Introduction
Inflammation is important in the pathology of
cerebral ischemic injury. Cerebral ischemia induces a marked
inflammatory response characterized by the activation and release
of cytokines, chemokines, adhesion molecules and endotoxins, which
exacerbate tissue damage (1).
Toll-like receptors (TLRs) have important functions in cerebral
ischemia/reperfusion injury (2).
TLRs and their ligands provide an important molecular interface
between microbes and immunity and experimental cerebral ischemia
upregulates TLR4 expression (3,4). In
addition, the brain damage and neurological deficits produced by
cerebral ischemia significantly decrease in TLR4-deficient mice
compared with wild-type mice (5).
Volatile anesthetic pretreatment significantly protects against
cerebral ischemia through several mechanisms (6,7).
Isoflurane (ISO) pretreatment inhibits excitotoxicity and reduces
glutamate release from anoxic brain slices in vitro
(8). ISO also inhibits
postsynaptic glutamate receptor-mediated responses in neocortical
and hippocampal cells (9–11) and rat brain slices (12). However, the exact mechanisms
underlying this protective effect remain to be elucidated.
Considering the function of the TLR4-mediated myeloid
differentiation primary response 88 (MyD88)-dependent signaling
pathway, the present study hypothesized that ISO pretreatment is
associated with the TLR4 signaling pathway during cerebral
ischemia/reperfusion damage. The association between the
TLR4-induced inflammatory response and ISO pretreatment has
attracted considerable interest in developing anti-inflammatory
therapies to combat ischemia-induced damage.
Astrocytes are the most abundant type of glial cell
within the brain. Although microglial cells are the first
inflammatory cells to respond to cerebral ischemia, the responses
of astrocytes to pro-inflammatory signals may also be relevant to
brain injury (13–15). However, the association between
inflammatory glial cells and TLR4 has not been fully elucidated.
The effects of ISO pretreatment on TLR4 expression in astrocytes
and microglia were investigated to increase understanding of the
association between glial cells and TLR4 signaling pathways during
inflammation. The anti-inflammatory effects associated with the
TLR4-mediated MyD88 activation pathway in the cerebral ischemic
brain were examined and the downstream molecules present in rats
with cerebral ischemia were further investigated.
Materials and methods
Animal surgical procedures
ISO was purchased from Abbott Laboratories (North
Chicago, IL, USA) and was stored in the dark. Pretreatment with 2%
ISO for 60 min was deemed sufficient to induce ischemic tolerance
(16,17). Male Sprague-Dawley rats weighing
between 250 and 270 g were provided by the Experimental Animal
Center of the Fourth Military Medical University (Xi’an, China).
The present study strictly followed the recommendations of the
Guide for the Care and Use of Laboratory Animals of the National
Institutes of Health (Bethesda, MD, USA). The animal use protocol
was reviewed and approved by the Institutional Animal Care and Use
Committee of the Fourth Military Medical University. Cerebral
ischemia was induced via middle cerebral artery occlusion (MCAO)
using the technique by Longa et al (18,19).
The right common carotid artery and the right external carotid
artery (ECA) were exposed through a ventral midline neck incision
and ligated proximally. A 3-0 nylon monofilament with a
silicone-coated tip was inserted from the distal end of the
isolated ECA and gently introduced to the internal carotid artery
(ICA). The microfilament was then advanced into the ICA ~18–20 mm
distal to the carotid bifurcation until mild resistance was felt.
The neck incision was closed with sutures and was left in place for
120 min to induce ischemia prior to reperfusion. The rectal
temperatures were monitored and maintained between 37.0 and 37.5°C
during the procedure.
Experimental procedures
The rats were randomly assigned into naive, sham
O2, sham ISO, MCAO-O2 and MCAO-ISO groups to
detect the effects of ISO pretreatment on the infarct volume, water
content, immunofluorescence staining and protein expression of
TLR4, MyD88 and nuclear factor (NF)-κB in cerebral ischemia. The
MCAO-O2 group received 60% O2/40%
N2 at 60 min and 24 h after the last pretreatment,
followed by the induction of focal cerebral ischemia. The MCAO-ISO
group received 2% ISO in O2 at 60 min and 24 h after the
last pretreatment, followed by the induction of focal cerebral
ischemia. The sham-O2 group received (60%
O2/40% N2) at 60 min and 24 h after the last
pretreatment, whereas the sham-ISO group received 2% ISO in
O2 at 60 min and 24 h after the last pretreatment. The
right common carotid artery and the right ECA were subsequently
exposed without the insertion of the monofilament into the artery.
The temperature was maintained at ~25°C during and following
surgery. The animals were exposed to incandescent lamps to maintain
their rectal temperatures at 37.0±0.5°C until pallanesthesia.
Neurological assessment
At 24 h after reperfusion, the rats in each group
(n=12) were neurologically assessed by an examiner in a blinded
manner. The deficits were scored using a modified scoring system
based on the system developed by Longa et al (19): 0, no deficits; 1, difficulty in
fully extending the contralateral forelimb; 2, unable to extend the
contralateral forelimb; 3, mild circling to the contralateral side;
4, severe circling and 5, falling to the contralateral side.
Measurement of infarct volume
The rats in each group (n=12) were anesthetized with
intraperitoneal sodium pentobarbital (50 mg/kg) 24 h after
ischemia. The brains were removed quickly and subsequently cut into
a total of seven 2 mm coronal sections. The sections were immersed
for 30 min in 2% triphenyltetrazolium chloride at 37°C and
subsequently fixed with 4% paraformaldehyde. At 24 h after
fixation, images were captured of the brain slices with a digital
camera (Sony A350; Sony Corporation, Tokyo, Japan) connected to a
personal computer.
Assessment of brain edema (brain water
content)
Brain water content was measured using the dry-wet
weight technique (20). A total of
12 rats in each group were sacrificed and their brain tissues were
quickly harvested. The 2 mm-thick frontal pole was removed. The 2
mm-thick brain tissue posterior to the frontal pole was selected to
measure water content. Following determination of the wet weight
(WW) using an electronic balance, the brain tissue was placed in an
oven at constant temperature (100±5°C) for 24 h. The dry weight
(DW) was subsequently measured. Water content was calculated based
on the following formula: (WW − DW) / WW × 100.
Immunohistochemistry
Immunofluorescence photomicro-graphs of the
lesion-associated cortex were obtained following the MCAO
treatment. After 24 h, the rats were anaesthetized using 6.0%
isoflurane and their brains were removed quickly and immersed in 4%
paraformaldehyde/0.1 M phosphate-buffered saline (PBS) for 2 h at
4°C. The cryostat sections (25 μm) in the first dish were
rinsed three times (10 min each) in 0.01 M PBS, pH 7.3 and
subsequently blocked with 2% goat serum in 0.01 M PBS containing
0.3% Triton X-100 for 1 h at room temperature. The samples were
then subjected to immunofluorescence staining.
To evaluate colocalization, a double
immunohistochemical analysis was performed, in which sections were
incubated for 48 h at 4°C with the primary antibodies TLR4 goat
polyclonal antibodies (1:200; Abcam, Cambridge, UK) with mouse
anti-cd11b clone OX-42 (1:300; Abcam) or mouse anti-glial
fibrillary acidic protein (GFAP; 1:5,000; Chemicon, Temecula, CA,
USA). The sections were washed three times in 0.01 M PBS (10 min
each) and then incubated for 4 h at room temperature with the
secondary antibodies: Fluorescein isothiocyanate-conjugated horse
anti-mouse immunoglobulin G (1:200; Vector, Burlingame, CA, USA)
and Alexa 594-conjugated donkey anti-rabbit IgG (1:800; Molecular
Probes, Rockford, IL, USA). For TLR4/GFAP and TLR4/OX-42 double
immunofluorescence, the sections were incubated with a mixture of
the two primary antibodies, followed by a mixture of the two
respective secondary antibodies. The staining specificities were
assessed on the sections in the second dish by omitting the
specific primary antibodies. No immunoreactive (IR) products were
identified on the sections (data not shown). Confocal images were
obtained using a confocal laser microscope (FV1000; Olympus, Tokyo,
Japan) and digital images were captured with the Fluoview 1000
microscope (Olympus).
Western blot analysis
The protein expression levels of MyD88, NF-κB and
TLR4 were detected using western blot analysis. The tissue samples
obtained from the right ischemic MCA were subjected to 120 min of
MCA occlusion, with reperfusion after 24, 48 and 72 h. The tissues
were subsequently lysed with radio-immunoprecipitation assay lysis
buffer, homogenized and then centrifuged at 12,000 rpm for 20 min
at 4°C. The protein content of the supernatants were quantified
using a bicinchoninic acid protein assay reagent kit (Beyotime
Institute of Biotechnology, Shanghai, China). The remaining
supernatants were boiled in sodium dodecyl sulfate (SDS) sample
buffer for 5 min. Equal quantities of the protein were run on
SDS/polyacrylamide gel electrophoresis and electrophoretically
transferred onto polyvinylidene fluoride membranes. Following
blocking, the blots were immersed overnight in TLR4 goat polyclonal
antibodies (1:200; Abcam), MyD88 rabbit polyclonal antibodies
(1:200; Abcam) or NF-κB rabbit polyclonal antibodies (1:400; Abcam)
at 4°C. The membranes were rinsed and further incubated for 1 h
with horseradish peroxidase-conjugated secondary antibodies
(1:1,000; Vector). The bound antibodies were exposed to an Amersham
Hyperfilm™ ECL (GE Healthcare, Amersham, UK). The relative
densities of the bands were analyzed and were corrected using the
values determined with anti-rat β-actin, which was used as the
internal control. The densities of the protein blots were analyzed
using Labworks Software Gel-Pro analyzer 4.0 (Ultra-Violet
Products, Cambridge, UK). The densities of TLR4, MyD88, NF-κB and
β-actin IR bands were quantified with background subtraction.
Squares of identical sizes were drawn surrounding each band to
measure density and the background near that band was subtracted.
The β-actin levels were used as loading controls as they did not
alter significantly following inflammation and nerve injury
(21). The expression levels of
TLR4, MyD88 and NF-κB were normalized against the β-actin levels
and were expressed as fold change relative to the control.
Quantification and statistical
analysis
All the data were collected and analyzed by
researchers in a blinded manner. A total of five nonadjacent
sections (25 μm) from the lesion-associated cortex sections
were randomly selected. In each group, 12 rats were used for
statistical analysis. The images were evaluated using a
computer-assisted image analysis program (MetaMorph 6.1; Universal
Imaging Corp., West Chester, PA, USA), which set the upper and
lower thresholds for the immunofluorescence intensity determined by
the signal. The images were collected using the same region and the
same field sizes within the same lamina to avoid variations in
staining between the laminae. The same configuration was used to
measure the cell areas in all the experimental groups. The measured
areas were automatically transferred to Microsoft Excel software
(Microsoft Campus, Redmond, WA, USA) for subsequent statistical
analysis. MetaMorph 6.1 was calibrated to standardize area
measurements. A standardized field area was sampled arbitrarily
from regions within the randomly selected lesion-associated cortex
sections.
The number of TLR4-positive cells within the same
areas was counted. The GFAP and OX-42 immunoreactivities and the
total number of TLR4-immunopositive cells within the superficial
cortex were averaged across the five spinal sections for each
experimental group. The number of cells double-labeled with TLR4
and GFAP or OX-42 in the superficial cortex were counted and the
proportion of double-labeled GFAP or OX-42 cells compared with the
total TLR4-positive cells was calculated. The data from the western
blot analysis are expressed as the mean ± standard deviation. A
repeated measures analysis of variance with the Bonferroni
confidence interval adjustment was conducted. P<0.05 was
considered to indicate a statistically significant difference.
Results
ISO pretreatment attenuates neurological
deficits, brain edema and cerebral infarct size following
ischemia/reperfusion
The neurological deficit scores of the MCAO-ISO
group at 24 h after reperfusion were lower than those of the
MCAO-O2 groups (Table
I). No changes were observed in the sham-O2, sham-ISO and naive
groups. The infarct volume of the MCAO-ISO group was significantly
reduced compared with that of the MCAO-O2 group 24 h
after reperfusion (9.12±0.07 vs. 16.23±0.05%; P<0.05). The
sham-O2, sham-ISO and naive groups had low infarct
volumes. The prevention of brain edema is critical for the
preservation of neurological function and survival following focal
cerebral ischemia/reperfusion. The brain water content was examined
in cerebral tissue from the left hemisphere 24 h after cerebral
ischemia/reperfusion. As shown in Fig.
1C, the brain water content was significantly increased in the
MCAO-O2 group compared with the sham-O2 group
(87.29±0.09 vs. 79.60±0.17%). However, the brain water content in
the MCAO-ISO group was significantly lower than that in the
MCAO-O2 group (82.61±0.08 vs. 87.29±0.09%; P<0.05).
No significant difference in brain water content was detected among
the sham-O2, sham-ISO and naive groups (Fig. 1).
 | Table INeurological deficit scores 24 h
after reperfusion from 120 min of middle cerebral artery occlusion
in the rat. |
Table I
Neurological deficit scores 24 h
after reperfusion from 120 min of middle cerebral artery occlusion
in the rat.
| Group | Neurological
deficit score
| Median
(range) |
|---|
| 0 | 1 | 2 | 3 | 4 | 5 |
|---|
| Naive | 9 | 3 | | | | | 0 (0–1) |
|
Sham-O2 | 8 | 4 | | | | | 0 (0–1) |
| Sham-ISO | 8 | 4 | | | | | 0 (0–1) |
|
MCAO-O2 | | | 2 | 5 | 5 | | 3 (2–4) |
| MCAO-ISO | | 5 | 7 | | | | 2 (1–2) |
ISO pretreatment reduces the protein
expression of TLR4 and significantly attenuates astrocytic and
microglial levels
Tissue sections of the right cortex were observed
and the cerebral histological alterations caused by MCAO in rats
were assessed using a confocal laser-scanning microscope.
Immunofluorescence histochemistry was employed to demonstrate TLR4
expression in the different groups. In order to rule out possible
grouping and treatment bias, the number of TLR4-IR cells was
normalized to that in naive rats 24 h after MCAO. MCAO
significantly increased TLR4 expression in the cerebral cortex,
which predominantly concentrated in the lesion-associated cortex.
However, ISO pretreatment significantly inhibited MCAO-induced TLR4
protein expression in the cerebral cortex. Statistical analysis
revealed that ISO pretreatment significantly affected the
MCAO-induced cerebral cortex TLR4 expression. The TLR4
immunodensity in the MCAO-ISO rats was notably decreased by ISO
pretreatment compared with that in the MCAO-O2 group. No
alterations were observed in the sham-O2, sham-ISO and
naive groups (Fig. 2).
Similarly to previous observations (22), the present results demonstrated
that MCAO significantly induced GFAP and OX-42 expression in the
lesion associated cortex. Immunohistochemistry indicated that the
activated astrocytes and microglia exhibited hypertrophied cell
bodies and thickened processes with enhanced GFAP immunoreactivity
and OX-42 immunoreactivity. Double immunofluorescence labeling
indicated that multiple GFAP-IR cells were positive for TLR4,
whereas fewer OX-42-IR cells were interspersed with activated
microglia (Fig. 3). The activated
astrocytes and microglia were predominantly distributed in the
superficial cortex.
ISO pretreatment affected MCAO-induced TLR4 protein
expression in the cerebral cortex. The immunodensities of GFAP and
OX-42 in the MCAO rats were notably decreased by ISO pretreatment
compared with those in the sham-O2 and sham-ISO groups
(Fig. 4).
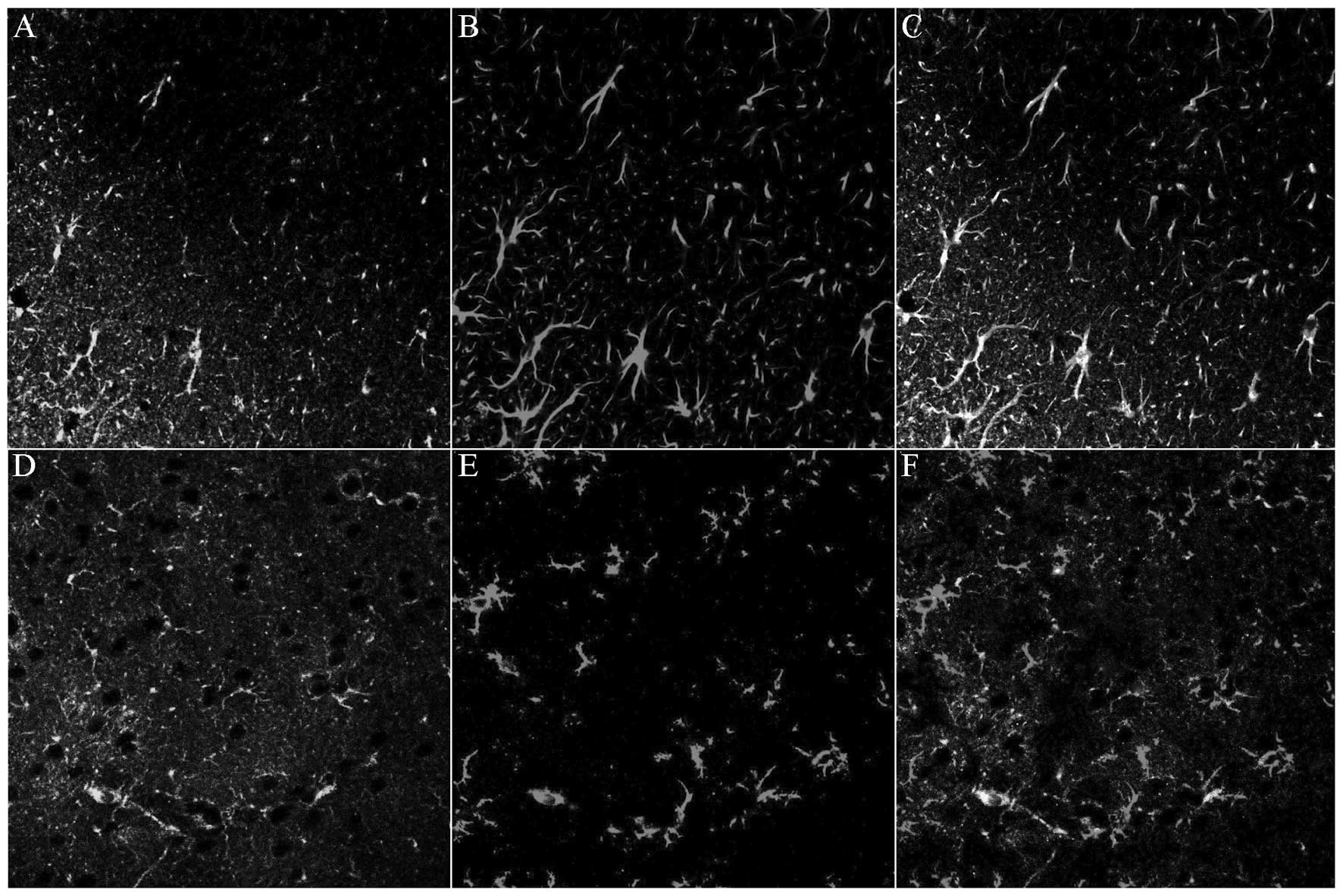
 | Figure 4Expression levels of (A–D) GFAP and
(E H) OX-42 in the lesion associated cortex in sham-O2, sham-ISO,
MCAO-O2 and MCAO-ISO groups, respectively, following
MCAO. Compared with the MCAO-O2-treated group, MCAO-ISO
produced a significant inhibitory effect on GFAP and OX-42 protein
expression, as well as on astrocyte and microglia inhibition in the
cerebral cortex. GFAP and OX-42 labeled astrocytes and microglia
observed in the cerebral cortex were counted. (I and J) Statistical
results of these comparisons. TLR4, toll-like receptor 4; GFAP,
glial fibrillary acidic protein; MCAO, middle cerebral artery
occlusion; ISO, isoflurane. |
The data suggested that TLR4 is predominantly
expressed in the astrocytes and microglia within the
lesion-associated region. ISO pretreatment also suppressed
MCAO-induced GFAP and OX-42 expression and inhibited astrocytic and
microglial activation.
ISO pretreatment attenuates the protein
expression levels of TLR4, MyD88 and NF-κB
TLR4-mediated NF-κB signaling contributes to
myocardial ischemia-reperfusion injury (23). Quantitative studies using western
blot analysis were performed to further observe whether ISO
pretreatment affected the expression levels of TLR4, MyD88 and
NF-κB.
The protein expression levels of TLR4, MyD88 and
NF-κB were examined in the right cortex using western blot analysis
at 24, 48 and 72 h. As shown in Fig.
5, ISO pretreatment significantly inhibited MCAO-induced TLR4
expression compared with that in the MCAO-O2 group. The
protein level of TLR4 peaked at 24 h and lasted up to 48 h.
Thereafter, the protein level of TLR4 gradually decreased until 72
h, although it remained significantly higher than those in the
sham-O2, sham-ISO and naive groups. No significant difference in
TLR4 expression was observed among the sham-O2, sham-ISO and naive
groups. Furthermore, in accordance with the immunohistochemical
results, the differences between the MCAO-O2 and
MCAO-ISO groups were significant (P<0.05; Fig. 4).
Similar to the effect of ISO pretreatment on Myd88
and NF-κB expression in the MCAO model, ISO pretreatment
significantly downregulated Myd88 expression following MCAO
compared with that in the MCAO-O2 group (P<0.05). The
protein level of Myd88 peaked at 48 h and then declined at 72 h,
although it remained significantly higher than those in the
sham-O2, sham-ISO and naive groups. No significant difference in
Myd88 expression was observed among the sham-O2,
sham-ISO and naive groups (Fig.
6). ISO pretreatment significantly inhibited MCAO-induced NF-κB
expression compared with that in the MCAO-O2 group
(P<0.05). The protein level of NF-κB peaked at 48 h and then
declined at 72 h, although it remained significantly higher than
those in the sham-O2, sham-ISO and naive groups.
Furthermore, no significant difference in NF-κB expression was
observed among the sham-O2, sham-ISO and naive groups (Fig. 7).
Discussion
In the present study, the effects of ISO
pretreatment on MCAO-induced TLR4, Myd88 and NF-κB expression were
examined, as well as on astrocytic and microglial activation. The
present findings established the following: i) ISO pretreatment
attenuated the neurological deficits, brain edema and cerebral
infarct volume caused by ischemia/reperfusion; ii) ISO pretreatment
potently inhibits TLR4 expression and significantly inhibited
astrocytic and microglial activation in brain tissues and iii) ISO
pretreatment downregulates the protein expression of TLR4, MyD88
and NF-κB.
The anti inflammatory effects of ISO during cerebral
ischemia injury have attracted attention in previous years.
Repeated 1 h ISO treatment induces a dose-dependent neuroprotection
against subsequent ischemic injury (24). Additionally, ISO pretreatment
reduces the infarct size after a 48 h delay; however, the
protective effect of ISO pretreatment may decrease after 72 h
(25). In the present study, the
potential involvement of TLR4 in the anti-inflammatory activity
elicited by ISO pretreatment was examined. The present study
revealed that pretreatment with 2% ISO (1 h/day) for 5 days
provided significant neuroprotection against the neurological
injury induced by MCAO. ISO pretreatment also reduced the water
content and volume of the infarct in the ischemic brain tissue.
Cerebral infarction is a complex pathophysiological
process, which involves multiple factors (26). Injured tissue releases endogenous
molecules, which in turn induce an inflammatory immune response. A
number of these endogenous components are ligands for TLR4
(27). TLR4 has important
functions in cerebral ischemia and in the induction of
immunoinflammatory reactions (28). TLR4, as a key component of the
innate immune system, functions as a pattern recognition receptor
that recognizes lipopolysaccharide (LPS). LPS is one of the most
immunostimulatory glycolipid components of the outer membrane of
Gram negative bacteria (29). The
TLR4-mediated MyD88-dependent signaling pathway is essential for
NF-κB activation (30). MyD88
recruitment to the toll interleukin receptor domain of TLR4
activates NF-κB, which is involved in cerebral ischemia/reperfusion
injury (31,32).
In the present study, MCAO significantly increased
TLR4 expression in the cerebral cortex and was more concentrated in
the lesion-associated cortex. However, TLR4 declined following ISO
pretreatment. In addition, ISO pretreatment potently inhibited TLR4
expression, which was correlated with a decrease in astrocytic and
microglial activation. In order to understand the mechanisms
involved in regulating the inflammatory response during cerebral
ischemia, TLR4 colocalization in astrocytes and microglial cells
was investigated. Astrocytes and microglial cells are involved in
the local innate immune response triggered by various stressors
(33,22). Certain functions of astrocytes and
microglia are ambiguous (34).
When activated, microglial cells rapidly migrate to the sites of
brain damage and clear debris to maintain the integrity of the
central nervous system (35,36).
Astrocytes secrete soluble mediators, which affect the innate and
adaptive immune responses (37,15).
The two cell types produce neuroprotective growth factors and
neurotrophins, which repair brain tissue. However, microglial cells
can cause severe neuronal damage when pro-inflammatory immune
mediators are produced excessively (35). By contrast, astrocytes promote
inflammation through NF-κB-dependent pathways (38,39).
In the present study, immunohistochemical staining demonstrated
that activated astrocytes and microglia in the MCAO-O2
group exhibited hypertrophied cell bodies and thickened processes
with enhanced GFAP immunoreactivity and OX-42 immunereactivity.
Double immunofluorescence labeling indicated that several of the
GFAP-IR cells were positive for TLR4, whereas a few OX-42-IR cells
overlapped with TLR4. Notably, it was observed that the
immunodensity of GFAP and OX-42 in the MCAO rats significantly
decreased following ISO pretreatment compared with those in the
sham-O2 and sham-ISO groups.
The protein levels of TLR4, Myd88 and NF-κB in each
group were determined using western blot analysis to determine
whether or not ISO pretreatment protects rat brains against focal
ischemia through the TLR4-mediated MyD88-dependent signaling
pathway. The protein level of TLR4 in the MCAO-O2 group
was significantly higher than that in the MCAO-ISO group. The
expression level of TLR4 in the ischemic brains peaked at 24 h,
lasted until 48 h and then gradually decreased at 72 h. These
findings suggested that the inflammatory peak occurred after 24 h
of ischemia. The expression level of MyD88 in the ischemic brains
increased at 48 h but decreased at 72 h. In addition, the
expression level of NF-κB increased at 48 h but decreased 72 h
after MCAO. These results suggested that the level of TLR4 in the
right cortex is attenuated by ISO pretreatment during cerebral
ischemia. The pattern of MyD88 and NF-κB expression was similar to
that of TLR4 expression.
The protein expression levels of MyD88 and NF-κB
increased later than that of the TLR4 protein. This result can be
attributed to the involvement of MyD88 and NF-κB in the
TLR4-mediated signaling pathway activated by cerebral ischemia.
In conclusion, the present study confirmed that ISO
pretreatment protected the brain from the damage caused by MCAO.
The neuroprotective effect of ISO pretreatment may be associated
with downregulation of TLR4, MyD88 and NF-κB expression.
Inflammatory responses mediated mainly by the activation of the
TLR4-MyD88 signaling pathway may have an important function in the
pathogenesis of cerebral ischemia. The effect of ISO pretreatment
on the TLR4-MyD88 signaling pathway is important during the acute
phase of ischemia and may provide new perspectives for therapeutic
targets in patients with cerebral ischemia.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant no. 81271343) and by the Shaanxi
Science and Technology research fund (grant no.
2014K11-02-01-14).
References
|
1
|
Fan H and Cook JA: Molecular mechanisms of
endotoxin tolerance. J Endotoxin Res. 10:71–84. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aderem A and Ulevitch RJ: Toll-like
receptors in the induction of the innate immune response. Nature.
406:782–787. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chakravarty S and Herkenham M: Toll-like
receptor 4 on nonhematopoietic cells sustains CNS inflammation
during endotoxemia, independent of systemic cytokines. J Neurosci.
25:1788–1796. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kaisho T and Akira S: Toll-like receptors
as adjuvant receptors. Biochim Biophys Acta. 1589:1–13. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kilic U, Kilic E, Matter CM, Bassetti CL
and Hermann DM: TLR-4 deficiency protects against focal cerebral
ischemia and axotomy-induced neurodegeneration. Neurobiol Dis.
31:33–40. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kitano H, Young JM, Cheng J, Wang L, Hurn
PD and Murphy SJ: Gender-specific response to isoflurane
preconditioning in focal cerebral ischemia. J Cereb Blood Flow
Metab. 27:1377–1386. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang J, Yang ZJ, Klaus JA, Koehler RC and
Huang J: Delayed tolerance with repetitive transient focal ischemic
preconditioning in the mouse. Stroke. 39:967–974. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eilers H and Bickler PE: Hypothermia and
isoflurane similarly inhibit glutamate release evoked by chemical
anoxia in rat cortical brain slices. Anesthesiology. 85:600–607.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Becker K, Eder M, Ranft A, et al: Low dose
isoflurane exerts opposing effects on neuronal network excitability
in neocortex and hippocampus. PLoS One. 7:e393462012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Puil E, El-Beheiry H and Baimbridge KG:
Anesthetic effects on glutamate-stimulated increase in
intraneuronal calcium. J Pharmacol Exp Ther. 255:955–961.
1990.PubMed/NCBI
|
|
11
|
Puil E and El-Beheiry H: Anaesthetic
suppression of transmitter actions in neocortex. Br J Pharmacol.
101:61–66. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bickler PE, Buck LT and Hansen BM: Effects
of isoflurane and hypothermia on glutamate receptor-mediated
calcium influx in brain slices. Anesthesiology. 81:1461–1469. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dong Y and Benveniste EN: Immune function
of astrocytes. Glia. 36:180–190. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ladeby R, Wirenfeldt M, Garcia-Ovejero D,
et al: Microglial cell population dynamics in the injured adult
central nervous system. Brain Res Brain Res Rev. 48:196–206. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song JH, Bellail A, Tse MC, Yong VW and
Hao C: Human astrocytes are resistant to Fas ligand and tumor
necrosis factor-related apoptosis-inducing ligand-induced
apoptosis. J Neurosci. 26:3299–3308. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li H, Yin J, Li L, Deng J, Feng C and Zuo
Z: Isoflurane post-conditioning reduces ischemia-induced nuclear
factor-kappaB activation and interleukin 1beta production to
provide neuroprotection in rats and mice. Neurobiol Dis.
54:216–224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bedirli N, Bagriacik EU, Emmez H, Yilmaz
G, Unal Y and Ozkose Z: Sevoflurane and isoflurane preconditioning
provides neuroprotection by inhibition of apoptosis-related mRNA
expression in a rat model of focal cerebral ischemia. J Neurosurg
Anesthesiol. 24:336–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen J, Graham SH, Zhu RL and Simon RP:
Stress proteins and tolerance to focal cerebral ischemia. J Cereb
Blood Flow Metab. 16:566–577. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shimamura N, Matchett G, Solaroglu I,
Tsubokawa T, Ohkuma H and Zhang J: Inhibition of integrin
alphavbeta3 reduces blood-brain barrier breakdown in focal ischemia
in rats. J Neurosci Res. 84:1837–1847. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo W, Wang H, Watanabe M, et al:
Glial-cytokine neuronal interactions underlying the mechanisms of
persistent pain. J Neurosci. 27:6006–6018. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang SC, Arumugam TV, Xu X, et al: Pivotal
role for neuronal Toll-like receptors in ischemic brain injury and
functional deficits. Proc Natl Acad Sci USA. 104:13798–13803. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hua F, Ma J, Ha T, et al: Activation of
Toll-like receptor 4 signaling contributes to hippocampal neuronal
death following global cerebral ischemia/reperfusion. J
Neuroimmunol. 190:101–111. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiong L, Zheng Y, Wu M, et al:
Preconditioning with isoflurane produces dose-dependent
neuroprotection via activation of adenosine triphosphate-regulated
potassium channels after focal cerebral ischemia in rats. Anesth
Analg. 96:233–237. 2003.
|
|
25
|
Kawaguchi M, Drummond JC, Cole DJ, Kelly
PJ, Spurlock MP and Patel PM: Effect of isoflurane on neuronal
apoptosis in rats subjected to focal cerebral ischemia. Anesth
Analg. 98:798–805. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hanisch UK and Kettenmann H: Microglia:
active sensor and versatile effector cells in the normal and
pathologic brain. Nat Neurosci. 10:1387–1394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kapinya KJ, Lowl D, Futterer C, et al:
Tolerance against ischemic neuronal injury can be induced by
volatile anesthetics and is inducible NO synthase dependent.
Stroke. 33:1889–1898. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee SJ and Lee S: Toll-like receptors and
inflammation in the CNS. Curr Drug Targets Inflamm Allergy.
1:181–191. 2002. View Article : Google Scholar
|
|
29
|
Beg AA: Endogenous ligands of Toll-like
receptors: implications for regulating inflammatory and immune
responses. Trends Immunol. 23:509–512. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Doyle SL and O’Neill LA: Toll-like
receptors: from the discovery of NFkappaB to new insights into
transcriptional regulations in innate immunity. Biochem Pharmacol.
72:1102–1113. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Baeuerle PA and Henkel T: Function and
activation of NF-kappaB in the immune system. Annu Rev Immunol.
12:141–179. 1994. View Article : Google Scholar
|
|
32
|
Seki E, Tsutsui H, Iimuro Y, et al:
Contribution of Toll-like receptor/myeloid differentiation factor
88 signaling to murine liver regeneration. Hepatology. 41:443–450.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Farina C, Aloisi F and Meinl E: Astrocytes
are active players in cerebral innate immunity. Trends Immunol.
28:138–145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Glezer I, Simard AR and Rivest S:
Neuroprotective role of the innate immune system by microglia.
Neuroscience. 147:867–883. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Aloisi F: Immune function of microglia.
Glia. 36:165–179. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Streit WJ, Conde JR, Fendrick SE, Flanary
BE and Mariani CL: Role of microglia in the central nervous
system’s immune response. Neurol Res. 27:685–691. 2005.PubMed/NCBI
|
|
37
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bowman CC, Rasley A, Tranguch SL and
Marriott I: Cultured astrocytes express toll-like receptors for
bacterial products. Glia. 43:281–291. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hang CH, Shi JX, Li JS, Wu W and Yin HX:
Concomitant upregulation of nuclear factor-κB activity,
proinflammatory cytokines and ICAM-1 in the injured brain after
cortical contusion trauma in a rat model. Neurol India. 53:312–317.
2005. View Article : Google Scholar : PubMed/NCBI
|