Introduction
Progressive pseudorheumatoid dysplasia (PPD; OMIM
#208230) is also termed spondyloepiphyseal dysplasia tarda with
progressive arthropathy, or arthropathy progressive
pseudorheumatoid of childhood. PPD is a rare autosomal recessive
hereditary disease, which is caused by mutations in the
Wntl-inducible signaling pathway protein 3 (WISP3) gene
(1,2). The WISP3 gene is located on
chromosome 6q22 and is composed of five coding exons, which encode
a 354-amino acid protein. WISP3 is maintains the stability of
cartilage by regulating the synthesis of type II collagen and
aggrecan, two major matrix components of articular cartilage, in
chondrocytes. The incidence rate of PPD in the UK is 1/1,000,000
(3). The disease is asymptomatic
at birth; however, progressive enlargement of numerous joints,
deformity and limitation of activity emerge with age, without
specificity in laboratory examination (4). PPD is often clinically misdiagnosed,
and should be distinguished from spondyloepiphyseal dysplasia tarda
and juvenile idiopathic arthritis in the early stages of the
disease. The aim of the present study was to report on the main
clinical and radiographic features of patients referred to our
center, and explore the genetic mutation spectrum of WISP3
and the association between phenotype and genotype of PPD in
Chinese patients with PPD. The clinical data of three patients with
PPD was analyzed, in order to summarize the associated clinical
characteristics, and the WISP3 genotype of the patients with
PPD was detected.
Patients and methods
Patients
The present study was approved by the Medical Ethics
Committee of Shengjing Hospital Affiliated to China Medical
University (Shenyang, China). The three patients with PPD from
three different families were referred from the Department of
Developing Pediatrics of Shengjing Hospital for genetic testing.
PPD was determined in the three patients by examining their medical
history, laboratory examinations and detection of radiological
abnormalities. The laboratory evaluation included routine blood,
liver and renal function tests, determination of serum calcium
phosphorous and alkaline phosphatase levels, as well as
measurements of C-reactive protein, rheumatoid factors,
antistreptolysin O and antinuclear antibodies, in order to exclude
connective tissue disease. Radiological investigations (X-rays of
the spine, hip, knee and hand) were performed, in order to make a
radiological diagnosis of PPD. The clinical and imaging
characteristics of each proband were reviewed by two clinical
geneticists and two radiologists, respectively. The medical
histories and examination results were systematically recorded, and
the skeletal X-ray films were reviewed. The patients were screened
for WISP3 gene mutations only after informed consent had
been obtained from the family members of the patients. All of the
available family members were then subjected to restriction
analysis. A total of 60 unrelated Han Chinese participants (40
males, 20 females; 5.2–22 years old) who were outpatients at the
Shengjing Hospital affiliated to China Medical University over a
period of two years, were used as normal controls. Written informed
consent was also obtained from the parents of the patients with PPD
and the 60 controls prior to blood sampling and DNA analysis.
Altogether, 67 DNA samples, including 3 affected individuals, 4
unaffected individuals from two families, and 60 healthy donors
were analyzed. DNA was extracted from peripheral white blood cells
with conventional methods. The entire coding region (five exons
with several alternatively spliced exons) of the WISP3 gene
(GeneBank accession number: NC_000006) were sequenced in order to
screen mutations using 6 pairs of primers.
Mutation analysis
A total of 2 ml peripheral venous blood was
collected from the pediatric patients. Genomic DNA was extracted
from the peripheral blood of the pediatric patients, their family
members, and the normal controls using the Universal Genomic DNA
Extraction kit Ver. 3.0 (Takara Biotechnology Co., Ltd., Dalian,
China). Five exons of WISP3 and the exon-intron borders were
amplified by polymerase chain reaction (PCR) (TP600; Takara
Biotechnology Co., Ltd.). The total volume used for the PCR
reaction was 50 μl, including 25 μl 2X GC buffer I, 8
μl dNTP (2.5 mmol/l), 2.5 μl upstream and downstream
primers respectively (10 pmol/l), and 0.5 μl LA Taq (5
U/μl; Takara Biotechnology Co., Ltd.) The primers were
synthesized by Sangon Biological Engineering Technology &
Services Co., Ltd. (Shanghai, China), the primer sequences are
shown in Table I. Approximately 40
ng genomic DNA was diluted to 50 μl with sterile water. The
PCR reaction conditions were set as follows: Predegeneration at
95°C for 5 min, followed by degeneration at 94°C for 30 sec. After
annealing for 30 sec at 58°C, extension was performed at 72°C for 1
min. After 35 cycles, a final extension was performed at 72°C for
10 min. The PCR products were then sent to Shanghai Sangon
Biological Engineering Technology & Services Co., Ltd. for
further analysis.
 | Table IPrimer sequences for the polymerase
chain reaction amplification of the WISP3 gene. |
Table I
Primer sequences for the polymerase
chain reaction amplification of the WISP3 gene.
| Amplicon | Primer sequence | Tm | GC content | Amplicon size
(bp) | Annealing temp.
(°C) |
|---|
|
WISP3-5′UTR&EXON1 |
F-GTACGTGAGGGTGAAGCTGGA | 59.9 | 57.1 | | |
|
R-GGAGAGACACTGTTTCCCGCA | 62.3 | 87.1 | 850 | 58 |
|
WISP3-EXON2 |
F-TAAAGGAGGAGTAAGAGTGGA | 51.2 | 42.9 | | |
|
R-CGGATTGTTTTTGACAGTATT | 52.5 | 33.3 | 868 | 50 |
|
WISP3-EXON3 |
F-GGTGATTTTACAGGGTCTTTAC | 53 | 40.9 | | |
|
R-ATTTATCCCTGTCTGAGGC | 51.2 | 47.4 | 667 | 50 |
|
WISP3-EXON4 |
F-GAAAGAGGGAGATAGAGTGATA | 50 | 40.9 | | |
|
R-GAAGTTAGAATCTGCTCTGGTT | 52.8 | 40.9 | 569 | 50 |
|
WISP3-EXON5&3′UTR |
F-GGTAAAGAGAGTGCTGGAAATC | 55.4 | 45.5 | | |
|
R-TGCTTAGATGGTAACAAATGTC | 52.7 | 36.4 | 683 | 52 |
Shanghai Sangon Biological Engineering Technology
& Services Co., Ltd. (http://genome-test.cse.ucsc.edu/cgi-bin/hgBlat?command=start)
sequenced the PCR products, following detection by 1% agarose gel
electrophoresis. The sequencing results were compared against
genomic sequences contained in the UCSC database (http://genome.ucsc.edu/), in order to identify genetic
mutations. In addition, the Human Gene Mutation Database
(www.genecards.org) was used to determine whether
each site was a novel mutation site.
Results
Clinical manifestion
All three of the patients with PPD were asymptomatic
after birth with normal intellectual development. They possessed no
distinctive facial features. The probands of families 1 and 3 were
both the only male child of non-consanguineous healthy parents, and
were delivered without complication at full-term. Their birth
weight and length were within the normal limits. Stature was normal
at onset of symptoms; however, a short trunk was observed at
diagnosis in all three patients. The patient of family 2 was a
fostered female child with unknown personal and family histories.
In the patients from families 1 and 3 the initial symptoms were
gait abnormalities and a degree of knee deformity. The initial
exhibiting symptom in the patient of family 2 was swelling of the
multiple interphalangeal joints. These three patients (two males
and one female) were included in this study over a period of two
years and nine months. These patients visited the clinic at the age
of 11–21 years for short stature (current height, near or less than
the standard deviation) and motion limitation of the joints. The
patients were followed up for six to nineteen months. During this
period, physical examinations and the routine blood tests (i.e.,
erythrocyte sedimentation rate, C-reactive protein, rheumatoid
factor, serum calcium levels, serum phosphorus levels, alkaline
phosphatase,parathyroid hormone, vitamin D status and insulin-like
growth factor 1) were conducted. At the final follow-up
appointment, all of the joints (small and large, including the
spine) were progressively limited in movement. The family members
of all three patients, including their parents, were not shown to
exhibit any abnormal clinical, biochemical or radiographic
manifestations. The detailed clinical data of the patients are
presented in Table II.
| Table IIBasic clinical data of three patients
with progressive pseudorheumatoid dysplasia. |
Table II
Basic clinical data of three patients
with progressive pseudorheumatoid dysplasia.
| Characteristic | Patient
|
|---|
| 1 | 2 | 3 |
|---|
| Gender | Male | Female | Male |
| Family history | − | Unknown | − |
| Age (years) | 13 | 11 | 21 |
| Age at onset
(years) | 6 | 4 | 3 |
| Site of
pathological changes | Knee joint | Interphalangeal
joint | Knee joint |
| Height (cm) | 153.7
(−1.2SDS) | 135.3
(−2.2SDS) | 155 160
(<2SDS) |
| Arthralgia | − | + | + |
| Spinal nerve
involvement | − | − | + |
| Enlargement and
stiffness of joint | + | ++ | +++ |
| Activity
limitation | Hip joint | Hip and knee
joints | All joints |
| Spine
malformation | Lumbar
lordosis | Scoliosis | Scoliosis |
| Muscle atrophy | Lower limbs | Lower limbs | − |
| Narrowing of
intervertebral and joint space | + | ++ | +++ |
| Vertebral
deformities | + | ++ | +++ |
| Acetabular fossa
deformation | + | ++ | +++ |
| Plain X-ray film of
the skull | − | − | − |
| Shortening of
column femoris | + | ++ | +++ |
Detection of mutations
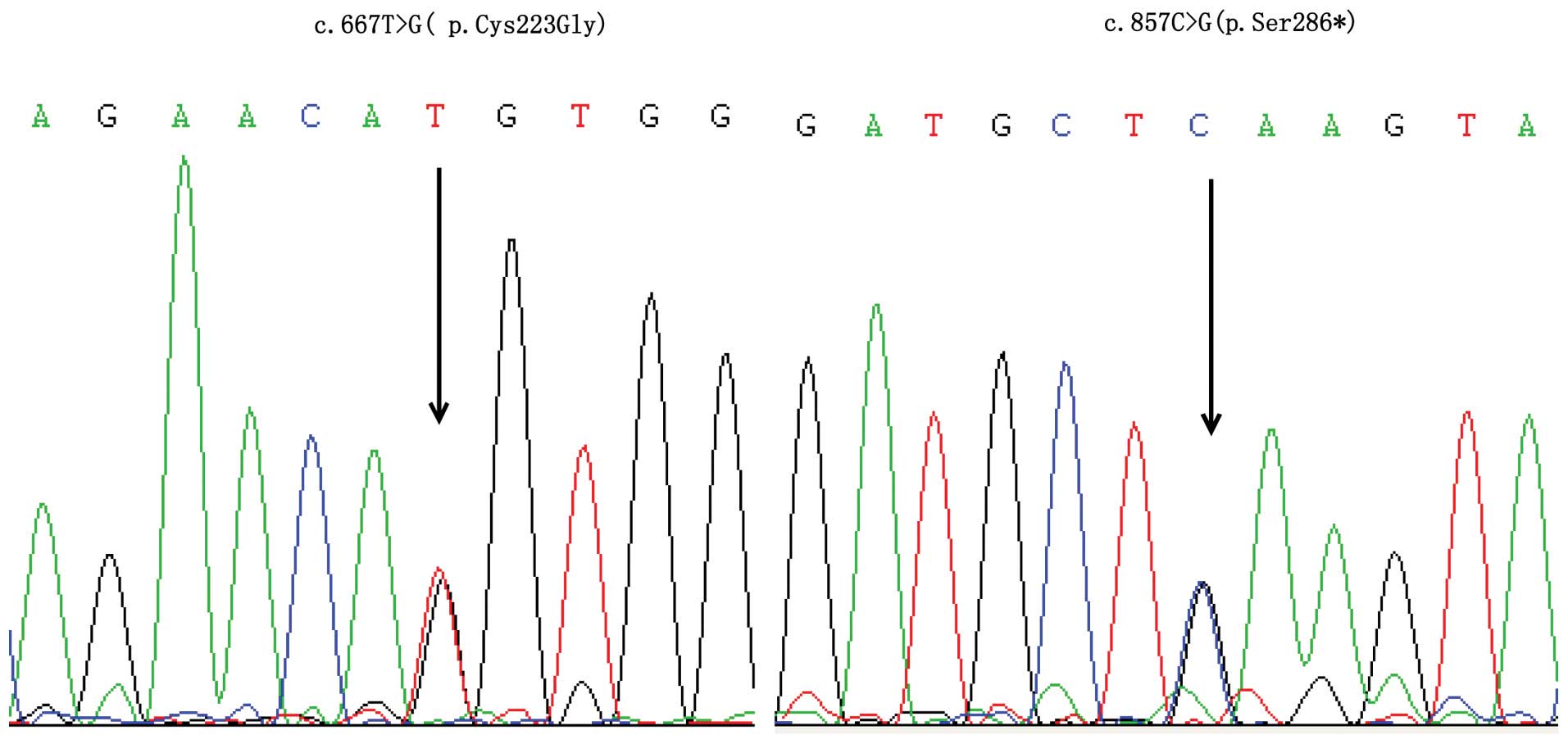
Five WISP3 mutations were detected (two
missense mutations, two nonsense mutations and one frameshift
mutation). In family 1, a compound heterozygous mutation was
detected; the proband carried a novel missense mutation:
c.667T>G (p.Cys223Gly) of the fourth exon in the maternal allele
and a nonsense mutation: c.857C>G (p.Ser286*) of the fifth exon
in the paternal allele (Fig. 2). A
homozygous missense mutation: c.342T>G (p.Cys114Trp) was
detected in the third exon of the patient from family 2. A
frameshift mutation and a nonsense mutation were detected in the
fifth exon: c.866dupA (p.Ser290Glufs*13) and the second exon:
c.136C>T (p.Gln46*) of the patient from family 3, respectively.
These two mutations formed a compound heterozygous mutation.
Discussion
The results of the present study are concordant with
the findings of a previous study (5), with similar clinical manifestations.
The clinical characteristics of PPD have been summarized in
previous studies (4–8) and have been shown to worsen over
time. As compared with the majority of skeletal dysplasias,
prenatal skeletal growth and morphogenesis are not altered in PPD
(2), and affected individuals are
asymptomatic after birth, and usually suffer from the disease
between the ages of 3 and 8 years old (4). The intellectual development of
patients with PPD is normal. A normal life expectancy is estimated;
however, joint contractures, fractures and spinal stenosis are
major prognostic factors associated with PPD (3,4,7).
Gait abnormality is often observed in the initial period, and is
followed by non-inflammatory and symmetrical joint enlargement,
accompanied by pain, ankylosis, and limitation of activity, which
may result in muscle atrophy, and finally disability (3,4).
Spine involvement develops after the age of 15 and adult height is
usually below the 3rd percentile. Progressive, non-inflammatory
multi-joint enlargement predominantly manifests as platyspondyly,
epiphyseal (metaphysis) expansion and joint stenosis in medical
imaging, without abnormalities detected by laboratory examination.
X-ray appearance of the head is normal (3,4).
Due to the similar clinical symptoms and
radiographic features, PPD is often clinically misdiagnosed, and
should be distinguished from spondyloepiphyseal dysplasia tarda and
juvenile idiopathic arthritis in the early stages of the disease
(Table III) (9).
| Table IIIKey similarities and differences
between PPD, SEDT and JIA. |
Table III
Key similarities and differences
between PPD, SEDT and JIA.
| PPD
|
|---|
| Similarities | Differences |
|---|
| SEDT | Short trunk, joint
pain and activity limitation, gait abnormality, similar vertebral
manifestation in X-ray film | Femoral epiphysis
becomes smaller instead of larger, which is accompanied by the
enlargement of the little toe joint. |
| JIA | Arthralgia,
swelling, activity limitation, growth delay | There is no
spondyloepiphyseal dysplasia. Joint involvement may be asymmetric
and accompanied by iridocyclitis, laboratory examination
abnormalities. Anti-inflammatory and immunosuppressive therapies
are effective. |
PPD is a rare genetic skeletal or cartilaginous
dysplasia characterized by continuous loss of cartilage and bone
destruction (10), due to the
functional loss or abnormality of the WISP3 protein. PDD was
initially reported in 1982 (1). In
1998, WISP3 was first cloned (11) and localized to the 6q22-6q23
chromosomal region. In 1999, using positional candidate cloning for
the first time, Hurvitz et al (2) demonstrated that PPD was caused by
WISP3 mutations. However, as compared with humans,
WISP3-null mice and mice overexpressing WISP3 exhibit
no gross abnormal phenotypes (12). WISP3 is an extracellular matrix
(ECM)-associated protein; it is a small secreted cysteine
rich-protein that is a member of the CCN protein family.
WISP3 contains five exons and four introns, which encode 354
amino acids. CCN proteins are secreted cysteine-rich heparin
binding glycoproteins that associate with the cell surface and
extracellular matrix (13). WISP3
is composed of an N-terminal signal peptide; followed by four
conserved cysteine-rich modular domains (which is known to be the
unifying feature of the CCN protein family); an insulin-like growth
factor binding protein domain (IGFBP) encoded by exon 2; a von
Willebrand type C repeat (VWC) encoded by exon 3, in which WISP3,
unlike all of the other identified CCN family proteins, lacks 4 of
10 conserved cysteine residues (14); a thrombospondin type I domain
(TSP-1) encoded by exon 4; and a cysteine knot carboxyl terminal
(CT) encoded by exon 5. WISP3 triggers signal transduction in cell
adhesion, migration, proliferation, differentiation and survival,
through direct binding to specific integrin receptors and heparan
sulfate proteoglycans (15). CCN
proteins simultaneously integrate and modulate the signals of
integrins, bone morphogenetic protein (BMP), vascular endothelial
growth factor, Wnt and Notch by direct binding. The WISP3
gene usually inhibits cell growth (14); however, the role of WISP3 in
the pathogenesis of PPD remains to be elucidated. Nakamura et
al (16) revealed that
WISP3 is involved in zebrafish cartilage development, and
suggested that dysregulation of BMP and/or Wnt signaling may
contribute to cartilage degeneration in humans with PPD (16). WISP3 is able to maintain
cartilage stability by regulating the synthesis of type II collagen
and aggrecan, two major matrix components of articular cartilage,
in chondrocytes (17).
Furthermore, it has been demonstrated that collagen II expression
is downregulated in chondrocytes possessing a WISP3
mutation. Mutated WISP3 leads to decreased intracellular
collagen II content, and secretion of extracellular collagen II
with a delayed secretion peak, which is an important mechanism
underlying the reduction of collagen size and density in the
cartilage of patients with PPD (17,18).
In addition, WISP3 mutations may result in continuous
degeneration and loss of articular cartilage, which only affects
the growth and differentiation of postnatal chondrocytes, without
affecting fetal cartilage growth (2). This may partly explain the
pathogenesis of PPD. Recent studies (18,19)
have shown that WISP3 may have a regulatory role through
inhibiting the proliferation of chondrocytes, and promoting the
differentiation of chondrogenic progenitor cells. Mutated WISP3
protein increased proliferative activity and decreased the rate of
apoptosis of C-20/A4 cells (18),
as well as aggregating abnormally in the cytoplasm. Therefore, it
may be suggested that WISP3 mutations cause functional
disorders, such as inhibition of chondrocyte proliferation and
dysregulation of type II collagen expression, which may be involved
in the pathogenesis of PPD (18).
Currently, ~56 different WISP3 mutations have
been reported globally, which are distributed in all coding exons
of the WISP3 gene (4–8,20).
The mutations localized in exon 2 (15 types) and exon 5 (15 types)
are predominant, followed by those in exon 4 (11 types). The
mutation in exon 1 (1 type) is the least frequent. Another five
types of mutations are localized in introns 1, 2 and 3. The most
common mutations are missense (23/56, 41%) and frameshift mutations
(20/56, 36%). P.Cys52* may be considered the hotspot mutation, and
is detected in various ethnic populations in the Middle East and
Europe with the highest frequency (4,5,6).
However, this particular mutation has not been detected in China
(7,8,20).
In the present study, two missense mutations, two
nonsense mutations and one frameshift mutation were detected.
Compound heterozygosity (c.667T>G/c.857C>G) of WISP3
was identified in family 1, where the proband possessed a novel
missense mutation c.667T>G (p.Cys223Gly) in the fourth exon of
the maternal allele, and a nonsense mutation c.857C>G
(p.Ser286*) in the fifth exon of the paternal allele. This sequence
variation was not present in any of the control samples, which
indicates that this variation is not a single nucleotide
polymorphism. As a novel missense mutation c.667T>G, T at
position 667 was replaced by G due to this mutation. This resulted
in codon 223 changing from TGT (coding Cys) to GGT (coding Gly),
which led to PPD in a compound heterozygous mutation state. The
WISP3 protein also has a series of primary structures consisting of
34 cysteines, which is similar to other proteins in the CCN family
(14). These primary structures
are conserved in number and position, as part of the sequence motif
forming each structural domain of WISP3. A change in any
cysteine residue can seriously interfere with the biological
functions of the protein; for example, regulation of BMP and/or Wnt
signaling, resulting in PPD when present in the homozygote or
compound heterozygote state (16).
The unique feature of these CCN family proteins is a multidomain
structure with an N-terminal secretory signal peptide and four
functional domains (IGFBP, VWC, TSP-1 and CT) (14). This novel missense mutation is
located in the TSP-1 domain, which binds heparin or sulfated
proteoglycans in order to modulate cell adhesion and maintain ECM
composition. The nonsense mutation c.857C>G (p.Ser286*),
produces a truncated WISP3 protein (p.Ser286*) composed of 285
amino acid residues, was detected for the first time in Chinese
patients in the present study. The nonsense mutation c.857C>G
has only previously been reported to be homozygous in an affected
individual in Turkey, and patients carrying a missense mutation in
one of the two alleles did not show any significant difference in
severity of the PPD phenotype, as compared with patients possessing
two nonsense mutations in both alleles (4). The phenotype of the male from family
1 in this study was no milder than that of the Turkish patient.
This nonsense mutation belongs to the fourth (CT) domain, which is
involved in disulfide-linked dimerization and is required for dimer
formation in the endoplasmic reticulum, an important function for
the establishment and maintenance of the normal 3D-conformation of
WISP3 protein (18). CT
domain-mediated dimerization has been suggested to act in synergy
with TSP-domain-mediated oligomerization, in order to give rise to
the large CCN oligomers, and may be an important factor in
directing how CCN proteins control and manipulate adhesion
processes and ECM composition, and mediate cell adhesion (1). Therefore, the present study
hypothesized that the compound heterozygous mutation formed a
truncated WISP3 protein and Cys223Gly mutated protein, and that the
two mutated WISP3 proteins possibly aggregate abnormally in the
cytoplasm, inducing morphological transformation of chondrocytes.
It has been reported worldwide in exons 4 and 5, that the majority
of mutations are either frameshift or missense mutations (26/56,
46%), or those involving cysteine residues (6). These findings indicate that the
inhibitory effect of WISP3 on cell proliferation requires
the expression of a full length protein and proper protein folding.
The present study also detected the following three recurrent
mutations: Homozygous missense mutation {c.342T>G (p.Cys114Trp)}
of the patient from family 2, and a compound heterozygous mutation,
the nonsense mutation c.136C>T (p.Gln46*) and the duplication
mutation c.866dupA (p.Ser290Glufs*13) of the proband from family 3.
The pathogenesis described above requires confirmation through
further studies regarding protein expression.
A total of 12 types of mutations (six frameshift
mutations, five missense mutations and two nonsense mutations) have
previously been detected in patients with PPD in China (8,20).
These include four different mutations (three frameshift mutations
and one missense mutation) localized in exon 4 and in exon 5 (two
missense mutations, one frameshift mutation and one nonsense
mutation) respectively, as well as three mutations in exon 2 (two
missense mutations and one nonsense mutation). However, there have
been no WISP3 mutations reported in exon 3 in Chinese
patients. Currently, the most common mutations are frameshift and
missense (10/12, 83%). In addition, the mutation c.624_625dupA in
exon 4, two mutations c.866dupA and c.1000T>C in exon 5, and
mutation c.136C>T in exon 2 are most commonly reported (7,8,20).
Therefore, it may be hypothesized that these four types of
mutations may be hot spot mutations of WISP3 in Chinese
populations.
There are currently no effective treatments for PPD,
which is the reason for the high disability rate associated with
the disease, despite its rarity. The disease can be diagnosed based
on typical clinical manifestations, and X-ray and biochemical
examination. A definitive diagnosis can be made at the molecular
biology level through analysis of WISP3 mutations. In the
present study, the three patients possessed similar clinical
phenotypes, and no specific correlation was observed between
genotype and phenotype. These results support the conclusion that
various WISP3 mutations may result in similar clinical
phenotypes (4).
In conclusion, a novel mutation c.667T>G
(p.Cys223Gly) and the c.857C>G (p.Ser286*) mutation were
detected in three Chinese patients with PPD, alongside three
recurrent mutations. The clinical phenotypes of PPD were relatively
consistent, and the patients exhibited the characteristic clinical
and radiographic findings associated with PPD. Therefore, imaging
examinations have diagnostic significance for PPD; however, the
final diagnosis must rely on the detection of WISP3
mutations.
Acknowledgments
The authors would like to thank the patients and
their family members for actively participating in the study. They
are particularly grateful to the patients and their family members
who provided images for the present study.
Abbreviations:
|
PPD
|
progressive pseudorheumatoid
dysplasia
|
|
WISP3
|
Wntl-inducible signaling pathway
protein 3
|
|
CCN
|
cysteine-rich 61/connective tissue
growth factor/nephroblastoma overexpressed
|
|
PCR
|
polymerase chain reaction
|
|
SEDT
|
spondyloepiphyseal dysplasia tarda
|
|
JIA
|
juvenile idiopathic arthritis
|
|
IGFBP
|
insulin-like growth factor binding
protein domain
|
|
VWC
|
Von Willebrand type C repeat
|
|
TSP-1
|
thrombospondin type I domain
|
|
CT
|
cysteine knot carboxyl terminal
|
|
BMP
|
bone morphogenetic protein
|
|
ECM
|
extracellular matrix
|
|
SDS
|
height standard deviation score
|
References
|
1
|
Wynne-Davies R, Hall C and Ansell BM:
Spondylo-epiphysial dysplasia tarda with Progressive arthropathy. A
“new” disorder of autosomal recessive inheritance. J Bone Joint
Surg Br. 64:442–445. 1982.
|
|
2
|
Hurvitz JR, Suwairi WM, Van Hul W, et al:
Mutations in the CCN gene family member WISP3 cause progressive
pseudorheumatoid dysplasia. Nat Genet. 23:94–98. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bennani L, Amine B, Ichchou L, Lazrak N
and Hajjaj-Hassouni N: Progressive pseudorheumatoid dysplasia:
three cases in one family. Joint Bone Spine. 74:393–395. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garcia Segarra N, Mittaz L, Campos-Xavier
AB, et al: The diagnostic challenge of progressive pseudorheumatoid
dysplasia (PPRD): a review of clinical features, radiographic
features, and WISP3 mutations in 63 affected individuals. Am J Med
Genet C Semin Med Genet. 160C:217–229. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Delague V, Chouery E, Corbani S, et al:
Molecular study of WISP3 in nine families originating from the
Middle-East and presenting with progressive pseudorheumatoid
dysplasia: identification of two novel mutations and description of
a founder effect. Am J Med Genet A. 138A:118–126. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dalal A, Bhavani GSL, Togarrati PP, et al:
Analysis of the WISP3 gene in Indian families with progressive
pseudorheumatoid dysplasia. Am J Med Genet A. 158A:2820–2828. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun J, Xia W, He S, et al: Novel and
recurrent mutations of WISP3 in two Chinese families with
progressive pseudorheumatoid dysplasia. PLoS One. 7:e386432012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ye J, Zhang HW, Wang T, et al: Clinical
diagnosis and WISP3 gene mutation analysis for progressive
pseudorheumatoid dysplasia. Zhonghua Er Ke Za Zhi. 48:194–198.
2010.In Chinese. PubMed/NCBI
|
|
9
|
Shivanand G, Jain V and Lal H: Progressive
pseudorheumatoid chondrodysplasia of childhood. Singapore Med J.
48:e151–e153. 2007.PubMed/NCBI
|
|
10
|
No authors listed. International
nomenclature and classification of the osteohondrodysplasias
(1997): International Working Group on Constitutional Diseases of
Bone. Am J Med Genet. 79:376–382. 1998. View Article : Google Scholar
|
|
11
|
Pennica D, Swanson TA, Welsh JW, et al:
WISP3 genes are members of the connective tissue growth factor
family that are up-regulated in wnt-1-transformed cells and
aberrantly expressed in human colon tumors. Proc Natl Acad Sci USA.
95:14717–14722. 1998. View Article : Google Scholar
|
|
12
|
Kutz WE, Gong Y and Warman ML: WISP3, the
gene responsible for the human skeletal disease progressive
pseudorheumatoid dysplasia, is not essential for skeletal function
in mice. Mol Cell Biol. 25:414–421. 2005. View Article : Google Scholar :
|
|
13
|
Zuo GW, Kohls CD, He BC, et al: The CCN
proteins: important signaling mediators in stem cell
differentiation and tumorigenesis. Histol Histopathol. 25:795–806.
2010.PubMed/NCBI
|
|
14
|
Holbourn KP, Acharya KR and Perbal B: The
CCN family of proteins: structure-function relationships. Trends
Biochem Sci. 33:461–473. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen PC, Cheng HC, Yang SF, Lin CW and
Tang CH: The CCN family proteins: modulators of bone development
and novel targets in bone-associated tumors. Biomed Res Int.
2014:4370962014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakamura Y, Weidinger G, Liang JO, et al:
The CCN family member Wisp3, mutant in progressive pseudorheumatoid
dysplasia, modulates BMP and Wnt signaling. J Clin Invest.
117:3075–3086. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sen M, Cheng YH, Goldring MB, Lotz MK and
Carson DA: WISP3-dependent regulation of type II collagen and
aggrecan production in chondrocytes. Arthritis Rheum. 50:488–497.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang M, Man XF, Liu YQ, et al: Dysfunction
of collagen synthesis and secretion in chondrocytes induced by
wisp3 mutation. Int J Endocrinol. 2013:6797632013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang M, Peng YQ, Zhou HD, Zhai MX, He YL
and Xie H: Construction of WISP3 gene’s mutants in SEDT-PA and
their expression in COS-7 cells. Zhong Nan Da Xue Xue Bao Yi Xue
Ban. 33:8–15. 2008.In Chinese. PubMed/NCBI
|
|
20
|
Yue H, Zhang ZL and He JW: Identification
of novel mutations in WISP3 gene in two unrelated Chinese families
with progressive pseudorheumatoid dysplas. Bone. 44:547–554. 2009.
View Article : Google Scholar
|
|
21
|
Ye J, Zhang HW, Qiu WJ, et al: Patients
with progressive pseudorheumatoid dysplasia: from clinical
diagnosis to molecular studies. Mol Med Rep. 5:190–195. 2012.
|