Introduction
Myocardial infarction (MI) is the rapid development
of myocardial necrosis, which is caused by a critical imbalance
between oxygen supply and demand of the myocardium (1). MI is caused by interruption of the
blood supply to the heart caused by atherosclerosis (2). Atherosclerosis is a series of
consecutively developed stages that is initiated by endothelial
dysfunction, followed by the generation of foam cells and fatty
streaks, plaque formation, plaque rupture and thrombosis (3). A common feature of coronary
atherosclerosis is intimal plaque calcification, which is followed
by the generation of smooth muscle cell layer calcification. During
calcification calcium (Ca2+) and phosphate
(PO4) ions precipitate inside the coronary artery wall
(4). Various calcification
inhibitors, which are present in the circulating blood stream and
at locally, are responsible for the prevention of mineralization.
The balance between Ca2+ and P ions can be altered due
to deficiencies of these inhibitors, leading to unwanted
precipitation inside vessel walls (5). An important calcification inhibitor
is fetuin-A, which acts as a central regulator extracellularly and
in the circulation (6).
α-2 Heremans Schmid glycoprotein (AHSG), also termed
human fetuin, is a major serum glycoprotein that was initially
identified in 1944 (7).
AHSG/fetuin-A is a 46 kDa serum glycoprotein that is commonly
synthesized by hepatocytes, and consists of two polypeptide chains.
Following digestion, these chains are linked by disulfide bonds
(8). Until recently the exact
function of this abundant serum protein remained unclear; however,
there is now more knowledge regarding its multifunctional roles.
Fetuin-A has been shown to remodel skeletal bones by capturing
Ca2+ ions in the circulation, and depositing them in the
extracellular region of bone marrow (9). Furthermore, fetuin-A inhibits the
effects of insulin receptors, by auto-phosphorylation and tyrosine
kinase activity destruction (10).
Besides the potential effects of fetuin-A on insulin signaling, it
has also been shown to hinder insulin action on adipocytes, by
adiponectin production (11). Like
albumin, fetuin-A is considered to be a negative acute phase
reactant protein (12). In
addition, it has been reported that knocking down fetuin-A
(AHSG−/−) expression led to organ calcification and
myocardial dysfunction in mice transgenic models (13).
The gene that encodes fetuin-A is localized to human
chromosome 3q27 position (14),
and the genomic structure consists of seven exons and six introns
(15). The fetuin-A gene is highly
polymorphic and various single nucleotide polymorphisms (SNPs) have
been identified. One of these SNPs is a C/T alteration at the 742
position (rs4917), which leads to a missense mutation (T248 M).
Another SNP within the fetuin-A gene is a 766 C/G substitution
(rs4918) that results in a Thr256Ser missense mutation (16). Furthermore, it has been reported
that fetuin-A serum levels are correlated with certain SNPs within
the AHSG gene; and the fetuin-A 766 C/G SNP is associated with
decreased circulating levels of fetuin-A (17). Previous studies have hypothesized
that mortality due to cardiovascular disease and MI may be
correlated with plasma levels of fetuin-A (18–20).
Risk analyses have evaluated the association between fetuin-A
polymorphisms and progression of MI in Western populations
(21); however, further
investigation is required to determine the potential role of
fetuin-A (742 and 766) gene polymorphisms on MI, with regards to
age, in Eastern populations. The present study aimed to determine
the association between fetuin-A 742 C/T and 766 C/G polymorphisms
and MI with regards to age onset.
Materials and methods
Subjects
A total of 146 patients with MI and 146 healthy
controls were enrolled in the present study. MI was defined using
the standard CHS criteria (22),
which includes: History of chest pain, cardiac enzyme levels and
characteristic changes on serial electrocardiograms. The control
subjects were recruited from patients visiting the Siyami Ersek
Chest, Heart and Vessel Education and Research Hospital due to
chest pain or for a general health check-up. Patients with the
following characteristics were excluded from the study: Pregnancy,
a previous clinical history for vascular heart disease, atrial
fibrillation, acute or chronic infections, immunological
conditions, malignancies, neoplasm, coagulation disorders or
chronic renal failure. All patients and healthy controls were
informed prior to their involvement in the study.
Ethical approval
The present study was approved by the Ethics
Committee of Marmara University (Istanbul, Turkey). The human
rights of the subjects were protected and any necessary approval
was secured from the Ethics Committee. All experiments performed on
human subjects were conducted in accordance with the Declaration of
Helsinki. All procedures were conducted with adequate understanding
and written consent from all of the subjects.
Risk factor assessment
Age, gender and smoking habits of the subjects were
obtained using a questionnaire form. Weight and height were
measured, and body mass index was calculated from these
measurements. Systolic and diastolic blood pressure was measured.
The presence of diabetes mellitus (DM) was defined by a repeated
fasting glucose level >126 mg/dl, the use of antidiabetic drugs
or both. Total cholesterol, and high and low density lipoprotein
cholesterol levels were determined enzymatically, and were also
measured enzymatically following dextrane sulfate magnesium
precipitation (23). Fibrinogen
and C-reactive protein (CRP) were measured at the Biochemistry
Laboratory of the Siyami Ersek Chest, Heart and Vessel Education
and Research Hospital using ELISA kits [Fibronectin Human ELISA
Kit, ab108847; CRP Human SimpleStep ELISA™ Kit, ab181416; Abcam,
Cambridge, MA, USA] according to the manufacturer’s
instructions.
Genotyping
All patients and healthy controls enrolled in the
present study provided 2–3 ml venous blood samples. All blood
samples were added to EDTA blood collection tubes containing 1.8
mg/ml EDTA and stored at −20°C. Genomic DNA was then isolated from
nucleated cells through harvesting using the ammonium acetate
salting out method (24). Fetuin
742 C/T and 766 C/G polymorphisms were genotyped according to
restriction enzyme digestions of polymerase chain reaction (PCR)
products. The fetuin 742 C/T and 766 C/G genotypes were determined
using PCR, followed by restriction digestion with SacI and
NlaIII enzymes respectively. The following PCR primers were
used: Forward: 5′-CCTCCCAAGCAGAAAC-3′ and reverse:
5′-TGATGATTCCGCATACCC-3′ for the Fetuin 742 region; and forward:
5′-GTCACCCCTCCTTGTAAC-3′ and reverse: 5′-CCCCAATGAGACCAC-3′ for
fetuin including the 766 C/G SNP. All primer sequences were
designed by the authors and synthesized by İontek Company localized
in İstanbul Technical University Techno-Park (İstanbul, Turkey) The
PCR reaction volume was 25 μl, containing 500 ng DNA, 2.5
μM forward and reverse primer, 0.2 μM dNTP (Thermo
Fisher Scientific, Waltham, MA, USA), 0.2 Units Taq DNA Polymerase
(18038-042; Thermo Fisher Scientific) and 2.5 mM of
MgCl2 (Thermo Fisher Scientific). The PCR protocol
included an initial denaturation step at 94°C for 3 min, followed
by 30 cycles of 30 sec denaturation at 94°C, 30 sec annealing at
59°C and 45 sec elongation at 72°C, followed by a final elongation
step at 72°C for 10 min. Each 10 μl PCR product was digested
using 5 units SacI (FD1133; Thermo Fisher Scientific) and
NlaIII (FD1834; Thermo Fisher Scientific) enzymes overnight
at 37°C. The digestion products were then subjected to 3% agarose
gel electrophoresis.
Statistical analysis
For comparison of the groups according to MI risk
factors Mann-Whitney U and χ2 tests were used. Allele
and genotype frequencies among the patients with MI and the control
subjects were compared with Hardy-Weinberg predictions, using
Fisher χ2-analysis. The results were expressed as odds
ratio and 95% confidence intervals. P<0.05 (2-sided) was
considered to indicate a statistically significant difference. SPSS
version 13.0 software (SPSS Inc., Chicago, IL, USA) was used for
all statistical analyses.
Results
Clinical characteristics
The clinical characteristics of the subjects from
all of the groups are presented in Table I. There was a significant
difference between the patients with MI and the control subjects,
with regards to gender, smoking habits, and CRP and fibrinogen
levels. Fetuin-A 742 T allele was determined by 165 and 201 bp
digestion products, whereas the C allele remained undigested.
Fetuin-A 766 G allele yielded 193 and 212 bp fragments, whereas the
C allele remained undigested by SacI restriction enzyme (Fig. 1). The genotype distributions and
allele frequencies of fetuin-A 742 C/T and 766 C/G SNPs within the
MI and control groups are presented in Table II. Only fetuin-A 742 C/T genotype
distribution was significantly different between the MI and control
groups (P=0.028). Conversely, there was no significant difference
between the genotype distribution of fetuin-A 766 C/G in the MI and
control groups (P=0.172). The fetuin-A 742 and 766 alleles were
compared between the MI and control subjects, and only the 742 C
allele was shown to be associated with a high risk of MI (P=0.034;
Table II).
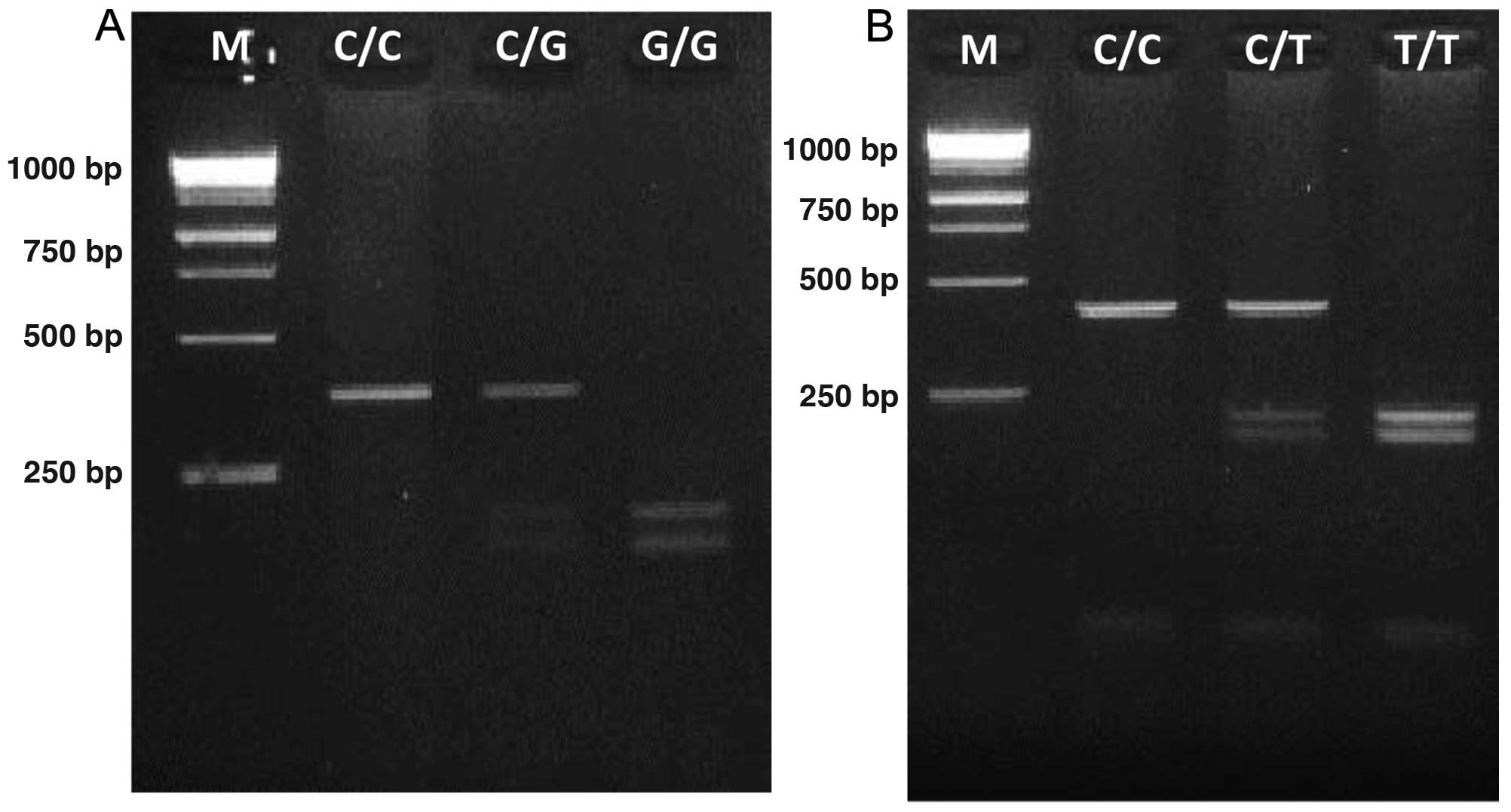 | Figure 1Genotyping of fetuin-A polymorphisms.
(A) Genotyping of fetuin-A (+766 C/G) gene polymorphism. M, 1 kb
DNA Marker; lane 2, homozygous for allele C (405 bp); lane 3,
heterozygous for allele C and allele G (405 bp, 212 bp and 193 bp);
lane 4, homozygous for allele G (212 bp and 193 bp). (B) Genotyping
of fetuin-A (+742 C/T) gene polymorphism. Lane 1 (M), 1 kb DNA
Marker; lane 2, homozygous for allele C (366 bp); lane 3, is
heterozygous for allele C and allele T (366 bp, 201 bp and 165 bp);
lane 4, homozygous for allele T (201 bp and 165 bp). |
 | Table IBaseline characteristics of the
patients with MI and the control subjects. |
Table I
Baseline characteristics of the
patients with MI and the control subjects.
| Characteristic | Control
(n=146) | MI (n=146) | MI (≤40)
(n=56) | MI (≥40)
(n=90) |
|---|
| Age (years) | 52.8±9.1 | 54.7±9.6 | 38.9±7.5b | 59.3±9.1 |
| Gender (%)
male | 40 | 72b | 69b | 76b |
| BMI
(kg/m2) | 28.2±3.5 | 28.5±3.8 | 28.2±3.1 | 28.7±3.8 |
| SBP (mmHg) | 132.1±17.4 | 140.7±15.9 | 138.5±16.1 | 142.5±17.0 |
| DBP (mmHg) | 78.5±12.3 | 86.6±10.2 | 86.4±10.1 | 88.5±11.2 |
| DM (%) | 10.7 | 12.7 | 12.1 | 12.9 |
| Smoker/former
smoker | 23 | 51a | 48a | 52a |
| HDL (mg/dl) | 40.8±4.9 | 42.1±4.4 | 41.9±4.6 | 43.1±5.2 |
| LDL (mg/dl) | 128.7±28.3 | 142.3±22.1 | 142.4±22.8 | 142.1±22.5 |
| TC (mg/dl) | 195±37.9 | 206.2±35.7 | 203.9±32.6 | 208±38.2 |
| CRP (mg/L) | 1.1 | 3.5a | 3.2a | 3.8a |
| Fibrinogen
(mg/dl) | 348.8±79.2 | 410.6±76.4a | 408.7±80.2a | 411.1±75.3a |
| Table IIGenotype distribution and allele
frequencies of Fetuin 742 C/T and Fetuin 766 C/G single nucleotide
polymorphisms between patients with MI and control subjects. |
Table II
Genotype distribution and allele
frequencies of Fetuin 742 C/T and Fetuin 766 C/G single nucleotide
polymorphisms between patients with MI and control subjects.
| Variable | MI (n=146) | Controls
(n=146) | P-value | OR at 95% CI |
|---|
| Fetuin 742 | | | | |
| CC | 81 (55.5%) | 102 (69.9%) | | |
| CT | 57 (39.0%) | 36 (24.7%) | 0.028 | 0.025–0.032 |
| TT | 8 (5.5%) | 8 (5.5%) | | |
| Allele C | 0.75 | 0.822 | 0.034 | 0.650
[0.435–0.969] |
| Allele T | 0.25 | 0.178 | | 0.810
[0.685–0.976] |
| Fetuin 766 | | | | |
| CC | 80 (54.8%) | 95 (65.1%) | | |
| CG | 58 (39.7%) | 43 (29.5%) | 0.172 | 0.169–0.184 |
| GG | 8 (5.5%) | 8 (5.5%) | | |
| Allele C | 0.747 | 0.798 | 0.139 | 0.868
[0.726–1.039] |
| Allele G | 0.253 | 0.202 | | 0.746
[0.506–1.101] |
Distribution of polymorphisms between
patient groups
The present study also investigated the distribution
of the polymorphic sites between subgroups of the MI group, as
compared with the control group. The most significant subgroup was
age-related case distribution. A cut-off value of 40 years old was
applied to the MI group, and the patients were divided into two
groups: MI≤40 years old or MI≥40 years old. The genotype
distributions and allele frequencies of fetuin-A 742 and 766 gene
polymorphisms in the younger and older MI groups and the healthy
controls are presented in Table
III and IV. Neither fetuin-A
742 C/T (P=0.519) nor fetuin-A 766 C/G (P= 0.653) genotype
distributions were significantly different between the young
patients with MI and the healthy control subjects. Similar to this
result, the allele frequencies of fetuin-A [742 (C/T) and 766
(G/C)] were not significantly different between the young patients
with MI and the control subjects (P=0.259 and 0.639, respectively;
Table III). However, fetuin-A
(742 and 766) gene polymorphisms had a significantly increased
presence in the older patients with MI, as compared with control
subjects (P=0.004 and P=0.017, respectively). However, in the older
patient subgroup, fetuin-A 742 (C/T) allele frequency was
significantly higher, as compared with the control group (P=0.036),
yet fetuin-A 766 (C/T) was not (P=0.078; Table IV).
| Table IIIGenotype distribution and allele
frequencies of Fetuin 742 C/T and 766 C/G single nucleotide
polymorphisms between young patients with MI and control
subjects. |
Table III
Genotype distribution and allele
frequencies of Fetuin 742 C/T and 766 C/G single nucleotide
polymorphisms between young patients with MI and control
subjects.
| Variable | MI ≤40 (n=56) | Controls
(n=146) | P-value | OR at 95% CI |
|---|
| Fetuin 742 | | | | |
| CC | 35 (62.5%) | 102 (69.9%) | | |
| CT | 16 (28.6%) | 36 (24.7%) | 0.519 | 0.508–0.528 |
| TT | 5 (8.9%) | 8 (5.5%) | | |
| Allele C | 0.768 | 0.822 | 0.259 | 0.719
[0.550–1.137] |
| Allele T | 0.232 | 0.178 | | 0.716
[0.422–1.219] |
| Fetuin 766 | | | | |
| CC | 36 (64.3 %) | 95 (65.1%) | | |
| CG | 15 (26.8 %) | 43 (29.5%) | 0.653 | 0.672–0.690 |
| GG | 5 (8.9 %) | 8 (5.5%) | | |
| Allele C | 0.777 | 0.798 | 0.639 | 0.919
[0.628–1.328] |
| Allele G | 0.223 | 0.202 | | 0.882
[0.519–1.495] |
| Table IVGenotype distributions and allele
frequencies of Fetuin 742 C/T and 766 C/G single nucleotide
polymorphisms between older patients with MI and control
subjects. |
Table IV
Genotype distributions and allele
frequencies of Fetuin 742 C/T and 766 C/G single nucleotide
polymorphisms between older patients with MI and control
subjects.
| Variable | MI ≥40 (n=90) | Controls
(n=146) | P-value | OR at 95% CI |
|---|
| Fetuin 742 | | | | |
| CC | 46 (51.1%) | 102 (69.9%) | | |
| CT | 41 (45.6%) | 36 (24.7%) | 0.004 | 0.002–0.005 |
| TT | 3 (3.3%) | 8 (5.5%) | | |
| Allele C | 0.738 | 0.822 | 0.036 | 0.752
[0.586–0.963] |
| Allele T | 0.262 | 0.178 | | 0.613
[0.392–0.962] |
| Fetuin 766 | | | | |
| CC | 44 (48.9%) | 95 (65.1%) | | |
| CG | 43 (47.8%) | 43 (29.5%) | 0.017 | 0.012–0.017 |
| GG | 3 (3.3%) | 8 (5.5%) | | |
| Allele C | 0.728 | 0.798 | 0.078 | 0.793
[0.618–1.017] |
| Allele G | 0.272 | 0.202 | | 0.677
[0.433–1.046] |
Carrier status for the C allele at exon 6 or 7 of
fetuin-A 742 or 766 respectively was associated with susceptibility
to MI [allele carrier frequencies; MI=57.5%, controls=77.4%
(P=0.0003)]. The lack of two C alleles in the fetuin-A gene was
significantly more likely in the older patients with MI. However,
the absence of C alleles was shown to increase the risk of MI at a
young age (OR=1.902, 95% CI=0.998–2.444, P= 0.0579).
Discussion
Cardiovascular disease (CVD) is the most common
cause of mortality worldwide (25). Although CVD-associated mortality
rates have declined in Western countries due to preventative
strategies, there are increasing numbers of high-risk groups for
CVD in developing countries (26).
There are various clinical outcomes of CVD, such as stroke, angina
pectoris and MI. CVD is a multifactorial polygenic disease that is
controlled by genetic and environmental risk factors (26). According to various case-control
studies, age, gender (male), smoking, family history, obesity,
hypertension and DM are all known risk factors for MI (27).
The etiology of MI is atherosclerosis of the
coronary arteries. Atherosclerosis is a chronic inflammatory
disease of the arterial wall that is characterized by an
accumulation of inflammatory cytokines and cells, which migrate
across the arterial wall. Migration of various inflammatory cells,
such as T-lymphocytes and macrophages, into the intermediate layer
of the artery wall due to epithelial dysfunction, occurs in the
initiation of atherosclerosis (3).
Following cell migration the cells, particularly macrophages,
engulf oxidized LDL and become foam cells. An accumulation of foam
cells and activation of inflammatory cells leads to the release of
pro-inflammatory cytokines, such as interleukin (IL)-1 and tumor
necrosis factor (TNF), which leads to plaque formation (28). Plaque enlargement through the
artery lumen causes shear stress and plaque instability (29). Plaque diameter, localization on the
artery as well as plaque rupture or thrombosis may result in
clinical symptoms, including coronary artery disease, angina
pectoris or MI (30). According to
coronary angiographic morphologies coronary artery plaque lesions
can be classified into four types: Concentric, type I eccentric,
type II eccentric and multiple irregular lesions. Among these
categories, type II eccentric lesions are clinically detected in
angina pectoris and MI (31).
Doherty et al (32),
suggested that coronary artery calcification, a major result of
Ca2+ ion deposits within the coronary artery wall, is a
protective process against the rupture of vulnerable plaques. Non-
or mildly-calcified coronary arteries are observed in MI, whereas
extremely calcified plaques are generally observed in cases of
stable angina. The amount of coronary calcification has been shown
to increase with age, and is more common in males of all ages
(33). According to previous
electron-beam computed tomography results, coronary artery plaque
calcification was reported as an important scale for MI progression
(34). The clinical outcome of MI
is determined according to degree and time course of obstruction,
collateral blood flow, and myocardial oxygen demand following
thrombosis of the disrupted coronary plaques; however, coronary
artery calcification is the most common symptom of MI regardless of
age and gender (35).
Fetuin-A is a circulating protein that inhibits
calcification by binding CaXPO4 ions and preventing
precipitation (36). Knock out of
fetuin-A expression in mice led to vascular calcification, due to
increased transforming growth factor-β, collagen and fibronectin
expression levels in cardiac cells (37). In vivo fetuin-A has been
shown to induce the expression of pro-inflammatory cytokines,
including IL-6 and TNF, in adipose tissues when liver fat
accumulation has been observed (38). Furthermore, increased fetuin-A
expression was previously shown to cause insulin resistance by
suppressing insulin receptor autophosphorylation and tyrosine
kinase activity (10). Fetuin-A is
associated with various metabolic syndromes, including
atherosclerosis. The role of fetuin-A on atherosclerosis is
suggested to be linked with the deactivation of macrophages, by
inhibition of pro-inflammatory cytokine expression, due to its
Ca2+ binding potential (2). Therefore, the association between
plasma fetuin-A levels and MI risk has been investigated in certain
studies (36). However,
contradictory results have been obtained from these studies.
Ketteler et al (39)
demonstrated that low levels of plasma fetuin-A were correlated
with cardiovascular-associated mortality in hemodialysis patients.
Furthermore, low fetuin-A levels were shown to be linked with
mitral and aortic calcification and stenosis, in Western European
countries (18–21,40–42).
In the Turkish population, decreased fetuin-A levels were
determined in patients with acute coronary syndrome, as compared
with healthy controls (42).
Conversely, coronary artery calcification has been shown to be
associated with increased plasma fetuin-A levels in diabetic
nephropathy (19). Increased
plasma levels of fetuin-A were also established in patients with
MI, regardless of MI-associated risk factors, such as age, smoking
habit and CRP levels (18). In
addition, fetuin-A has been indicated as a predictor of mortality,
in ST-elevated MI cases (20).
These results suggest that serum plasma fetuin-A levels may be a
predictor of coronary artery calcification, or MI-associated
mortality. As serum protein levels may differ according to disease
stage, time of measurement or other metabolic syndrome involvement,
investigation of the association between MI and fetuin-A levels may
be a potential diagnostic tool. Fetuin-A is encoded by the ASHG
gene localized at the 3q27 chromosome region, and is highly
polymorphic (14,15). A total of 30 SNPs have been
identified in the fetuin-A gene, and the most frequently observed
SNPs that are linked with fetuin-A levels were 742 C/T and 766 C/G
SNPs (16). It has previously been
reported that fetuin-A 766 Ser allele carriers have lower fetuin-A
levels, as compared with Thr allele carriers (17). Recently, the EPIC-Postdam study
showed that fetuin-A 742 C (rs4917) allele is associated with high
plasma fetuin-A levels and MI risk (21). According to these findings,
although fetuin-A SNPs may determine fetuin-A plasma levels, the
levels could also change due to renal disease, diabetes mellitus or
ethnic origin. Instead of measuring fetuin-A plasma levels in MI
risk determination, SNPs within the fetuin-A gene may have
potential in the determination of MI risk. The present study
determined that fetuin-A 742 C/T and 766 C/G genotypes were
associated with MI in older patients. The results of the present
study demonstrated that fetuin-A 742 T allele is linked with MI
susceptibility and older patients with MI were more likely to carry
both 742 T and 766 G alleles. This is the first report, to the best
of out knowledge, to demonstrate that carriage of two alleles in
both fetuin-A 742 C/T and 766 C/G SNPs may be a genetic marker for
the susceptibility to MI at an older age.
Acknowledgments
The present study was supported by the Istanbul
Kultur University Research Fund.
References
|
1
|
Pouleur AC, Barkoudah E, Uno H, et al:
Pathogenesis of sudden unexpected death in a clinical trial of
patients with myocardial infarction and left ventricular
dysfunction, heart failure, or both. Circulation. 122:597–602.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang H and Eitzman DT: Acute myocardial
infarction leads to acceleration of atherosclerosis.
Atherosclerosis. 229:18–22. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ross R: Atherosclerosis is an inflammatory
disease. Am Heart J. 138:419–420. 1999. View Article : Google Scholar
|
|
4
|
Hong C, Zhu F, Du D, Pilgram TK, Sicard GA
and Bae KT: Coronary artery calcification and risk factors for
atherosclerosis in patients with venous thromboembolism.
Atherosclerosis. 183:169–174. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McGeachie M, Ramoni RL, Mychaleckyj JC, et
al: Integrative predictive model of coronary artery calcification
in atherosclerosis. Circulation. 120:2448–2454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Westenfeld R, Schäfer C, Smeets R, et al:
Fetuin-A (AHSG) prevents extraosseous calcification induced by
uraemia and phosphate challenge in mice. Nephrol Dial Transplant.
22:1537–1546. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anderson L and Anderson NG: High
resolution two-dimensional electrophoresis of human plasma
proteins. Proc Natl Acad Sci USA. 74:5421–5425. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee CC, Bowman BH and Yang FM: Human alpha
2-HS-glycoprotein: the A and B chains with a connecting sequence
are encoded by a single mRNA transcript. Proc Natl Acad Sci USA.
84:4403–4407. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jahnen-Dechent W, Schinke T, Trindl A, et
al: Cloning and targeted deletion of the mouse fetuin gene. J Biol
Chem. 272:31496–31503. 1997. View Article : Google Scholar
|
|
10
|
Auberger P, Falquerho L, Contreres JO, et
al: Characterization of a natural inhibitor of the insulin receptor
tyrosine kinase: cDNA cloning, purification and anti-mitogenic
activity. Cell. 58:631–640. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dahlman I, Eriksson P, Kaaman M, et al:
alpha2-Heremans-Schmid glycoprotein gene polymorphisms are
associated with adipocyte insulin action. Diabetologia.
47:1974–1979. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lebreton JP, Joisel F, Raoult JP, Lannuzel
B, Rogez JP and Humbert G: Serum concentration of human alpha 2 HS
glyco-protein during the inflammatory process: evidence that alpha
2 HS glycoprotein is a negative acute-phase reactant. J Clin
Invest. 64:1118–1129. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schafer C, Heiss A, Schwarz A, et al: The
serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a
systemically acting inhibitor of ectopic calcification. J Clin
Invest. 112:357–366. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rizzu P and Baldini A: Three members of
the human cystatin gene superfamily, AHSG, HRG and KNG, map within
one megabase of genomic DNA at 3q27. Cytogenet Cell Genet.
70:26–28. 1995. View Article : Google Scholar
|
|
15
|
Osawa M, Umetsu K, Sato M, et al:
Structure of the gene encoding human alpha 2-HS glycoprotein
(AHSG). Gene. 196:121–125. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Osawa M, Yuasa I, Kitano T, et al:
Haplotype analysis of the human alpha2-HS glycoprotein (fetuin)
gene. Ann Hum Genet. 65:27–34. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stenvinkel P, Pecoits-Filho R, Lindholm B
and DialGene C: Gene polymorphism association studies in dialysis:
the nutrition-inflammation axis. Semin Dial. 18:322–330. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weikert C, Stefan N, Schulze MB, et al:
Plasma fetuin-a levels and the risk of myocardial infarction and
ischemic stroke. Circulation. 118:2555–2562. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mehrotra R, Westenfeld R, Christenson P,
et al: Serum fetuin-A in nondialyzed patients with diabetic
nephropathy: relationship with coronary artery calcification.
Kidney Int. 67:1070–1077. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lim P, Collet JP, Moutereau S, et al:
Fetuin-A is an independent predictor of death after ST-elevation
myocardial infarction. Clin Chem. 53:1835–1840. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fisher E, Stefan N, Saar K, et al:
Association of AHSG gene polymorphisms with fetuin-A plasma levels
and cardiovascular diseases in the EPIC-Potsdam study. Circ
Cardiovasc Genet. 2:607–613. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fried LP, Borhani NO, Enright P, et al:
The cardiovascular health study: design and rationale. Ann
Epidemiol. 1:263–276. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Friedewald WT, Levy RI and Fredrickson DS:
Estimation of the concentration of low-density lipoprotein
cholesterol in plasma, without use of the preparative
ultracentrifuge. Clin Chem. 18:499–502. 1972.PubMed/NCBI
|
|
24
|
Chen W: Rapid isolation of DNA from human
peripheral blood. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 14:141–142.
1992.In Chinese. PubMed/NCBI
|
|
25
|
Murray CJ and Lopez AD: Evidence-based
health policy-lessons from the global burden of disease study.
Science. 274:740–743. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yusuf S, Reddy S, Ounpuu S and Anand S:
Global burden of cardiovascular diseases: part I: general
considerations, the epidemiologic transition, risk factors and
impact of urbanization. Circulation. 104:2746–2753. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wilson PW, D’Agostino RB, Levy D, Belanger
AM, Silbershatz H and Kannel WB: Prediction of coronary heart
disease using risk factor categories. Circulation. 97:1837–1847.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maseri A and Fuster V: Is there a
vulnerable plaque? Circulation. 107:2068–2071. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Holmes DR Jr, Hartzler GO, Smith HC and
Fuster V: Coronary artery thrombosis in patients with unstable
angina. Br Heart J. 45:411–416. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mandelkorn JB, Wolf NM, Singh S, et al:
Intracoronary thrombus in nontransmural myocardial infarction and
in unstable angina pectoris. Am J Cardiol. 52:1–6. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ambrose JA, Winters SL, Arora RR, et al:
Coronary angiographic morphology in myocardial infarction: a link
between the pathogenesis of unstable angina and myocardial
infarction. J Am Coll Cardiol. 6:1233–1238. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Doherty TM, Detrano RC, Mautner SL,
Mautner GC and Shavelle RM: Coronary calcium: the good, the bad and
the uncertain. Am Heart J. 137:806–814. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shemesh J, Stroh CI, Tenenbaum A, et al:
Comparison of coronary calcium in stable angina pectoris and in
first acute myocardial infarction utilizing double helical
computerized tomography. Am J Cardiol. 81:271–275. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Raggi P, Cooil B, Shaw LJ, et al:
Progression of coronary calcium on serial electron beam tomographic
scanning is greater in patients with future myocardial infarction.
Am J Cardiol. 92:827–829. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vliegenthart R, Oudkerk M, Song B, van der
Kuip DA, Hofman A and Witteman JC: Coronary calcification detected
by electron-beam computed tomography and myocardial infarction. The
rotterdam coronary calcification study. Eur Heart J. 23:1596–1603.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schinke T, Amendt C, Trindl A, Pöpschke O,
Müller-Esterl W and Jahnen-Dechent W: The serum protein alpha2-HS
glycoprotein/fetuin inhibits apatite formation in vitro and in
mineralizing calvaria cells. A possible role in mineralization and
calcium homeostasis. J Biol Chem. 271:20789–20796. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Merx MW, Schäfer C, Westenfeld R, et al:
Myocardial stiffness, cardiac remodeling and diastolic dysfunction
in calcification-prone fetuin-A-deficient mice. J Am Soc Nephrol.
16:3357–3364. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stefan N, Hennige AM, Staiger H, et al:
Alpha2-Heremans-Schmid glycoprotein/fetuin-A is associated with
insulin resistance and fat accumulation in the liver in humans.
Diabetes Care. 29:853–857. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ketteler M, Bongartz P, Westenfeld R, et
al: Association of low fetuin-A (AHSG) concentrations in serum with
cardiovascular mortality in patients on dialysis: a cross-sectional
study. Lancet. 361:827–833. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kaden JJ, Reinöhl JO, Blesch B, et al:
Systemic and local levels of fetuin-A in calcific aortic valve
stenosis. Int J Mol Med. 20:193–197. 2007.PubMed/NCBI
|
|
41
|
Ix JH, Chertow GM, Shlipak MG, Brandenburg
VM, Ketteler M and Whooley MA: Association of fetuin-A with mitral
annular calcification and aortic stenosis among persons with
coronary heart disease: data from the Heart and Soul Study.
Circulation. 115:2533–2539. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Afsar CU, Uzun H, Yurdakul S, et al:
Association of serum fetuin-A levels with heart valve calcification
and other biomarkers of inflammation among persons with acute
coronary syndrome. Clin Invest Med. 35:E206–E215. 2012.PubMed/NCBI
|














