Introduction
Sepsis is a potentially fatal condition and affects
millions of individuals worldwide, with a high rate of mortality
(1). Cardiac dysfunction is well
recognized in sepsis and septic shock (2,3) and
sepsis-induced myocardial dysfunction (SIMD) is a key contributor
to morbidity (40%) and mortality (30~60%) in patients with sepsis
(4–6). Thus, it is one of the predictors of
poor outcome in patients with sepsis (7). At present, the prognosis for patients
with SIMD remains poor, thus, a strategy to protect against SIMD is
crucial.
The etiopathogenesis of SIMD is yet to be fully
elucidated (7,8), however it is known to be
characterized by complex mechanisms involving the attenuation of
the adrenergic response at the cardiomyocyte level, alterations in
the structure and function of atrial ion channels, mitochondrial
dysfunction/ineffective oxygen utilization and blunted calcium
sensitivity of contractile proteins (9–11).
Several studies have indicated that the inflammatory reaction
during sepsis is important in the pathogenesis of SIMD (12–14).
A number of studies have demonstrated that specific cytokines are
associated with SIMD, including tumor necrosis factor-α, peroxisome
proliferator-activated receptor-γ coactivator-1 α and β,
interleukin (IL)-6, IL-10, IL-1β, and also transforming growth
factor-β1 (TGF-β1) (15–18). TGF-β1 is an immunomodulatory
cytokine with a broad range of anti-inflammatory effects, and has
been indicated to ameliorate the adverse effects of
pro-inflammatory cytokines on myocardial cells (19). As an early inflammatory factor in
SIMD, previous studies have hypothesized that TGF-β1 may have the
potential to be used therapeutically for SIMD in humans (18).
Additionally, impaired mitochondrial function has
been hypothesized to contribute to SIMD (20). In a model of sepsis, the oxidation
of myocardial mitochondrial proteins and lipids were assessed,
which led to the suggestion that damage to the outer mitochondrial
membrane (OMM) had occurred (21).
Disturbance of the OMM leads to permeabilization, and thus, there
is a large release of cytochrome c (22). Certain studies have indicated that
cytochrome c oxidase (COX) is inhibited in the septic heart,
and one study noted a competitive inhibition of myocardial COX
early in sepsis, which progresses to become non-competitively
inhibited during the late hypodynamic phase (23,24).
Whether exogenous cytochrome c has therapeutic value in
sepsis and sepsis-associated organ dysfunction remains unclear
(11), however, it is clear that
cytochrome c (endogenous/exogenous) serves an important
function in sepsis via the inhibition of COX (23).
At present, the effect of exogenous cytochrome
c on SIMD via TGF-β1 remains to be fully elucidated, thus in
the current study, this was investigated using a mouse model.
Materials and methods
Animal preparation and construction of
the SIMD model
A total of 75 male Kunming mice (body weight, 20–24
g) obtained from the Laboratory Animal Center of Chongqing Medical
University (Chongqing, China) were randomly divided into the
following five groups: Normal (N, n=15); sham-operation (SHAM,
n=15); sepsis by cecal ligation puncture (CLP, n=15); normal saline
(NS, n=15); and cytochrome c (Cytc, n=15). Following normal
feeding for one week, a sepsis model for the CLP, Cytc and NS
groups was constructed using a previously described CLP procedure
(25) in which the cecum is
extruded and ligated. In the SHAM group, the cecum was flipped
subsequent to opening the abdomen, and no ligation was performed.
At the conclusion of the operation, 30 ml/kg saline (Kelun
Pharmaceutical Co., Ltd, Sichuan, China) was injected under the
skin of the mice in the SHAM, CLP, Cytc and NS groups, in order to
replenish the fluids lost during the operation. Animals in the Cytc
group received a 20 mg/kg intraperitoneal injection of Cytc
(dissolved in 0.15 ml normal saline; Sigma-Aldrich, St. Louis, MO,
USA), whereas mice in the NS group received an intra-peritoneal
injection of an equal volume of normal saline. In every group there
were three subgroups, in each of which five animals were sacrificed
at 0 (subgroup 1), 6 (subgroup 2) and 12 h (subgroup 3) following a
30-min resting period subsequent to injection, non-injected mice
were sacrificed at these time-points without the 30 min resting
period. All experimental procedures were approved by the
Institutional Animal Care and Use Committee of Chongqing Medical
University (Chongqing, China).
Transmission electron microscopy
The mice were anesthetized by 10% chloral hydrate
(Sangon Biotech Co., Ltd, Shanghai, China) injection into the
cavum abdominis (3 ml/kg). The middle lobes of the
myocardium were cut using a Paraffin Slicing Machine (Shanghai
Medical Equipment Works Co., Ltd., Shanghai, China) into 1–2
mm-cubes and fixed in 2.5% glutaraldehyde (Sangon Biotech Co., Ltd)
at 4°C, then the samples were treated with osmium tetroxide
(Sigma-Aldrich) and dehydrated in serial concentrations of ethanol
(Sangon Biotech Co., Ltd). The samples were then treated with
propylene oxide (Befar Group Co., Ltd, Shanghai, China) for 10 min
and with a propylene oxide/epoxy resin mixture (Sinopec Group,
Beijing, China) for 1 h. These were left overnight with caps
removed from the vials to allow for the evaporation of any
remaining propylene oxide. Subsequently, the samples were embedded
in labeled capsules with freshly prepared resin. Electron
photomicrographs were captured using a Hitachi H-7500 Transmission
Electron Microscope (Hitachi, Ltd., Tokyo, Japan).
Histopathological examination
Formalin-fixed (Sangon Biotech Co., Ltd),
paraffin-embeded (Sigma-Aldrich) myocardial sections (2 µm
thick) were cut and stained with hematoxylin & eosin (H&E;
Huayueyang Biotechnology Co., Ltd., Beijing, China). The samples
were visualized using an electron microscope (H-7560, Hitachi,
Tokyo, Japan).
RNA isolation and analysis
A total of 100 mg myocardial mouse tissue was
homogenized with 2 ml TRIzol (Invitrogen Life Technologies,
Carlsbad, CA, USA), with 20µl diethylpyrocarbonate water
(Beyotime Institute of Biotechnology, Shanghai, China) per
Eppendorf tube, 100 µl tissue homogenates was added to each
tube. Reverse transcription (RT) was completed using the Reverse
Transcription System (Promega Corporation, Madison, WI, USA) and
treated with DNase I (CWBIO, Beijing, China) to eliminate genomic
DNA contamnination. RNA was quantified using the RNA 6000 Nano kit
and the 2100 Bioanalyzer system (Agilent Technologies, Inc., Santa
Clara, CA, USA) with the RNA ladder as the standard.
RT-quantitative polymerase chain reaction
(qPCR)
The RT-qPCR reaction was performed with a final
volume of 25 µl [2.5 µl 10X LA Taq buffer (Takara
Biotechnology Co., Ltd., Dalian, China); 0.4 mM deoxyribonucleotide
triphosphate (Takara Biotechnology Co., Ltd.); 0.4 pM each of the
5′ and 3′ primers (Takara Biotechnology Co., Ltd); 1.25 µl
20X SYBR Green I buffer (OPE, Shanghai, China) and 1.25 U LA Taq
DNA Polymerase (Takara Biotechnology Co., Ltd.)]. GAPDH was used as
internal control to normalize the aromatase mRNA level. The primer
sequences used were as follows: GAPDH, F 5′-CAT CAC TGC CAC CCA GAA
GA-3′ and R 5′-GCT GTA GCC AAA TTC GTT GT-3′; TGF-β1, F 5′-TGG TGG
ACC GCA ACA AC-3′ and R 5′-AGC CAC TCA GGC GTA TCA G-3′. The
thermal cycling profile consisted of an initial 2 min at 94°C
followed by 40 cycles of 94°C for 20 sec, 56°C for 20 sec and 72°C
for 30 sec. Melting curve analysis was subsequently performed. The
experiments were repeated three times with separate samples using a
Stratagene Mx3000PTM RT-qPCR system (Agilent Technologies,
Inc.).
The sensitivity of RT-qPCR was determined from the
Ct values obtained with known quantities of TGF-β1 and GAPDH
standards. The variability between results of different PCR runs
was calculated from duplicate samples for all of the targets, and
identified an average standard deviation for the threshold cycle
(Ct) of the fluorescence signal. Gene expression analysis was
conducted using the comparative 2−ΔΔCt method.
Enzyme-linked immunosorbent assay
(ELISA)
Under anesthesia with chloral hydrate, blood samples
were collected via the abdominal aorta in EDTA tubes
(Becton-Dickinson, Franklin Lakes, NJ, USA) and were centrifuged at
1,500 × g for 10 min at 4°C. The resulting plasma supernatant was
stored at −80°C until required for analysis, and the concentrations
of TGF-β1 were quantified using a Mouse TBF-β ELISA kit (R&D
Systems), according to the manufacturer’s instructions.
Protein determination
Myocardial tissue specimens were homogenized in 1 ml
ice-cold lysate buffer (CWBIO) consisting of 100 mM
Tris-hydrochloric acid; 300 mM sodium chloride; 2% (v/v) Tween-20;
0.4% NP-40; 20% glycerol; protease inhibitor cocktail; and
phosphatase inhibitors. The homogenates were centrifuged at 12,000
× g for 20 min at 4°C and the concentrations of the extracted
proteins were determined using the Bicinchonic Acid Protein Assay
kit (Sigma-Aldrich). The proteins were then stored at −70°C.
Western blot analysis
Protein samples (50 µg) were diluted with 2X
loading buffer (Beyotime Institute of Biotechnology) and heated in
boiling water for 5 min. The proteins were separated by 10%
SDS-polyacrylamide gel electrophoresis [10% separating gel and 5%
stacking gel (CWBIO); and Protean II xi/XL electrophoresis machine
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and were
transferred to a polyvinylidine difluoride membrane (Pall Corp.,
Port Washington, NY, USA). The membranes were then blocked with 5%
skimmed milk for 1–2 h at 37°C, were incubated with the following
primary antibodies for 1–2 h at 37°C: Mouse anti-human IgG2B
polyclonal antibody (1:1,000; R&D Systems), mouse anti-human
GAPDH polyclonal antibody (1:2,000; R&D Systems), and then
washed three times with phosphate-buffered saline with Tween-20
(Sangon Biotech Co., Ltd). Following further incubation with the
secondary antibody (goat anti-rabbit/mouse polyclonal IgG; 1:2,000;
BIOSS, Beijing, China) for 1 h at 37°C, the membrane was stained by
ECL chemiluminescence using BeyoECL Plus (Beyotime Institute of
Biotechnology) and visualized using ChemiDoc MP (Bio-Rad
Laboratories, Inc.). This experiment was repeated three time using
separate samples.
Statistical analysis
Data are presented as the mean ± standard deviation.
SPSS, version 10.0 (SPSS, Inc., Chicago, IL, USA) was used for data
analysis. TGF-β1 and SMAD1/5/8 expression were compared using
analysis of variance. Comparison between groups was assessed with
the Student-Newman-Keuls method, P<0.05 was considered to
indicate a statistically significant difference.
Results
Ultrastructural organization of
myocardial tissues
Myocardial tissues from mice in the SHAM group
exhibited the following characteristics: i) A large number of rows
of mitochondria; ii) smooth and complete mitochondrial cristae; and
iii) neat myocardial fibers (Fig.
1A). Isolated myocardial tissues in mice in the CLP 12-h group
exhibited the characteristics of cardiomyocyte damage (Fig. 1B and C), including marked exudation
of dissolved filaments; increased lipid droplets; mitochondrial
disorder and increased swelling of the mitochondria and vacuoles;
individual mitochondria; and bleeding of red blood cells in the
gaps between cells. Samples from the Cytc 12-h group, however,
demonstrated characteristics of protection (Fig. 1D), including no significant
increase, swelling or vacuolization of mitochondria; no widening of
the perinuclear space of cardiomyocytes; and no dissolved
filaments. The results from isolated myocardial tissues suggest
that cytochrome c treatment may protect against SIMD, as
certain areas of the tissues exhibited normal myocardial
histological features.
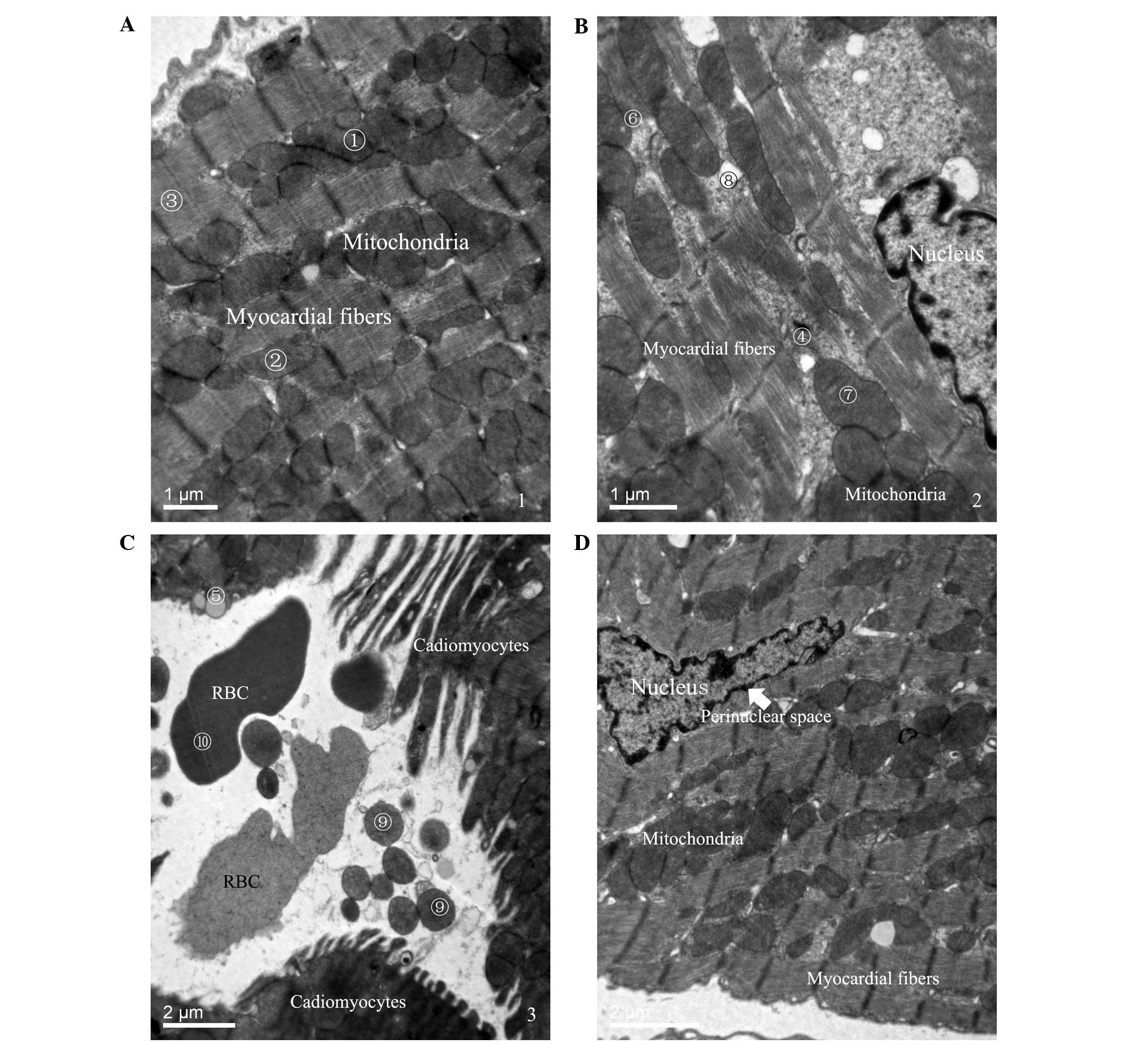 | Figure 1Ultrastructural organization of
myocardial tissues. Following electron microscopy, (A) the SHAM
group exhibited various characteristics, including (1) a large number of rows of mitochondria,
(2) smooth and complete
mitochondria cristae, and (3) neat
myocardial fibers (Scale bar, 1 µm). Isolated myocardial
tissues in mice in the (B) CLP (Scale bar, 1 µm) and (C) NS
groups (Scale bar, 2 µm) demonstrated characteristics of
cardiomyocyte damage at 12 h subsequent to the induction of sepsis,
including (4) marked exudation of
dissolved filaments, (5) increased
lipid droplets, (6) mitochondrial
disorder and increased swelling of the (7) mitochondria and (8) vacuoles, (9) individual mitochondria and (10) bleeding of red blood cells in the
gaps between cells. (D) The Cytc group exhibited characteristics of
protection 12 h subsequent to the induction of sepsis, including no
significant increase, swelling or vacuolization of mitochondria, no
widening of the perinuclear space of cardiomyocytes and no
dissolved filaments (Scale bar, 2 µm). RBC, red blood cell;
SHAM, sham-operation; CLP, sepsis; Cytc, cytochrome c. |
Histopathological examination
Histological assessment of the effects of cytochrome
c on SIMD was conducted at 12 h. Myocardial tissues (n=5)
from each experimental group were processed for histological
evaluation with H&E staining (magnification, ×400). As
presented in Fig. 2A and B,
myocardial tissues from the SHAM group exhibited normal
histological features; these features included the myocardial cell
arrangement, which consisted cross-striations or a coarse linear
pattern, interconnected into a network of oval-shaped nuclei
centrally located within the cytoplasm-rich myocyte. Isolated
myocardial tissues from the CLP group displayed characteristics of
acute cardiomyocyte damage and acute inflammation, including tissue
edema (Fig. 2C) and focal
myocardial necrosis (Fig. 2D),
vacuole degeneration (Fig. 2E) and
myocardial fiber breakage (Fig.
2F), myofascial cell hyperplasia and interstitial vascular
congestion (Fig. 2G). These
alterations were observed in all myocardial tissues from this
group. Furthermore, the data from isolated myocardial tissues in
the Cytc group suggested that cytochrome c treatment in part
prevented sepsis-induced damage (Fig.
2H and I).
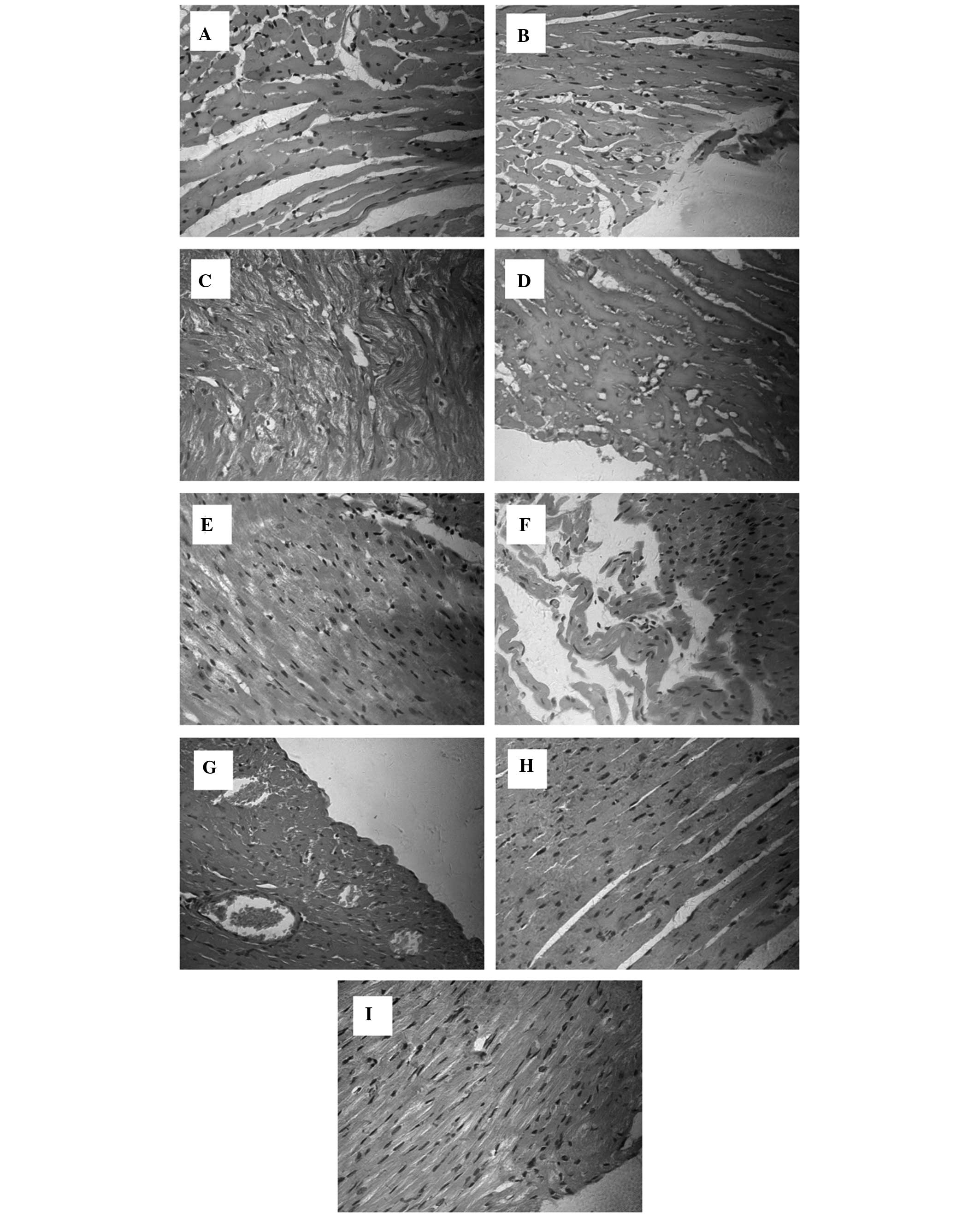 | Figure 2Histopathological analysis of the
effects of cytochrome c on SIMD was performed at 12 h
following injection of normal saline or Cytc. Myocardial tissues
from each experimental group were processed for histological
evaluation subsequent to H&E staining (magnification, x400). (A
and B) The SHAM group exhibited normal histological features,
including normal arrangements of myocardial fibers, no infiltration
of inflammatory cells in the stroma and no myocardial cell
swelling. The CLP group displayed characteristics of acute
cardiomyocyte damage and acute inflammation, including (C) tissue
edema and (D) focal myocardial necrosis, (E) granular degeneration
and (F) myocardial fiber breakage in myocardial cells, (G)
myofascial cell hyperplasia and interstitial vascular congestion.
The Cytc group demonstrated that cytochrome c treatment in
part prevents against sepsis-induced damage, and (H) certain parts
of the tissues demonstrated normal myocardial histological
features, and (I) no clear manifestations of intravascular
congestion. SIMD, sepsis-induced myocardial dysfunction; H&E,
hematoxylin and eosin; SHAM, sham-operation; CLP, sepsis; Cytc,
cytochrome c. |
Effects of cytochrome c injection on
TGF-β1 gene expression
RT-qPCR demonstrated that the expression levels of
TGF-β1 were significantly higher in the CLP 6-h group than in the
SHAM 6-h group (P<0.05) (Fig.
3B). In addition, the level of TGF-β1 was observed to be
significantly downregulated compared with the CLP 6-h group
following 6 and 12-h Cytc treatment (P<0.05).
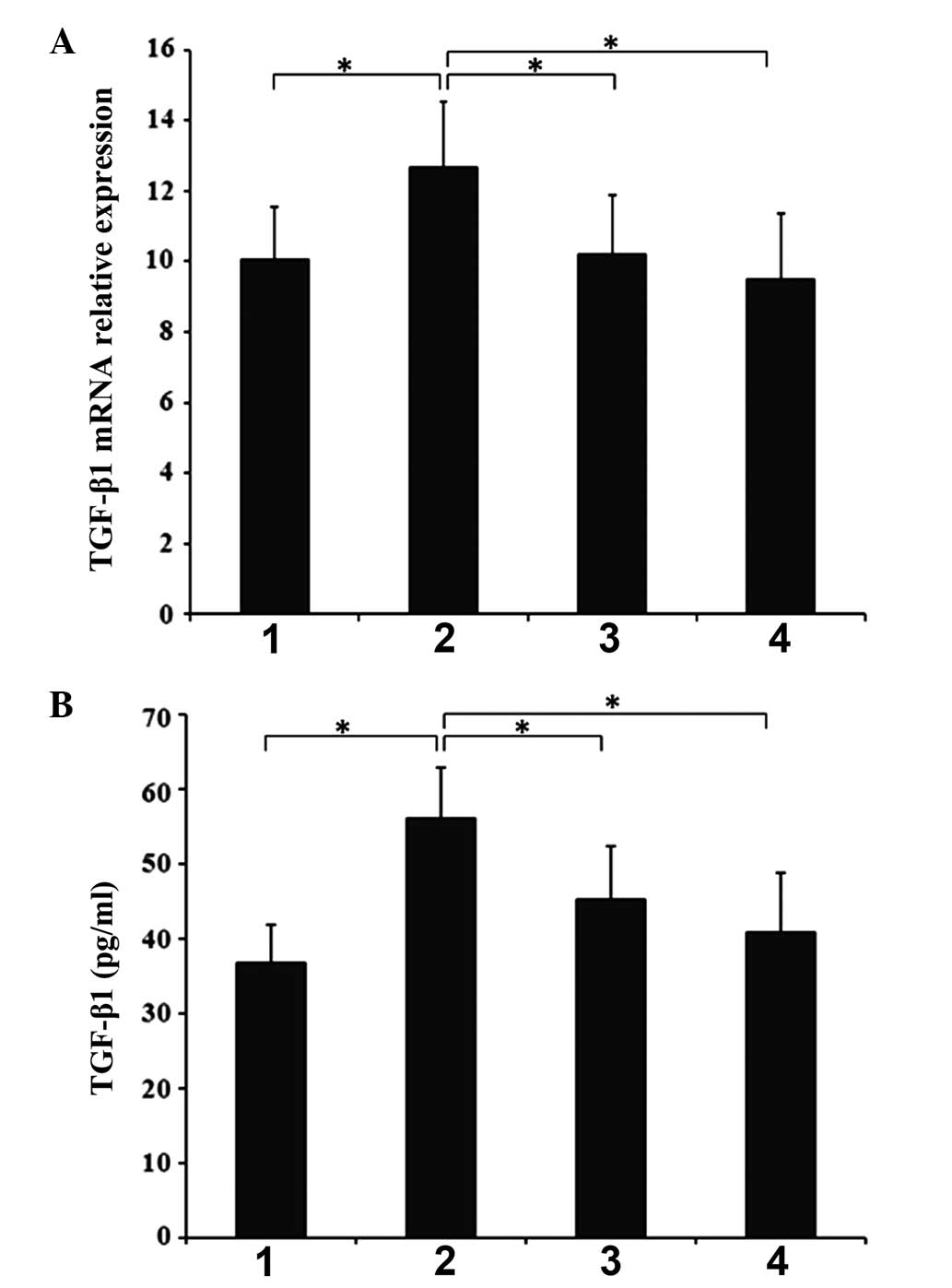 | Figure 3(A) Effects of cytochrome c on
gene and protein expression of TGF-β1 in SIMD. (B) Effects of
cytochrome c on protein plasma levels of TGF-β1 in SIMD.
Myocardial tissues and plasma were harvested at 6 and 12 h
subsequent to intraperitoneal injection of cytochrome c, and
levels of TGF-β1 gene expression and protein production were
measured by RT-qPCR and ELISA, respectively. 1, SHAM 6-h group; 2,
CLP 6-h group; 3, Cytc 6-h group; 4, Cytc 12-h group. All results
are presented as the mean ± standard deviation from three
independent experiments; *P<0.05 between groups
denoted by horizontal lines. TGF-β1, transforming growth factor-β1;
SIMD, sepsis-induced myocardial dysfunction; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; SHAM,
sham-operation; CLP, sepsis; Cytc, cytochrome c. |
Effects of cytochrome c on protein plasma
levels of TGF-β1
The ELISA demonstrated that TGF-β1 levels in the
plasma were significantly higher in the CLP 6-h group than those in
the SHAM 6-h group (P<0.05) (Fig.
3B). Cytochrome c treatments for 6 and 12 h were also
observed to significantly downregulate the expression of TGF-β1
compared with the CLP 6-h group (P<0.05).
Effects of cytochrome c on protein
expression of TGF-β1 and SMAD1/5/8
Western blot analysis demonstrated that the protein
levels of TGF-β1 and the TGF-β1-activated SMAD1/5/8 were
significantly higher in the CLP 6-h group compared with the SHAM
6-h group (P<0.05) (Fig. 4). In
addition, cytochrome c treatment for 6 and 12 h
significantly downregulated the expression of TGF-β1 (P<0.01)
(Fig. 4B) and SMAD1/5/8
(P<0.01) (Fig. 4C) compared
with the CLP group.
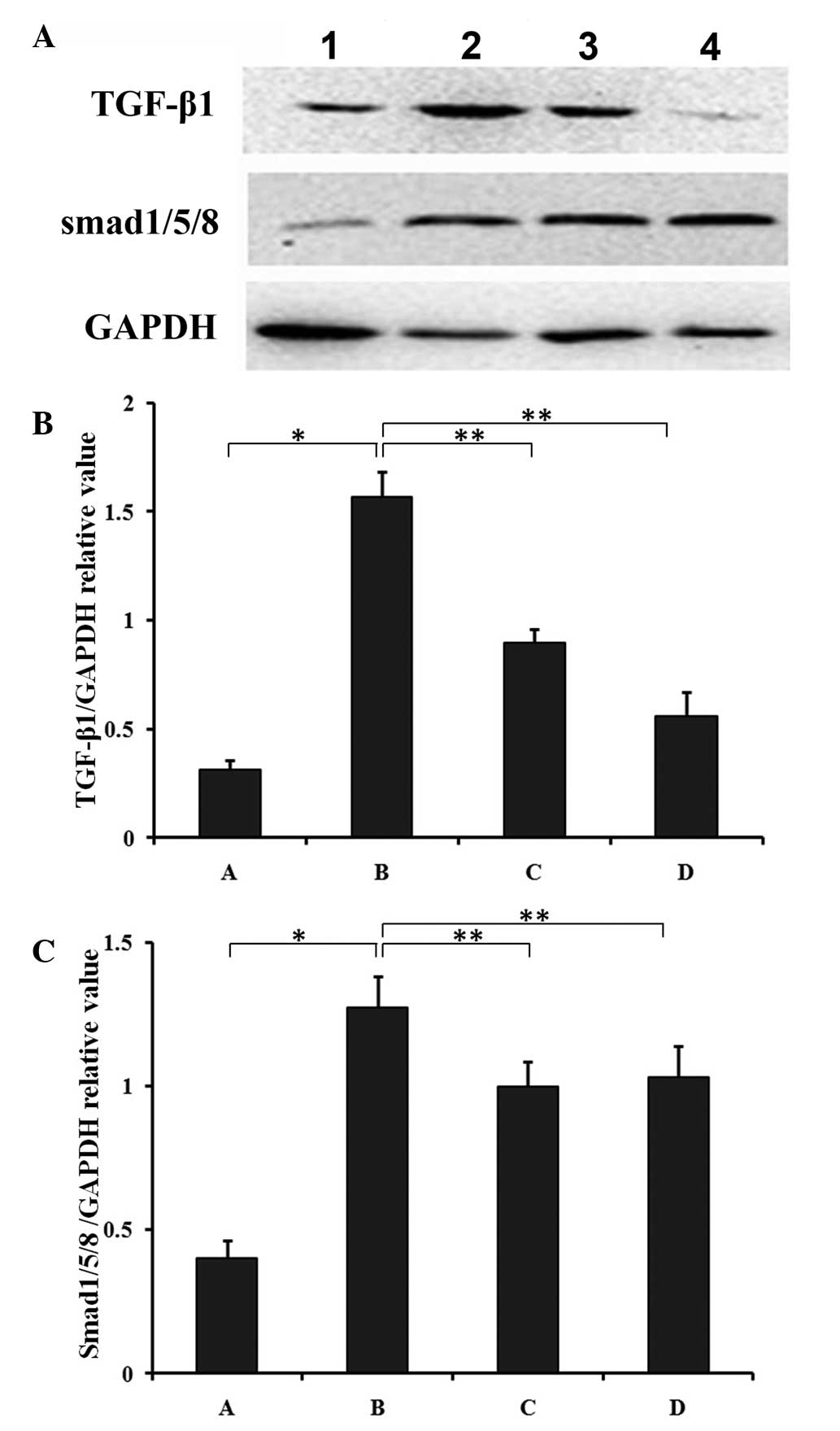 | Figure 4Effects of cytochrome c on
protein expression of TGF-β1 and SMAD1/5/8 in SIMD. (A) The
myocardial tissues were harvested at 6 and 12 h subsequent to
intraperitoneal injection of cytochrome c and underwent
western blotting. The protein expression levels of (B) TGF-β1 and
(C) SMAD1/5/8 in the different groups were quantified relative to
GAPDH expression. 1, SHAM 6-h group; 2, CLP 6-h group; 3, Cytc 6-h
group; 4, Cytc 12-h group. All results are presented as the mean ±
standard deviation from three independent experiments.
*P<0.05 and **P<0.01 between groups
denoted by horizontal lines. TGF-β1, transforming growth factor-β1;
SIMD, sepsis-induced myocardial dysfunction; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; SHAM,
sham-operation; CLP, sepsis; Cytc, cytochrome c. |
Discussion
In the current study, the main observations were as
follows: i) Exogenous cytochrome c inhibited the expression
of TGF-β1 within the myocardial tissues from mice with SIMD; and
ii) this function may proceed bia the dowregulation of
TGF-β1-activated SMAD1/5/8 expression.
Mitochondrial dysfunction has been suggested to be
involved in the pathogenesis of SIMD (23,26),
in the initial dysfunction and subsequent amplification. The
mitochondrial oxidative phosphorylation machinery has been
demonstrated to be essential for cell function, maintenance and
survival (27). Patients with
sepsis have been reported to exhibit increased mitochondrial
respiratory capacity in peripheral blood immune cells (28), and impaired oxidative
phosphorylation has been suggested to lead to SIMD (27). Failure of the cell to consume ATP
and provide adequate ADP at the adenine nucleotide transporter
during oxidative stress predisposes it to cytochrome c
release and the initiation of apoptosis (29). Cytochrome c and COX
represent the terminal step of the electron transport chain, which
is considered to be the rate-limiting step for metabolism in
mammals (27). They exhibit unique
regulatory features, including allosteric regulation, isoform
expression and regulation through cell signaling pathways (23). The role of cytochrome c and
COX phosphorylation has been investigated in various human
diseases, including cancer, asthma, ischemia/reperfusion injury,
inflammation and sepsis (27,30).
Inflammatory signaling can function as an off-switch, whereas
growth factor signaling leads to a general partial inhibition of
COX (31). In sepsis, COX has been
identified to be competitively inhibited early, which progresses to
non-competitive inhibition in the later stages (32). Therefore, the presence of COX
activity, and the level of activity, can be used as a predictive
biomarker for sepsis-associated mortality in humans (33). During sepsis and septic shock, the
endogenous Cytc of myocardial tissues in mitochondrion exudates
excess cytoplasm and the density of the mitochondrion markedly
decreases, resulting in the reduced inhibition of the functions of
mitochondria and heart. Previous studies have demonstrated that the
addition of exogenous cytochrome c may reverse myocardial
competitive COX inhibition, restore the activity of COX and improve
cardiac function during sepsis (32). It was reported that exogenous
cytochrome c repleted cardiac mitochondria, restored heme
c content and increased the kinetic activity of COX
(32,34). Thus, exogenous cytochrome c
administration is suggested for use in a novel therapeutic strategy
for SIMD.
TGF-β is an important cytokine that regulates
proliferation, differentiation, apoptosis, embryonic development,
angiogenesis, wound healing and other functions in various cell
types (35). Members of the TGF-β
superfamily, comprising the TGF-β and bone morphogenetic protein
(BMP) families, are released in pathophysiological conditions and
are the classical activators of SMAD proteins (36). SMAD proteins are the intracellular
effectors of TGF-β signaling and are activated by receptors, which
results the SMAD proteins forming complexes with each other and
translocating into the nucleus, where they regulate transcription
(37,38). TGF-β1 induces formation of a
complex of type I and II receptors, which results in the activation
of SMAD 2/3 and the SMAD and non-SMAD signaling pathways (39,40).
BMPs, including ALK 2/3/6, bind to BMP R-II and activates SMADs
1/5/8 (41). TGF-β1 activated its
downstream signaling (SMADs) and induced greater fibrotic and
oxidative stress to atrial compared with ventricular fibroblasts
(42). Thus, enhanced expression
of TGF-β1 and other family members in the heart under
pathophysiological conditions is an indicator of the activation of
SMAD signaling (41). A previous
study indicated that TGF-β may suppress the transcriptional
activity of genes associated with mitochondrial biogenesis or
function (43) and the
mitochondria have been demonstrated to modulate TGF-β1 signal
transduction (44). Loss of
activity of COX during conditions of stress has been suggested to
result in alterations to the mitochondrial membrane potential
(45). Thus, TGF-β1 is
hypothesized to increase mitochondrial oxygen consumption and ATP
generation in the presence of diverse energy substrates (46).
The current study observed that exogenous cytochrome
c was protective against SIMD in a mouse model. Exogenous
cytochrome c modulation of TGF-β1 signaling was associated
with reduced SMAD1/5/8 protein expression, significantly
downregulated expression of TGF-β1 and SMAD1/5/8.
Together, the observations of the current study
support the use of exogenous cytochrome c as a novel
therapeutic strategy for SIMD. One potential mechanism for this may
be via the inhibition of the TGF-β1/SMAD1/5/8 pathway.
Acknowledgments
The current study was supported by the Natural
Science Foundation of Chongqing (grant no. 2011jjA0479); and the
Medical Scientific Research Projects of Chongqing (grant no.
2012-1-023).
References
|
1
|
Wheeler DS: Death to sepsis: targeting
apoptosis pathways in sepsis. Crit Care. 13:10102009. View Article : Google Scholar
|
|
2
|
Rudiger A and Singer M: Mechanisms of
sepsis-induced cardiac dysfunction. Crit Care Med. 35:1599–1608.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sharma AC: Sepsis-induced myocardial
dysfunction. Shock. 28:265–269. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sips PY, Irie T, Zou L, et al: Reduction
of cardiomyocyte S-nitrosylation by S-nitrosoglutathione reductase
protects against sepsis-induced myocardial depression. Am J Physiol
Heart Circ Physiol. 304:H1134–H1146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Krishnagopalan S and Kumar A, Parrillo JE
and Kumar A: Myocardial dysfunction in the patient with sepsis.
Curr Opin Crit Care. 8:376–388. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Blanco J, Muriel-Bombin A, Sagredo V, et
al: Incidence, organ dysfunction and mortality in severe sepsis: a
Spanish multicentre study. Crit Care. 12:R1582008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Romero-Bermejo FJ, Ruiz-Bailen M,
Gil-Cebrian J and Huertos-Ranchal MJ: Sepsis-induced
cardiomyopathy. Curr Cardiol Rev. 7:163–183. 2011. View Article : Google Scholar
|
|
8
|
Takasu O, Gaut JP, Watanabe E, et al:
Mechanisms of cardiac and renal dysfunction in patients dying of
sepsis. Am J Respir Crit Care Med. 187:509–517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rudiger A and Singer M: The heart in
sepsis: from basic mechanisms to clinical management. Curr Vasc
Pharmacol. 11:187–195. 2013.PubMed/NCBI
|
|
10
|
Aoki Y, Hatakeyama N, Yamamoto S, et al:
Role of ion channels in sepsis-induced atrial tachyarrhythmias in
guinea pigs. Br J Pharmacol. 166:390–400. 2012. View Article : Google Scholar :
|
|
11
|
Levy RJ and Deutschman CS: Cytochrome c
oxidase dysfunction in sepsis. Crit Care Med. 35(Suppl): S468–S475.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yao YM, Luan YY, Zhang QH and Sheng ZY:
Pathophysiological aspects of sepsis: an overview. Methods Mol
Biol. 1237:5–15. 2015.
|
|
13
|
Cao J, Xu F, Lin S, et al: IL-27 controls
sepsis-induced impairment of lung antibacterial host defence.
Thorax. 69:926–937. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sharma D, Packiriswamy N, Malik A, et al:
Nonhematopoietic β-Arrestin-1 inhibits inflammation in a murine
model of polymicrobial sepsis. Am J Pathol. 184:2297–2309. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schilling J, Lai L, Sambandam N, Dey CE,
Leone TC and Kelly DP: Toll-like receptor-mediated inflammatory
signaling reprograms cardiac energy metabolism by repressing
peroxisome proliferator-activated receptor γ coactivator-1
signaling. Circ Heart Fail. 4:474–482. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hsu BG, Lee RP, Yang FL, Harn HJ and Chen
HI: Post-treatment with N-acetylcysteine ameliorates endotoxin
shock-induced organ damage in conscious rats. Life Sci.
79:2010–2016. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pathan N, Hemingway CA, Alizadeh AA, et
al: Role of interleukin 6 in myocardial dysfunction of
meningococcal septic shock. Lancet. 363:203–209. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kumar A, Kumar A, Paladugu B, Mensing J
and Parrillo JE: Transforming growth factor-beta1 blocks in vitro
cardiac myocyte depression induced by tumor necrosis factor-alpha,
interleukin-1beta, and human septic shock serum. Crit Care Med.
35:358–364. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rainer PP, Hao S, Vanhoutte D, et al:
Cardiomyocyte-specific transforming growth factor beta suppression
blocks neutrophil infiltration, augments multiple cytoprotective
cascades, and reduces early mortality after myocardial infarction.
Circ Res. 114:1246–1257. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
ElZarrad MK, Mukhopadhyay P, Mohan N, et
al: Trastuzumab alters the expression of genes essential for
cardiac function and induces ultrastructural changes of
cardiomyocytes in mice. PloS One. 8:e795432013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zang Q, Maass DL, Tsai SJ and Horton JW:
Cardiac mitochondrial damage and inflammation responses in sepsis.
Surg Infect (Larchmt). 8:41–54. 2007. View Article : Google Scholar
|
|
22
|
Bouchier-Hayes L, Lartigue L and Newmeyer
DD: Mitochondria: pharmacological manipulation of cell death. J
Clin Invest. 115:2640–2647. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Groening P, Huang Z, La Gamma EF and Levy
RJ: Glutamine restores myocardial cytochrome C oxidase activity and
improves cardiac function during experimental sepsis. JPEN J
Parenter Enteral Nutr. 35:249–254. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Levy RJ, Vijayasarathy C, Raj NR, Avadhani
NG and Deutschman CS: Competitive and noncompetitive inhibition of
myocardial cytochrome C oxidase in sepsis. Shock. 21:110–114. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Singleton KD, Serkova N, Beckey VE and
Wischmeyer PE: Glutamine attenuates lung injury and improves
survival after sepsis: role of enhanced heat shock protein
expression. Crit Care Med. 33:1206–1213. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chopra M, Golden HB, Mullapudi S, Dowhan
W, Dostal DE and Sharma AC: Modulation of myocardial mitochondrial
mechanisms during severe polymicrobial sepsis in the rat. PLoS One.
6:e212852011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hüttemann M, Lee I, Grossman LI, Doan JW
and Sanderson TH: Phosphorylation of mammalian cytochrome c and
cytochrome c oxidase in the regulation of cell destiny:
respiration, apoptosis, and human disease. Adv Exp Med Biol.
748:237–264. 2012.PubMed/NCBI
|
|
28
|
Sjövall F, Morota S, Persson J, Hansson MJ
and Elmér E: Patients with sepsis exhibit increased mitochondrial
respiratory capacity in peripheral blood immune cells. Crit Care.
17:R1522013. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kantrow SP, Tatro LG and Piantadosi CA:
Oxidative stress and adenine nucleotide control of mitochondrial
permeability transition. Free Radic Biol Med. 28:251–260. 2000.
View Article : Google Scholar
|
|
30
|
Hüttemann M, Lee I, Pecinova A, Pecina P,
Przyklenk K and Doan JW: Regulation of oxidative phosphorylation,
the mitochondrial membrane potential, and their role in human
disease. J Bioenerg Biomembr. 40:445–456. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hüttemann M, Helling S, Sanderson TH, et
al: Regulation of mitochondrial respiration and apoptosis through
cell signaling: cytochrome c oxidase and cytochrome c in
ischemia/reperfusion injury and inflammation. Biochim Biophys Acta.
1817:598–609. 2012. View Article : Google Scholar
|
|
32
|
Piel DA, Deutschman CS and Levy RJ:
Exogenous cytochrome C restores myocardial cytochrome oxidase
activity into the late phase of sepsis. Shock. 29:612–616.
2008.PubMed/NCBI
|
|
33
|
Lorente L, Martín MM, López-Gallardo E, et
al: Platelet cytochrome c oxidase activity and quantity in septic
patients. Crit Care Med. 39:1289–1294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Piel DA, Gruber PJ, Weinheimer CJ, et al:
Mitochondrial resuscitation with exogenous cytochrome c in the
septic heart. Crit Care Med. 35:2120–2127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Saas P and Perruche S: Functions of
TGF-β-exposed plasmacytoid dendritic cells. Crit Rev Immunol.
32:529–553. 2012. View Article : Google Scholar
|
|
36
|
Hegarty SV, O’Keeffe GW and Sullivan AM:
BMP-Smad 1/5/8 signalling in the development of the nervous system.
Prog Neurobiol. 109:28–41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Derynck R and Zhang YE: SMAD-dependent and
SMAD-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wrighton KH, Lin X and Feng XH:
Phosphocontrol of TGF-beta superfamily signaling. Cell Res.
19:8–20. 2009. View Article : Google Scholar
|
|
39
|
Voloshenyuk TG, Landesman ES, Khoutorova
E, et al: Induction of cardiac fibroblast lysyl oxidase by TGF-β1
requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine. 55:90–97.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Olieslagers S, Pardali E, Tchaikovski V,
et al: TGF-β1/ALK5-induced monocyte migration involves PI3K and p38
pathways and is not negatively affected by diabetes mellitus.
Cardiovasc Res. 91:510–518. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Euler-Taimor G and Heger J: The complex
pattern of SMAD signaling in the cardiovascular system. Cardiovasc
Res. 69:15–25. 2006. View Article : Google Scholar
|
|
42
|
Yeh YH, Kuo CT, Chang GJ, Qi XY, Nattel S
and Chen WJ: Nicotinamide adenine dinucleotide phosphate oxidase 4
mediates the differential responsiveness of atrial versus
ventricular fibroblasts to transforming growth factor-β. Circ
Arrhythm Electrophysiol. 6:790–798. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sohn EJ, Kim J, Hwang Y, Im S, Moon Y and
Kang DM: TGF-β suppresses the expression of genes related to
mitochondrial function in lung A549 cells. Cell Mol Biol
(Noisy-le-grand). 58(Suppl): OL1763–OL1767. 2012.
|
|
44
|
Corcoran JB, McCarthy S, Griffin B, et al:
IHG-1 must be localised to mitochondria to decrease Smad7
expression and amplify TGF-β1-induced fibrotic responses. Biochim
Biophys Acta. 1833:1969–1978. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Piplani H, Vaish V, Rana C, et al:
Up-regulation of p53 and mitochondrial signaling pathway in
apoptosis by a combination of COX-2 inhibitor, Celecoxib and
Dolastatin 15, a marine mollusk linear peptide in experimental
colon carcinogenesis. Mol Carcinog. 52:845–858. 2013. View Article : Google Scholar
|
|
46
|
Abe Y, Sakairi T, Beeson C and Kopp JB:
TGF-β1 stimulates mitochondrial oxidative phosphorylation and
generation of reactive oxygen species in cultured mouse podocytes,
mediated in part by the mTOR pathway. Am J Physiol Renal Physiol.
305:F1477–F1490. 2013. View Article : Google Scholar : PubMed/NCBI
|














