Introduction
Liver disease is a prevalent medical condition which
has numerous etiologies, including chemical exposure, alcohol,
lipid peroxidative products and viral infection (1). To date, several types of medications
have been investigated for the treatment of liver diseases
(2).
D-galactosamine (D-GaIN) is a well-known inducer of
hepatic injury in vitro and in vivo. D-GaIN treatment
results in the loss of uridine 5′-triphosphate, uridine
5′-diphosphate and uridine 5′-monophosphate as well as inhibition
of RNA and protein synthesis (3).
In addition, D-GaIN-induced oxidative stress is generated through
reactive hydroxyl radical damage to the cell membrane via
stimulation of lipid peroxidation (4). Several studies have shown that
D-GaIN-induced hepatocyte death was mediated through
mitogen-activated protein kinase (MAPK) and nuclear factor
erythroid 2-related factor 2 (Nrf2) signaling (5–7).
Nrf2 is a transcription factor which targets certain
genes, including those for nicotinamide adenine dinucleotide
phosphate hydrogen (NADPH), quinine oxidoreductase 1 (Nqo1),
glutathione (GSH) synthesis and glutathione-s-transferase (GST)
(8); in addition, Nrf2 has been
shown to have protective effects against oxidative stress (9). During oxidative stress, Nrf2
translocates to the nucleus from the cytosol, and as a result,
antioxidant enzymes become upregulated and oxidative stress damage
decreases (10).
MAPKs include c-jun N-terminal kinase (JNK), p38
MAPK and extracellular signal-regulated kinase (ERK). These
proteins have been reported to be phosphorylated via GaIN-induced
oxidative stress (5). Of note,
activated-JNK induces hepatocyte death and apoptosis via activation
of caspase-3 as well as liver cell necrosis (11).
Superoxide dismutase (SOD) and catalase (CAT) are
important cellular defense systems which transform superoxide into
oxygen and hydrogen peroxide for detoxification (12). In addition, GSTs are a superfamily
of enzymes that protect against chemical toxicity and oxidative
stress (13,14). Porphyra (P.) yezoensis is a
red algae found along the coasts of Korea, China and Japan. In the
present study, the protective effect of Porphyra yezoensis
glycoprotein (PYGP) on D-GaIN-induced cytotoxicity was investigated
in Hepa 1c1c7 cells.
Materials and methods
Preparation of PYGP
P. yezoensis was purchased in 2013 in the
Republic of Korea (Shuyup, Busan, Korea). P. yezoensis
powder (40 g) was suspended in 1 l distilled water and stirred for
4 h at room temperature. The suspension was centrifuged at 3,000 ×
g at 4°C for 20 min and vacuum filtered, followed by the addition
of triple volumes (total quantity of filtrate × 3) of ethanol.
Following 24 h, the solution was filtered and concentrated using
rotary evaporation at 40°C. The concentrated solution was divided
into 1.5-ml tubes, freeze-dried and stored at −70°C until further
use.
Cell culture
Normal mouse Hepa 1c1c7 hepatocyte cells (no. 22026)
were purchased from the Korean Cell Line Bank (Seoul, Korea). Cells
were maintained at 37°C in a 5% CO2 humidified
atmosphere. Hepa 1c1c-7 cells were cultured in minimum essential
medium α (Gibco-BRL, Grand Island, NY, USA) without nucleosides
supplemented with 10% fetal bovine serum (FBS; Hyclone
Laboratories, Inc., Logan, UT, USA) and antibiotics (Gibco-BRL).
The medium was replaced every two days.
Cell proliferation
Hepa 1c1c7 cell proliferation was measured using a
CellTiter 96 aqueous non-radioactivity cell proliferation assay
(Promega Corp., Madison, WI, USA). This assay determines cell
proliferation based on the cleavage of
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfonyl)-2H-tetrazolium
(MTS) into a formazan product, which is soluble in tissue culture
medium. Hepa 1c1c7 cells were seeded onto 96-well plates at a
density of 1.5×103 cells/well in 100 μl medium. Cells
were cultured for 24 h, following which the medium was replaced
with serum-free medium (SFM) containing PYGP (20 or 40 μg/ml) for
24 h. PYGP-treated Hepa 1c1c7 cells were then exposed to 20 mM
D-GaIN (Sigma-Aldrich, St. Louis, MO, USA) for 24 h. Subsequently,
cells were incubated in MTS solution for 30 min at 37°C. Cell
proliferation was measured at 490 nm using a Benchmark Plus 10730
microplate reader (Benchmark; Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Estimation of dehydrogenase release
Hepa 1c1c7 cell injury was quantitatively assessed
via determination of lactate dehydrogenase (LDH), which is released
from damaged or destroyed cells. Hepa 1c1c7 cells were seeded onto
96-well plates at a density of 1.5×103 cells/well in 100
μl medium. Cells were cultured for 24 h, following which the SFM
was replaced with PYGP (20 or 40 μg/ml) for 24 h. Hepa 1c1c7 cells
were then exposed to either D-GaIN (20 mM) with PYGP (20 or 40
μg/ml) for 24 h. LDH release was measured using an LDH cytotoxicity
assay kit according to the manufacturer’s instructions (Cayman
Chemical Co., Ann Arbor, MI, USA). Absorbance was then measured at
490 nm using a Benchmark Plus 10730 microplate reader (Bio-Rad
Laboratories, Inc.).
Lipid peroxidation measurement
Cells were collected in lysis buffer
(phosphate-buffered saline, 0.05% butyl hydroxyl toluene; Cell
Biolabs Inc., San Diego, CA, USA) and homogenized on ice using a
thiobarbituric acid reactive substances (TBARS) assay kit (Cell
Biolabs, Inc.) according to the manufacturer’s instructions.
Absorbance was then measured at 532 nm using a Benchmark Plus 10730
microplate reader (Bio-Rad Laboratories, Inc.).
Western blot analysis
Hepa 1c1c7 cells were plated onto 100-mm dishes.
Cells were cultured until they reached 60–80% confluence and were
then pre-treated with PYGP (20 or 40 μg/ml) for 24 h. Cells were
then exposed to D-GaIN (20 mM) with PYGP (20 or 40 μg/ml) for 24 h.
Cells were washed with ice-cold phosphate-buffered saline (PBS;
0.15 M sodium phosphate, 0.15 M sodium chloride, pH 7.4;
Gibco-BRL), following which lysis buffer [20 mM Tris-base (pH 7.5),
150 mM NaCl, 0.25% Na-deoxycholate, 1 mM EDTA, 1 mM ethylene glycol
tetraacetic acid, 1% Triton X-100 from iNtRON Biotechnology, Seoul,
Korea; containing 2.5 M sodium pyrophosphate, 1 mM
β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml
aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A and 1 mM
phenylmethylsulfonyl fluoride from Sigma-Aldrich] was added.
Protein content was determined using a bicinchoninic acid protein
assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA). Proteins
were separated using 10–15% SDS-PAGE and then transferred to a
polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA).
The transferred membrane was blocked at room temperature with 1%
bovine serum albumin (BSA) in Tris-buffered saline with Tween 20
[TBS-T; 10 mM Tris-HCl (pH 7.5), 150 mM NaCl and 0.1% Tween 20] and
then incubated, with agitation, with the indicated primary
antibodies: Rabbit anti-mouse ERK immunoglobulin G (IgG) polyclonal
antibody [diluted 1:1,000 with BSA/TBS-T; incubated for 4 h at room
temperature (RT)], rabbit anti-phosphorylated (p)-ERK IgG
polyclonal antibody (diluted 1:1,000 with BSA/TBS-T; incubated for
4 h at RT), mouse anti-mouse JNK IgG monoclonal antibody (diluted
1:1,000 with BSA/TBS-T; incubated for 4 h at RT), mouse anti-mouse
p-JNK IgG monoclonal antibody (diluted 1:1,000 with BSA/TBS-T;
incubated for 4 h at RT), rabbit anti-mouse p38 IgG polyclonal
antibody (diluted 1:1,000 with BSA/TBS-T; incubated for 4 h at RT),
mouse anti-mouse p-p38 IgG monoclonal antibody (diluted 1:1,000
with BSA/TBS-T; incubated for 4 h at RT), rabbit anti-mouse Nrf2
IgG polyclonal antibody (diluted 1:1,000 with BSA/TBS-T; incubated
for 4 h at RT), goat anti-mouse Nqo1 IgG polyclonal antibody
(diluted 1:1,000 with BSA/TBS-T; incubated for 4 h at RT), mouse
anti-mouse GST IgG monoclonal antibody (diluted 1:1,000 with
BSA/TBS-T; incubated for 4 h at RT), and rabbit anti-mouse heme
oxygenase (HO)-1 IgG polyclonal antibody (diluted 1:1,000 with
BSA/TBS-T; incubated for 4 h at RT), which were all purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The secondary
antibody was a peroxidase-conjugated goat, mouse and rabbit
antibody (1:10,000; GE Healthcare, Little Chalfont, UK). Super
Signal West Pico Stable Peroxide Solution and the Super Signal West
Pico Luminol/Enhancer solution (Thermo Fisher Scientific, Rockford,
IL, USA) were then added and the signal was monitored using X-ray
film (Kodak, Rochester, NY, USA) and a developer and fixer twin
pack (Kodak).
Antioxidant enzyme measurement
SOD (Superoxide dismutase assay kit; Cayman Chemical
Co., Ann Arbor, MI, USA), CAT (Catalase assay kit; Cayman Chemical
Co.) and GST (Glutathione s-transferase assay kit; Cayman Chemical
Co.) activities of Hepa 1c1c7 cells were measured according to the
manufacturer’s instructions. Absorbance was then measured using a
Benchmark microplate reader (Benchmark Plus 10730; Bio-Rad
Laboratories, Inc.).
Statistical analysis
Values are presented as the mean ± standard
deviation and data were analyzed with SPSS 10.0 software (SPSS,
Inc., Chicago, IL, USA) using an analysis of variance followed by a
Duncan’s multiple range test. P<0.05 was considered to indicate
a statistically significant difference between values.
Results
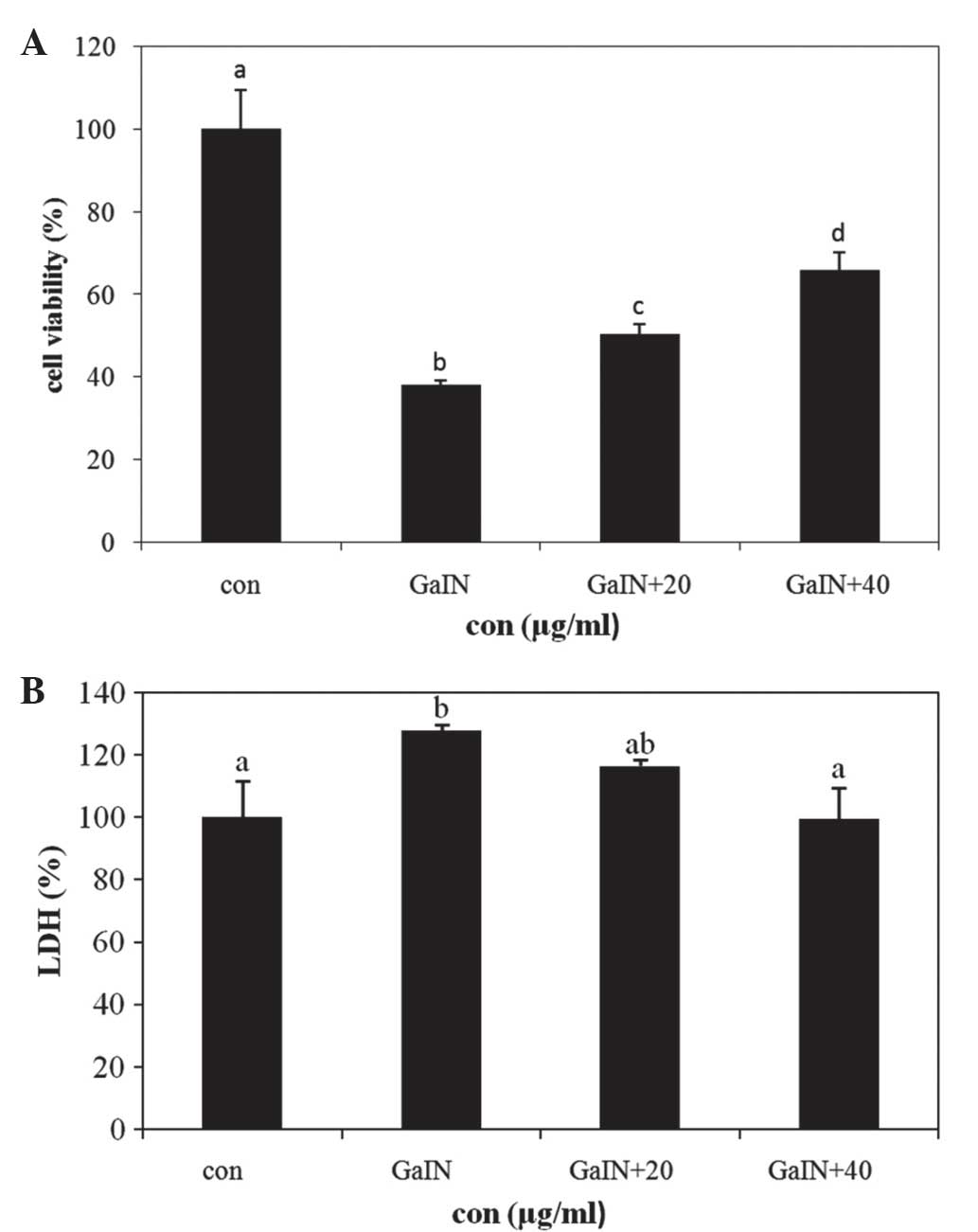
Protective effects of PYGP against
D-GaIN-induced injury in Hepa 1c1c7 cells
The protective effect of PYGP against D-GaIN-induced
hepatotoxicity in normal mouse liver cells was measured using the
MTS cell viability assay. Cells were pre-treated with PYGP for 24 h
and then incubated with D-GaIN and PYGP for 24 h. The results of
the MTS assay showed that D-GaIN treatment induced Hepa 1c1c7 cell
death, whereas pretreatment with PYGP significantly attenuated the
cytotoxic effects of D-GaIN (P<0.05; Fig. 1A).
LDH is a soluble enzyme located in the cytosol,
which is released into the surrounding culture medium upon cell
damage (15,16). The results of the present study
demonstrated that the D-GaIN-only treatment group showed increased
LDH levels compared with those of the control group; by contrast,
the PYGP pre-treatment groups showed significantly decreased LDH
enzyme release compared with that of the D-GaIN-only treatment
group (P<0.05; Fig. 1B).
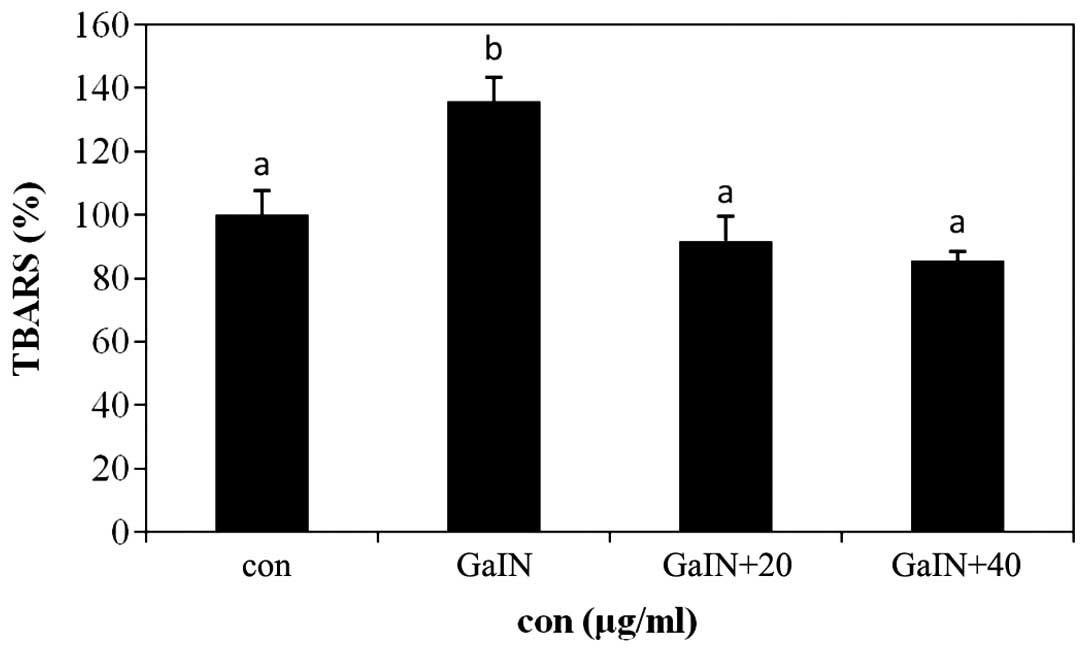
Inhibitory effect of Hepa 1c1c7 cells on
TBARS production using PYGP
Lipid peroxidation induces TBARS generation and cell
damage through oxidative stress; in addition, TBARS and lipid
peroxide produced during oxidative stress may cause or aggravate
diseases associated with aging and hepatotoxicity (17). The results of the present study
revealed that levels of TBARS were increased in the D-GaIN-only
treatment group compared with those of the control group. In
addition, pre-treatment with PYGP significantly inhibited the
D-GaIN-induced increase in TBARS levels (P<0.05; Fig. 2).
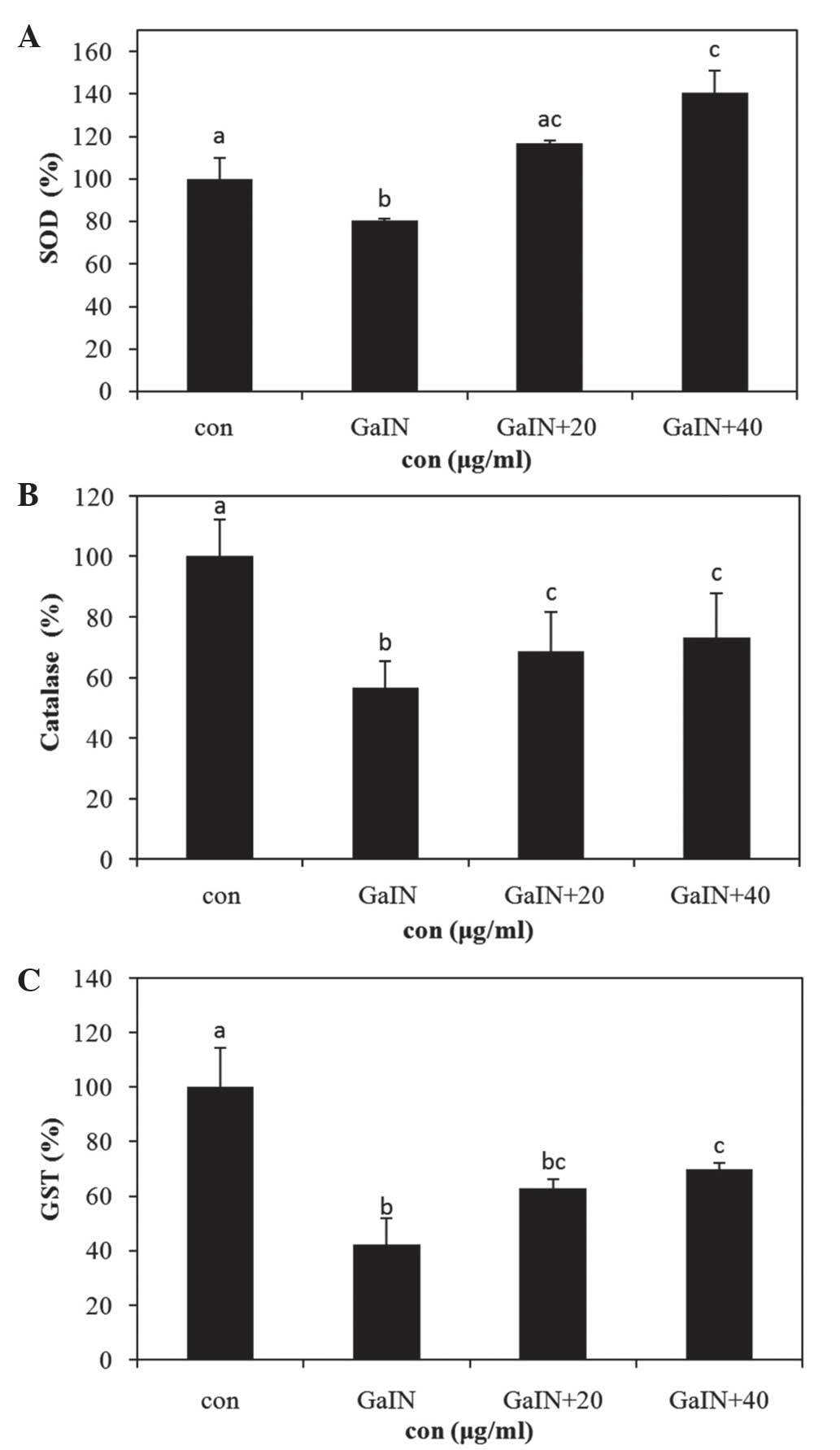
Increased antioxidant enzyme activity in
Hepa 1c1c7 cells in the presence of PYGP
CAT, SOD and GST are the primary defensive enzymatic
antioxidants in eukaryotic cells (18). The activity levels of these enzymes
were all found to be significantly inhibited in the D-GaIN-only
treatment group compared with those in the control group. By
contrast, the PYGP pre-treatment group showed increased antioxidant
enzyme activity compared with that in the D-GaIN-only treatment
group (P<0.05; Fig. 3).
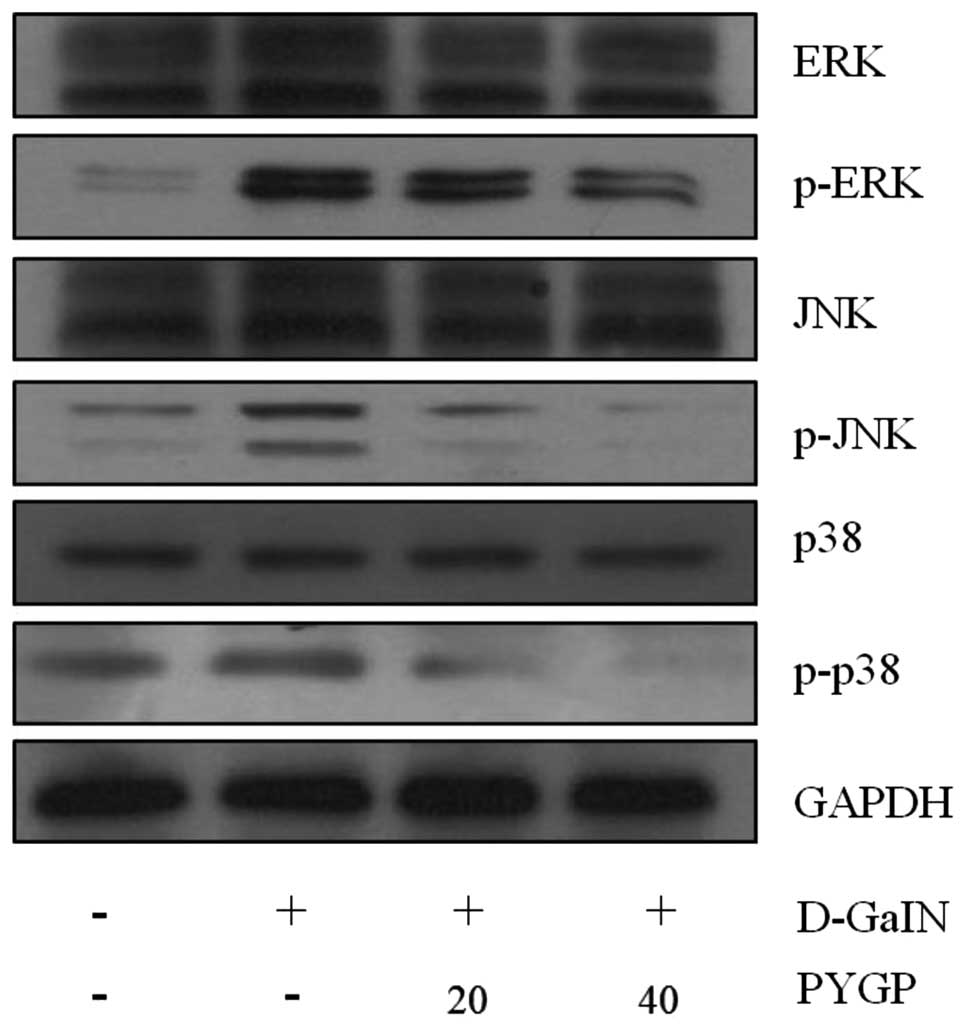
Effect of PYGP pre-treatment on
GaIN-induced expression and phosphorlyation of via MAPKs
ERK, JNK and p38 MAPK are known to be phosphorylated
and activated in response to D-GaIN (11). In the present study, the
phosphorylation of each MAPK was examined using western blot
analysis. The results revealed that PYGP suppressed the
D-GaIN-induced activation of each MAPK; in addition, treatment with
40 μg/ml PYGP was observed to have a more suppressive effect
compared with that of 20 μg/ml PYGP. However, total ERK, JNK and
p38 MAPK protein expression levels were not altered following
treatment with D-GaIN or PYGP pre-treatment (Fig. 4).
PYGP upregulates the Nrf2 signaling
pathway
Nrf2 has previously been reported to increase the
expression of various oxidant defense proteins, including HO-1,
Nqo1 and GST (7,8). In the present study, western blot
analysis was used to examine the expression of these proteins
following D-GaIN-induced cell injury and pre-treatment with PYGP.
Cells exposed to D-GaIN only showed reduced Nrf2, HO-1, Nqo1 and
GST protein expression levels compared with those of the untreated
cells. By contrast, the expression of these Nrf2-stimulated
proteins following PYGP pre-treatment were significantly increased
compared with those in the D-GaIN-only group (Fig. 5).
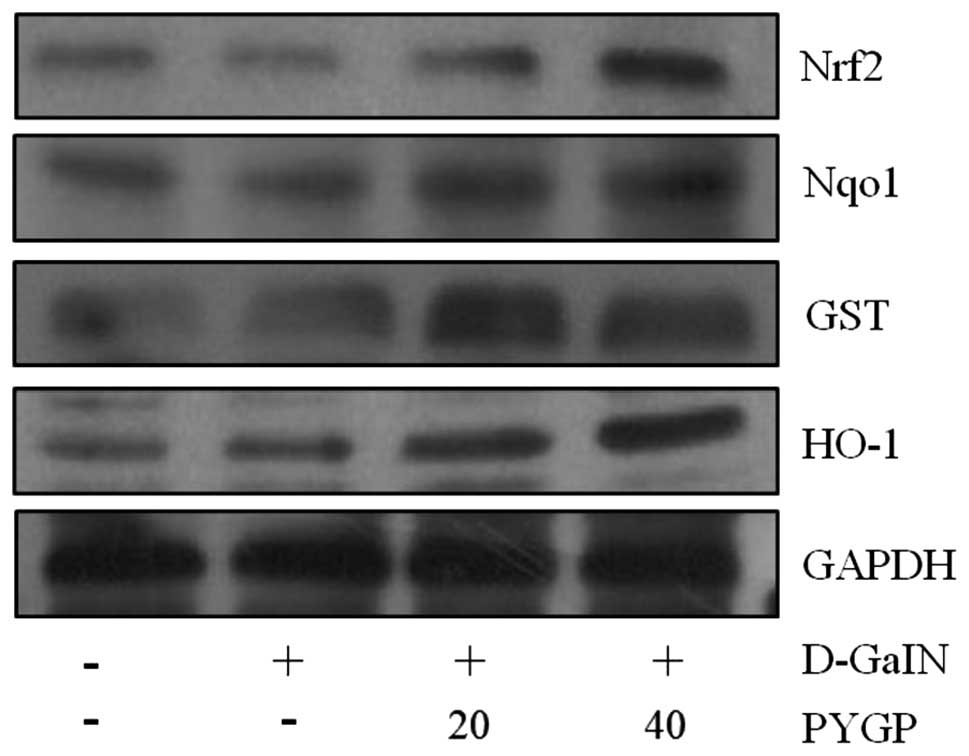 | Figure 5Effect of PYGP on D-GaIN-induced
expression of Nrf2, Nqo1, GST and HO-1 in Hepa 1c1c7 cells. Cells
were pre-treated with PYGP (20 and 40 μg/ml) for 24 h and then
administered 20 mM D-GaIN with PYGP (20 and 40 μg/ml) for 24 h.
Cell pellets were then collected using lysis buffer and western
blot analysis was performed in order to determine protein
expression levels of Nrf2, Nqol, GST and HO-1. PYGP, Porphyra
yezoensis glycoprotein; D-GaIN, D-galactosamine; Nrf2, nuclear
factor erythroid 2-related factor 2; Nqol, quinine oxidoreductase
1; GST, glutathione s-transferase; HO-1, heme oxygenase 1. |
Discussion
In the present study, cytotoxic injury was induced
in Hepa 1c1c7 cells using D-GaIN, which is a commonly used model
for screening anti-hepatotoxic and anti-hepatoxic activities of
drugs. D-GaIN-induced liver injury has previously been shown to
closely resemble acute viral hepatitis (19,20).
D-GaIN has direct and indirect roles, which affect the oxidative
stress properties of organs. Several studies have shown that D-GaIN
induced changes in liver antioxidant enzyme levels (17,21–23).
In addition, D-GaIN was reported to induce hepatotoxicity by
inhibiting RNA and protein synthesis as well as reducing uridine
5′-triphosphate, uridine 5′-diphosphate and uridine
5′-monophosphate levels (17,24,25).
Increased LDH release into the medium as a result of cell damage is
widely used as a measure of cytotoxicity (26). In the present study, D-GaIN
treatment was found to induce cytotoxicity in Hepa 1c1c7 cells, the
effect of which was attenuated in cells pre-treated with PYGP.
Peroxidation of endogenous lipids is a major factor
affecting the cytotoxic activity of D-GaIN (7). D-GaIN-induced oxidative stress damage
is generally attributed to the formation of highly reactive
hydroxyl radicals, such as superoxide anions, which stimulate lipid
peroxidation and damage cell membranes (4,27).
In the present study, TBARS was significantly increased following
D-GaIN-only treatment; however, pre-treatment with PYGP decreased
the oxidative damage in cells, based on the decreased levels of
TBARS compared to those in the D-GaIN-only group. SOD and CAT are
first-line cellular antioxidant defense enzymes. SOD reacts with
O2 in order to generate H2O2 and
H2O (13), while CAT
accelerates the dismutation reaction of H2O2
and the formation of H2O and O2 (28,29).
GST binds to numerous different lipophilic drugs and chemicals;
thus, it likely binds to D-GaIN and functions as an enzyme for GSH
conjugation reactions (30). In
the present study, lipid peroxidation induced by D-GaIN was
measured; following 24 h of exposure to D-GaIN, there was a
significant increase in TBARS levels compared with those of the
control group. However, the PYGP pre-treatment groups showed
reduced TBARS levels compared with those in the D-GaIN-only group.
Furthermore, D-GaIN significantly decreased the activity levels of
the antioxidant enzymes CAT, GST and SOD compared with those in the
control group, while pretreatment with PYGP increased these enzyme
levels compared with those in the D-GaIN-only treatment group.
The MAPK signaling pathway is an important signaling
pathway which regulates tumor necrosis factor (TNF)-α expression;
however, the detailed mechanism of this remains to be fully
elucidated (31,32). MAPKs have been confirmed to
participate in regulating cytokine production in response to a
broad range of stimuli (33,34).
MAPKs include three major proteins: ERK, JNK and p38 MAPK. These
proteins have important biological roles in cell proliferation,
differentiation, metabolism, survival and apoptosis (35). In the present study, levels of
their activated forms, p-ERK, p-JNK, and p-p38 MAPK, were observed
to be increased in the D-GaIN only treatment group compared with
those in the untreated control group; however, PYGP pre-treatment
attenuated the D-GaIN-mediated activation of ERK, JNK and p38
MAPK.
As a transcription factor, Nrf2 promotes the
translation of genes which have a protective effect against
oxidative/electrophilic stress (36,37).
Nrf2 exists as a binding repressor of kelch-like erythroid
cell-derived protein with CNC homology-associated protein 1 (Keap
1) in the cytoplasm (38). In
response to oxidative stress, Nrf2 dissociates from Keap 1 and
translocates to the nucleus in order to induce an array of
cytoprotective genes, including Nqo1, GST and HO-1 (39,40).
In the present study, western blot analysis revealed that Nrf2,
Nqo1, GST and HO-1 expression levels were decreased in the presence
of D-GaIN. However, PYGP pre-treatment increased Nrf2, Nqo1, GST
and HO-1 expression levels compared with those in the D-GaIN-only
treatment group. These results indicated that PYGP pre-treatment
upregulated Nrf2 protein levels and stimulated the activity of
antioxidants and phase II detoxifying enzymes.
In conclusion, the results of the present study
demonstrated that PYGP reduced D-GaIN-induced hepatotoxicity in
normal mouse Hepa 1c1c7 hepatocytes via upregulation of
antioxidative enzymes, MAPKs and the Nrf2 pathway. These findings
therefore indicated that PYGP may have potential for use for the
prevention of hepatotoxicity.
Acknowledgements
The present study was supported by the Fishery
Commercialization Technology Development Program through Korea
Institute of Planning and Evaluation for Technology in Food,
Agriculture, Forestry and Fisheries (iPET) funded by the Ministry
of Oceans and Fisheries (MOF; 2012300734).
References
|
1
|
Hwang JM, Tseng TH, Tsai YY, Lee HJ, Chou
FP, Wang CJ and Chu CY: Protective effects of baicalein on
tert-butyl hydroperoxide-induced hepatic toxicity in rat
hepatocytes. J Biomed Sci. 12:389–397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jose M, Javier C, Javier FP, et al:
S-Adenosylmethionine in alcoholic liver cirrhosis: a randomized,
placebo-controlled, double-blind, multicenter clinical trial. J
Hepatol. 30:1081–1089. 1999. View Article : Google Scholar
|
|
3
|
MacDonald JR, Thayer KJ and White C:
Inhibition of galactosamine cytotoxicity in an in vivo/in vitro
hepatocellular toxicity model. Toxicol Appl Pharmacol. 89:269–277.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sakaguchi S and Yokota K: Role of
Ca2+ on endotoxin-sensitivity by galactosamine
challenge: lipid peroxide formation and hepatotoxicity in zymosan
primed mice. Pharmacol Toxicol. 77:81–86. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hou CC, Huang CC and Shyur LF: Echinacea
alkamides prevent lipopolysaccharide/D-galactosamine-induced acute
hepatic injury through JNK pathway-mediated HO-1 expression. J
Agric Food Chem. 59:11966–11974. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cho HI, Park JH, Choi HS, Kwak JH, Lee DU,
Lee SK and Lee SM: Protective mechanisms of acacetin against
D-galactosamine and lipopolysaccharide-induced fulminant hepatic
failure in mice. J Nat Prod. 77:2497–2503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Das J, Ghosh J, Roy A and Sil PC:
Mangiferin exerts hepatoprotective activity against D-galactosamine
induced acute toxicity and oxidative/nitrosative stress via
Nrf2-NFκB pathways. Toxicol Appl Pharmacol. 260:35–47. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rajesh KT, Kim HM, Sorachai S, Thomas WK,
Masayuki Y and Shyam B: Identification of Nrf2-regulated genes
induced by the chemopreventive agent sulforaphane by
oligonucleotide microarray. Cancer Res. 62:5196–5203. 2002.
|
|
9
|
Martin E, Victoria B, Walter M, Andrea GL
and Patricia GV: The Nrf2-Keap1 cellular defense pathway and heat
shock protein 70 (Hsp70) response. Role in protection against
oxidative stress in early neonatal unilateral ureteral obstruction
(UUO). Cell Stress Chaperones. 16:57–68. 2011. View Article : Google Scholar
|
|
10
|
Dhakshinamoorthy S and Jaiswal AK:
Functional characterization and role of INrf2 in antioxidant
response element-mediated expression and antioxidant induction of
NAD (P) H:quinine oxidoreductase 1 gene. Oncogene. 20:3906–3917.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nishioka H, Kishioka T, Iida C, Fujii K,
Ichi I and Kojo S: Activation of mitogen activated protein kinase
(MAPK) during D-galactosamine intoxication in the rat liver. Bioorg
Med Chem Lett. 16:3019–3022. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sally KN, Swapan KB, Gary KG, Paul M and
Joe MM: The induction of human superoxide dismutase and catalase in
vivo: A fundamentally new approach to antioxidant therapy. Free
Radic Biol Med. 40:341–347. 2006. View Article : Google Scholar
|
|
13
|
Nirupama M and Friedrich HM: reactive
oxygen species: response of algal cells. J Plant Physiol.
157:183–193. 2000. View Article : Google Scholar
|
|
14
|
Chao ES, Dunbar D and Kaminsky LS:
Intracellular lactate dehydrogenase concentration as an index of
cytotoxicity in rat hepatocyte pri mary culture. Cell Biol Toxicol.
4:1–11. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jauregui HO, Hayner NT, Driscoll JL,
Williams-Holland R, Lipsky MH and Galletti PM: Trypan blue dye
uptake and lactate dehydrogenase in adult rat hepatocytes-freshly
isolated cells, cell suspension and primary mono layer cultures. In
Vitro. 17:1100–1110. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Allocati N, Federici L, Masulli M and Di
Ilio C: Glutathione transferases in bacteria. Febs J. 276:L58–L75.
2009. View Article : Google Scholar
|
|
17
|
Pushpavalli G, Kalaiarasi P, Veeramani C
and Pugalendi KV: Effect of chrysin on hepatoprotective and
antioxidant status in D-galactosamine-induced hepatitis in rat. Eur
J Pharmacol. 631:36–41. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Halliwell B: Biochemistry of oxidative
stress. Biochem Soc Trans. 35:1147–1150. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Decker K and Keppler D: Galactosamine
induced liver injury. Proq Liver Dis. 4:183–199. 1972.
|
|
20
|
Nakagiri R, Hashizume E, Kavahashi S,
Sakai Y and Kamiya T: Suppression by hydrangea Dulcis folium of
D-galactosamine-induced liver injury in vitro and in vivo. Biosci
Biotechnol Biochem. 67:2641–2643. 2003. View Article : Google Scholar
|
|
21
|
Han KH, Hashimoto N, Shimada K, Sekikawa
M, Noda T, Yamauchi H, Hashimoto M, Chiji H, Topping DL and
Fukushima M: Hepatoprotective effects of purple potato extract
against D-galactosamine-induced liver injury in rats. Biosci
Biotechnol Biochem. 70:1432–1437. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi Y, Sun J, He H, Guo H and Zhang S:
Hepatoprotective effects of Ganoderma lucidum peptides against
D-galactosamine-induced liver injury in mice. J Ethnopharmacol.
117:415–419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lim HK, Kim HS, Choi HS, Oh S, Jang CG,
Choi J, Kim SH and Chang MJ: Effects of acetylbergenin against
D-galactosamine-induced hepatotoxicity in rats. Pharmacol Res.
42:471–474. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang XH, Gao L, Gao J, Fan YM, Xu LZ, Zhao
XN and Xu Q: Mechanisms of hepatoprotection of Terminalia catappa
L. extract on D-galactosamine-induced liver damage. AM J Chin Med.
32:509–519. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aristatile B, Al-Numair KS, Al-Assaf AH
and Pugalendi KV: Pharmacological effect of carvacrol on
D-galactosamine-induced mitochondrial enzymes and DNA damage by
single-cell gel electrophoresis. J Nat Med. 65:568–577. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu L and Yeh YY: Inhibition of
cholesterol biosynthesis by organosulfur compounds derived from
garlic. Lipids. 35:2000. View Article : Google Scholar
|
|
27
|
Halliwell B and Gutteridge JMC: Oxygen
toxicity, oxygen radicals, transition metals and disease. Biochem
J. 219:1–14. 1984.PubMed/NCBI
|
|
28
|
Jones P and Suggett A: The
catalase-hydrogen peroxide system. A theoretical appraisal of the
mechanism of catalase action. Biochem J. 110:621–629.
1968.PubMed/NCBI
|
|
29
|
Halliwell B: Antioxidant defence
mechanisms: from the beginning to the end (of the beginning). Free
Radic Res. 31:261–272. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Anandan R, Devi KP, Devaki T and
Govindaraju P: Preventive effects of Picrorhiza kurroa on
D-galactosamine-induced hepatitis in rats. J Clin Biochem Nutr.
25:87–95. 1998. View Article : Google Scholar
|
|
31
|
Guha M and Mackman N: LPS induction of
gene expression in human monocytes. Cell Signal. 13:85–94. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kawai T and Akira S: TLR signaling. Semin
Immunol. 19:24–32. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guan KL: The mitogen activated protein
kinase signal transduction pathway: from the cell surface to the
nucleus. Cell Signal. 6:581–589. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Johnson GL, Dohlman HG and Graves LM: MAPK
kinase kinases (MKKKs) as a target class for small-molecule
inhibition to modulate signaling networks and gene expression. Curr
Opin Chem Biol. 9:325–331. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Klaassen CD and Reisman SA: Nrf2 the
rescue: effects of the antioxidative/electrophilic response on the
liver. Toxicol Appl Pharmacol. 244:57–65. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Inoue H, Maeda-Yamamoto M, Nesumi A and
Murakami A: Delphinidin-3-O-galactoside protects mouse hepatocytes
from (−)-epigallocatechin-3-gallate-induced cytotoxicity via
upregulation of heme oxygenase-1 and heat shock protein 70. Nutr
Res. 32:357–364. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jaiswal AK: Nrf2 signaling in coordinated
activation of antioxidant gene expression. Free Radic Biol Med.
36:1199–1207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kensler TW, Wakabayash N and Biswal S:
Cell survival responses to environmental stresses via the
Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 47:86–116.
2007. View Article : Google Scholar
|
|
40
|
Wu KC, Cui JY and Klaassen CD: Effect of
graded Nrf2 activation on phase-I and II drug metabolizing enzymes
and transporters in mouse liver. Plos One. 7:e390062012. View Article : Google Scholar
|