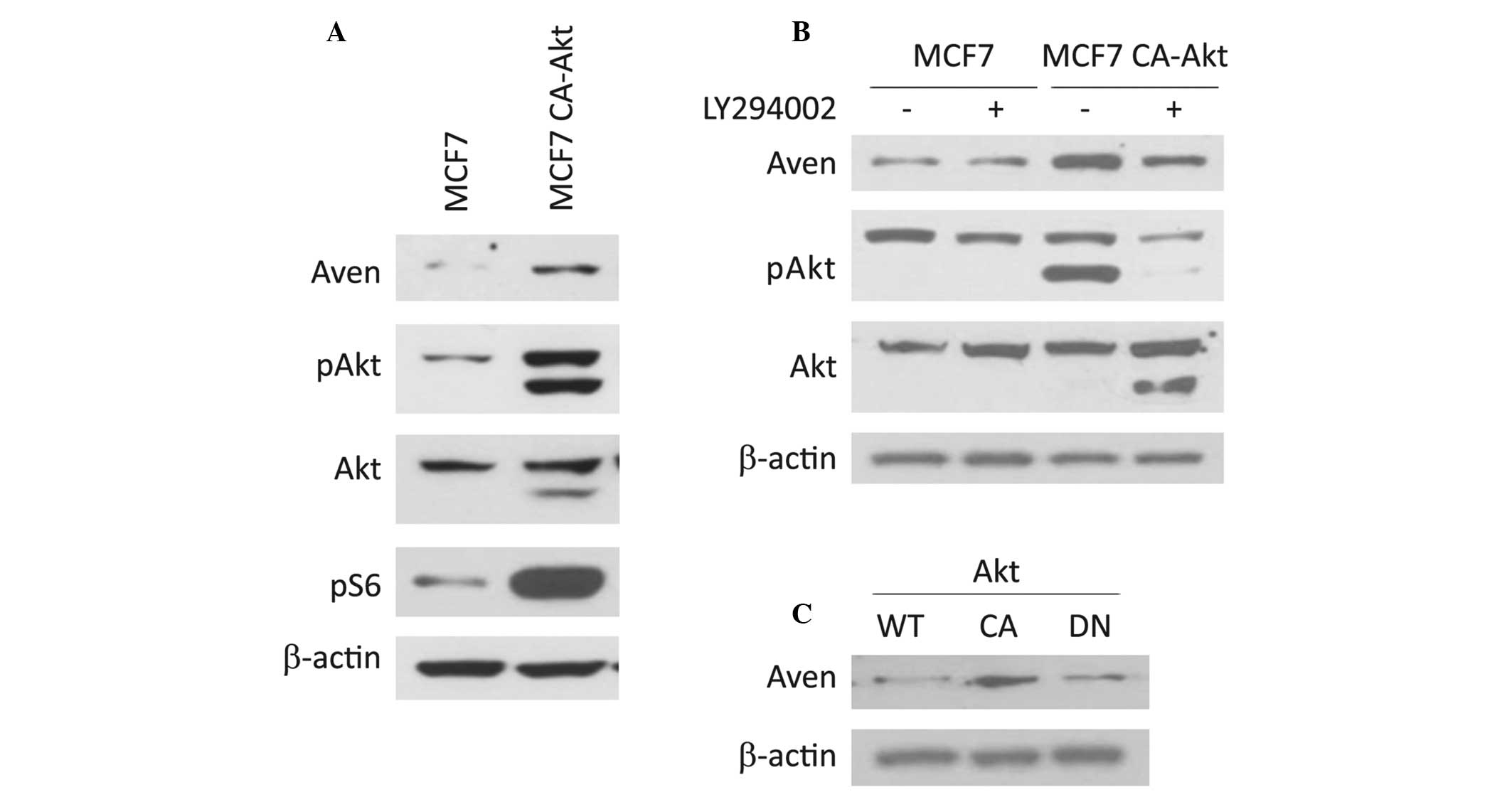
To identify the molecular mechanism involved in Aven
expression, the expression level of Aven in several cell lines was
analyzed. Notably, the expression level of Aven in MCF7 CA-Akt cell
lines was significantly higher than that of its parental MCF7 cells
(Fig. 1A). To confirm the possible
involvement of the Akt signaling pathway in Aven expression,
LY294002, an upstream kinase inhibitor of Akt, was administered to
MCF7 CA-Akt cells. As shown in Fig.
1B, Aven expression was significantly abrogated by LY294002
treatment, suggesting that the expression level of Aven is
associated with the Akt signaling pathway. In addition, this
hypothesis was further supported by the observation that ectopic
expression of the constitutively active form of Akt significantly
increases the level of Aven in MCF7 cells, but its dominant
negative form does not (Fig.
1C).
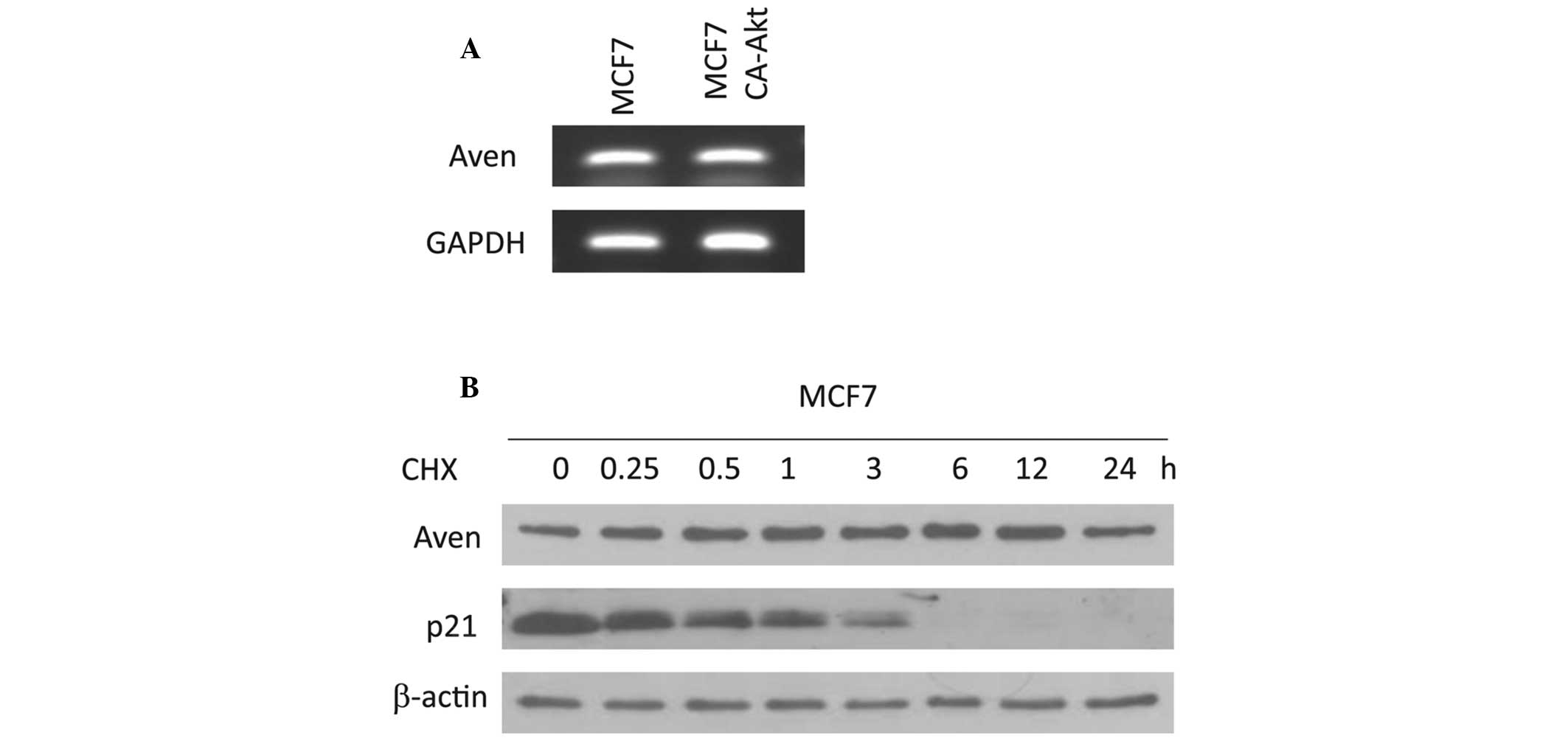
Expression of Aven is not affected by
transcriptional control or protein stability
To elucidate whether the level of Aven may be
regulated by a transcriptional process, the mRNA level of Aven was
analyzed in MCF7 and MCF7 CA-Akt cells. As shown in Fig. 2A, no significant difference was
observed in the mRNA levels of Aven between these two cells, which
was further confirmed by qPCR (data not shown). This finding
indicates that accumulation of Aven in MCF7 CA-Akt cells is
regulated in a transcription-independent manner. Subsequently, the
degradation rate of the Aven protein in MCF7 cells was determined
using western blot analysis along a time course following addition
of the protein synthesis inhibitor, cycloheximide. As shown in
Fig. 2B, Aven protein stability
was not significantly altered up to 24 h (Fig. 2B), indicating that Aven is a stable
protein in cellular systems. In addition, the protein level of Aven
in MCF7 CA-Akt cells was not altered during the 24 h time period
(data not shown). All these results suggest that accumulation of
Aven in MCF7 CA-Akt cells may not be affected by its stability.
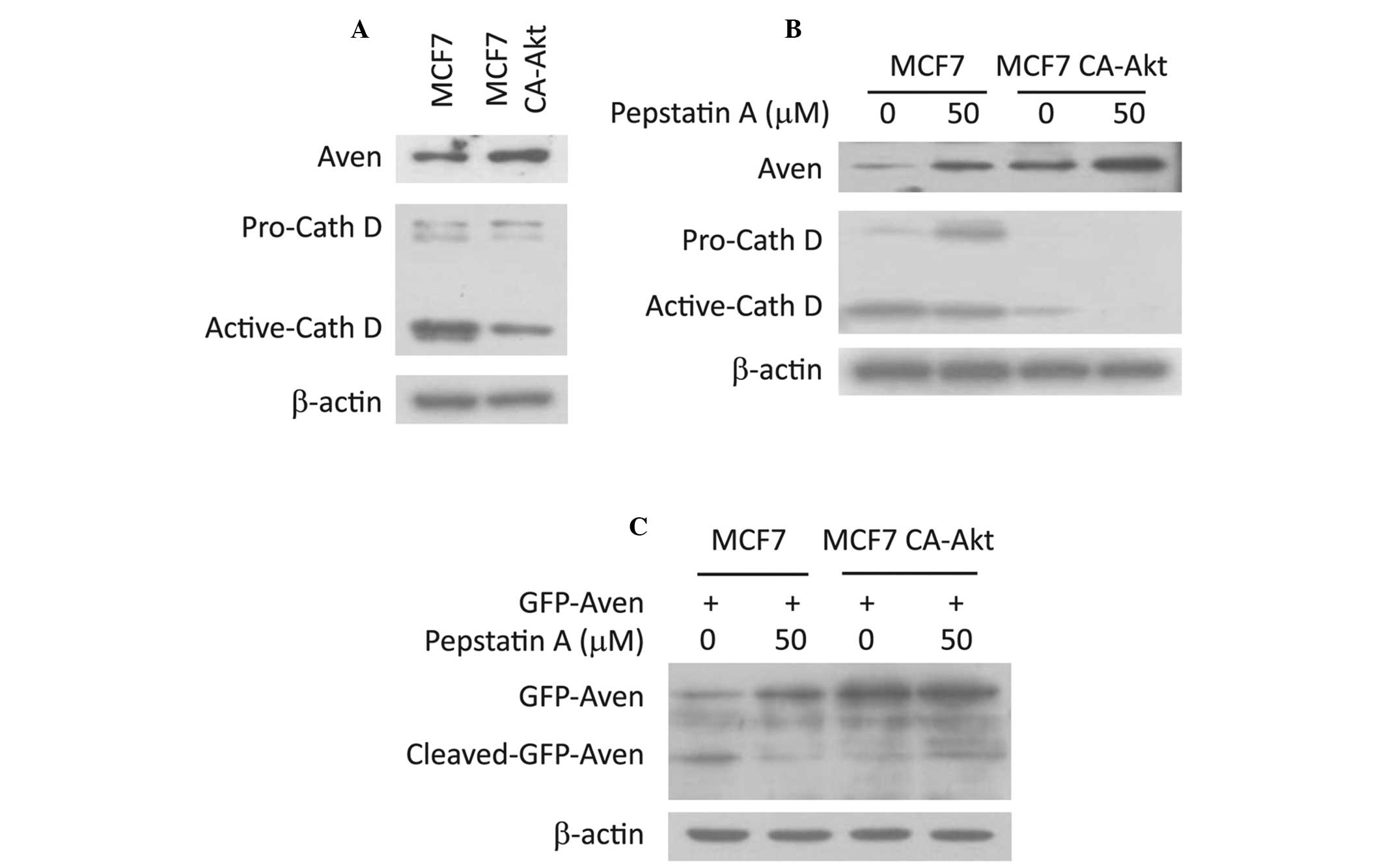
Cathepsin D is responsible for
maintaining the levels of full-length Aven
A previous study revealed that full-length Aven may
be cleaved by cathepsin D to unleash its anti-apoptotic potential
(10). There are two cleavage
sites between amino acid 144/145 and between amino acid 196/197 in
the Aven protein. Therefore, it was investigated whether cathepsin
D regulates the level of Aven. As shown in Fig. 3A, the expression level of cathepsin
D in MCF7 CA-Akt cells was lower than that of MCF7 cells, which was
inversely correlated with Aven levels, indicating that the
expression of cathepsin D may be a major determinant for
full-length Aven level in MCF7 CA-Akt cells. To confirm this
hypothesis, Aven levels were compared in the absence or presence of
pepstatin A, an inhibitor of cathepsin D. As shown in Fig. 3B, Aven levels were markedly
increased following treatment with pepstatin A, supporting the
possible role of cathepsin D in the regulation of Aven levels.
Subsequently, the present study aimed to detect cleaved Aven
fragment, which is generated by cathepsin D activity in MCF7 cells,
however, the Aven antibody was not able to detect the fragment
(data not shown). Thus, a GFP-Aven overexpression system was used.
When GFP-Aven was overexpressed in MCF7 cells, full-length GFP Aven
and cleaved GFP-Aven were clearly detected. However, the only form
of GFP-Aven in MCF7 CA-Akt cells was full-length and the cleaved
form was not detected (Fig. 3C).
In addition, pepstatin A treatment resulted in an evident increase
in the full-length form and a decrease in the cleaved form
(Fig. 3C). These findings support
the hypothesis that the cellular level of full-length Aven is
determined by cathepsin D activity, which may be regulated through
the Akt signaling pathway.
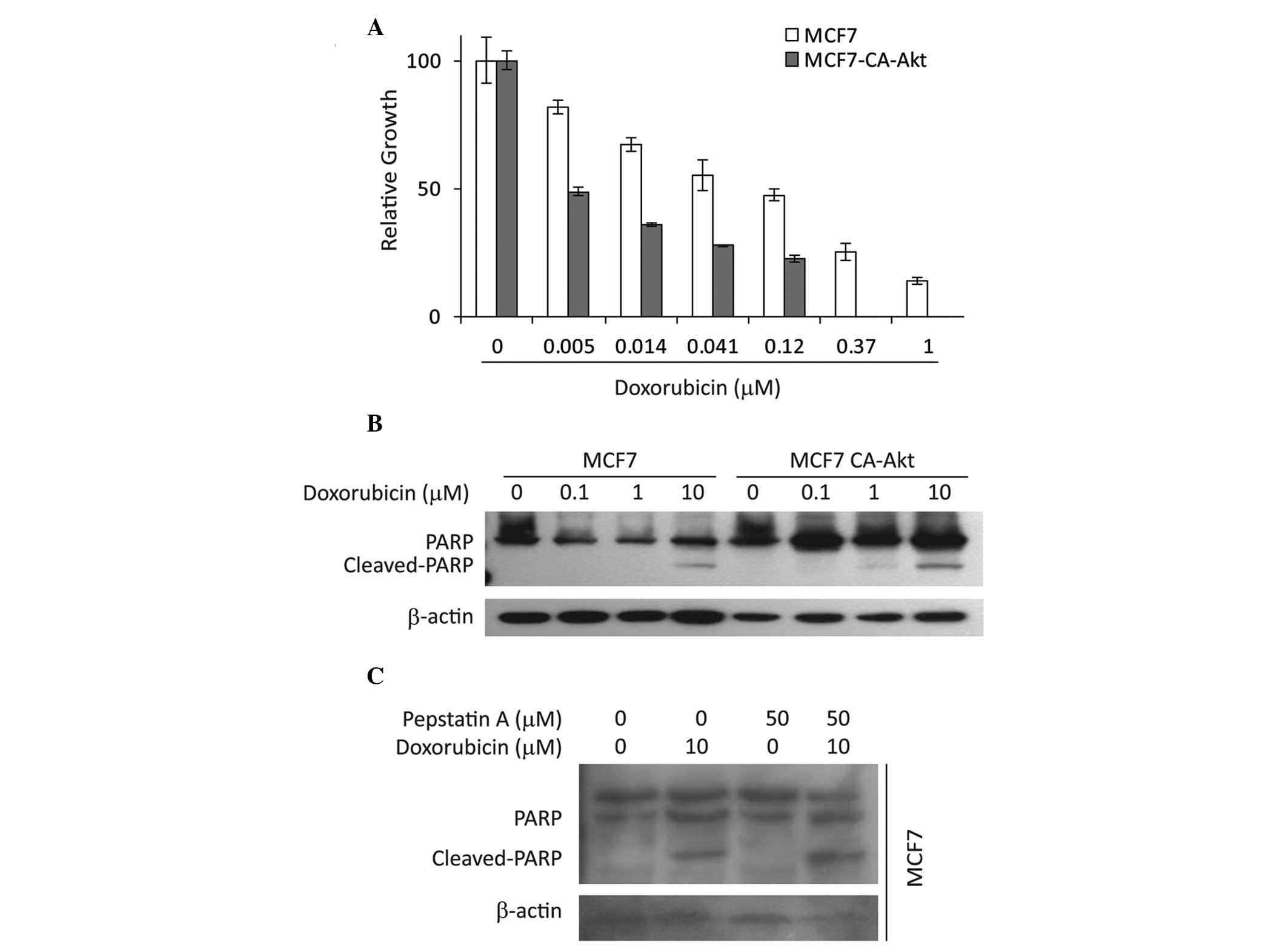
Cleaved-Aven affects drug resistance
It has previously been demonstrated that ΔN-Aven
cleaved by cathepsin D is the active form for the anti-apoptotic
functions of Aven (10). Thus, the
present study aimed to determine whether the expression levels of
Aven affect the sensitivity of cancer cells to chemotherapeutic
agents. The present data demonstrate that MCF7 CA-Akt cells are
more sensitive to doxorubicin than the parental MCF7 cells, which
was evidenced by a cell proliferation assay (Fig. 4A) and PARP cleavage (Fig. 4B). These results suggest that
higher levels of ΔN-Aven may contribute to doxorubicin resistance.
To further clarify this theory, the effect of pepstatin A on
doxorubicin-induced apoptosis in MCF7 cells was determined. As
shown in Fig. 4C, pepstatin A
treatment led to an increase in doxorubicin-induced PARP cleavage.
These results indicate that inhibition of ΔN-Aven formation by
pepstatin A is associated with increased apoptosis in MCF7 cells
and full-length Aven may be insufficient to inhibit apoptosis.
Discussion
Drug resistance to chemotherapy is a major obstacle
for the treatment of several types of tumor and is an important
cause of cancer chemotherapy failure. The cellular resistance of
cancer cells to chemotherapeutic agents may be divided into two
types of resistance; intrinsic resistance and extrinsic resistance
(17,18). Intrinsic resistance is
predominantly involved in cell differentiation or in genetic
alterations during tumor initiation. Extrinsic resistance results
from the expansion of rare genetic variants in a tumor cell
population. A good example is multidrug resistance (MDR) in which
cells are resistant to numerous structurally and functionally
unrelated chemotherapeutic agents (19,20).
Several molecular mechanisms have been proposed to explain MDR,
including tumor cell-specific mechanisms such as decreased cellular
drug accumulation, drug sequestration into intracellular vesicles,
DNA repair pathway activation, which counteracts the effects of the
drugs and evasion of apoptosis or cell cycle arrest (21–23).
In addition, genes that control cell death and survival signaling,
including the genes encoding B-cell lymphoma 2 and p53, may acquire
mutations that lead to drug resistance through modulation or
impairment of apoptosis. In addition, activation of alternative
signaling pathways that modulate cell migration, proliferation and
apoptosis may be involved in the development of drug-resistance
pathways (24,25).
Aven has previously been reported to act as an
intracellular anti-apoptotic molecule through regulating Bcl-xL and
Apaf-1 (8,9). Additionally, Aven may be cleaved at
L144/196 sites by aspartic protease cathepsin D and the cleaved
C-terminal of Aven, ΔN-Aven, is essential for its anti-apoptotic
function (10). These findings
indicate that intracellular levels of Aven may contribute to drug
resistance to chemotherapeutic agents. The current data reveal that
accumulation of full length Aven occurred through the Akt signaling
pathway (Fig. 1). In addition,
this accumulation was associated with inhibiting its proteolytic
cleavage by cathepsin D (Fig. 3),
indicating that the Akt signaling pathway may be associated with
the anti-apoptotic function of Aven.
It has been well demonstrated that the Akt signaling
pathway is associated with tumor cell survival, proliferation and
invasiveness (19,26). The activation of Akt is also one of
the most frequent alterations observed in human cancer and tumor
cells. MCF7 CA-Akt cells that are constantly active grow more
rapidly than their parental MCF7 cells (data not shown). However,
it was observed that MCF7 CA-Akt cells are more sensitive to
chemotherapeutic agents (Fig. 4).
Furthermore, inhibition of cathepsin D by pepstatin A resulted in
an increase in sensitivity to chemotherapeutic agents (Fig. 4). These results clearly indicate
that the ratio of ΔN-Aven to full length Aven is governed by
cathepsin D, which is regulated by the Akt signaling pathway.
In conclusion, the present data demonstrate the
regulatory mechanism underlying the expression of Aven through the
Akt signaling pathway and cathepsin D activity. Akt signaling and
cathepsin D activity are intracellular regulators for maintaining
the ratio of ΔN-Aven to full length Aven, which contribute to
resistance to chemotherapeutic agents.
Acknowledgements
This study was supported by a grant from the
National Research Foundation of Korea funded by the Korean
government (The Ministry of Science, ICT and Future Planning; grant
nos. 2011-0030074 and 2012R1A1A2043451).
References
|
1
|
Rich T, Allen RL and Wyllie AH: Defying
death after DNA damage. Nature. 407:777–783. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roos WP and Kaina B: DNA damage-induced
cell death by apoptosis. Trends Mol Med. 12:440–450. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bernstein C, Bernstein H, Payne CM and
Garewal H: DNA repair/pro-apoptotic dual-role proteins in five
major DNA repair pathways: fail-safe protection against
carcinogenesis. Mutat Res. 511:145–178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Herr I and Debatin KM: Cellular stress
response and apoptosis in cancer therapy. Blood. 98:2603–2614.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sanchez-Prieto R, Rojas JM, Taya Y and
Gutkind JS: A role for the p38 mitogen-acitvated protein kinase
pathway in the transcriptional activation of p53 on genotoxic
stress by chemotherapeutic agents. Cancer Res. 60:2464–2472.
2000.PubMed/NCBI
|
|
6
|
Okada H and Mak TW: Pathways of apoptotic
and non-apoptotic death in tumour cells. Nat Rev Cancer. 4:592–603.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maddika S, ande SR, Panigrahi S, et al:
Cell survival, cell death and cell cycle pathways are
interconnected: implications for cancer therapy. Drug Resist Updat.
10:13–29. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chau BN, Cheng EH, Kerr DA and Hardwick
JM: Aven, a novel inhibitor of caspase activation, binds Bcl-xL and
Apaf-1. Mol Cell. 6:31–40. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kutuk O, Temel SG, Tolunay S and Basaga H:
Aven blocks DNA damage-induced apoptosis by stabilising Bcl-xL. Eur
J Cancer. 46:2494–2505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Melzer IM, Fernandez SB, Bosser S, et al:
The Apaf-1-binding protein Aven is cleaved by Cathepsin D to
unleash its anti-apoptotic potential. Cell Death Differ.
19:1435–1445. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo JY, Yamada A, Kajino T, et al:
Aven-dependent activation of ATM following DNA damage. Curr Biol.
18:933–942. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Esmaili AM, Johnson EL, Thaivalappil SS,
Kuhn HM, Kornbluth S and Irusta PM: Regulation of the ATM-activator
protein Aven by CRM1-dependent nuclear export. Cell Cycle.
9:3913–3920. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Choi J, Hwang YK, Sung KW, et al: Aven
overexpression: association with poor prognosis in childhood acute
lymphoblastic leukemia. Leuk Res. 30:1019–1025. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eissmann M, Melzer IM, Fernandez SB, et
al: Overexpression of the anti-apoptotic protein AVEN contributes
to increased malignancy in hematopoietic neoplasms. Oncogene.
32:2586–2591. 2013. View Article : Google Scholar
|
|
15
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jo SK, Hong JY, Park HJ and Lee SK:
Anticancer activity of novel daphnane diterpenoids from daphne
genkwa through cell-cycle arrest and suppression of Akt/STAT/Src
signalings in human lung cancer cells. Biomol Ther (Seoul).
20:513–519. 2012. View Article : Google Scholar
|
|
17
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and Gonzalez-Baron M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Szakacs G, Paterson JK, Ludwig JA,
Booth-Genthe C and Gottesman MM: Targeting multidrug resistance in
cancer. Nat Rev Drug Discov. 5:219–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Krishna R and Mayer LD: Multidrug
resistance (MDR) in cancer. Mechanisms, reversal using modulators
of MDR and the role of MDR modulators in influencing the
pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 11:265–283.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lothstein L, Israel M and Sweatman TW:
Anthracycline drug targeting: cytoplasmic versus nuclear - a fork
in the road. Drug Resist Updat. 4:169–177. 2001. View Article : Google Scholar
|
|
23
|
Stavrovskaya AA: Cellular mechanisms of
multidrug resistance of tumor cells. Biochemistry (Mosc).
65:95–106. 2000.
|
|
24
|
El Maalouf G, Le Tourneau C, Batty GN,
Faivre S and Raymond E: Markers involved in resistance to
cytotoxics and targeted therapeutics in pancreatic cancer. Cancer
Treat Rev. 35:167–174. 2009. View Article : Google Scholar
|
|
25
|
Tamburrino A, Piro G, Carbone C, Tortora G
and Melisi D: Mechanisms of resistance to chemotherapeutic and
anti-angiogenic drugs as novel targets for pancreatic cancer
therapy. Front Pharmacol. 4:562013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|