Introduction
Protein A separated from the Staphylococcus
aureus cell wall (PA) can specifically combine with
immunoglobulin (Ig)G, IgM and IgA in most mammals (1). Protein A can combine with the Fc
fragment of immunoglobulin. The ligation of protein A with certain
genes was shown not to influence binding sites between antibody and
antigen (2). PA does not contain
disulfide bonds due to absence of cysteine and cystine; its
physical properties are therefore very steady (3). Under acidic conditions, even combined
with heating, the activity of PA does not change. PA exogenous
expression was implemented in E. coli (4). Separated or recombinant PA
specifically combines with Fc fragments of human IgG1, IgG2 and
IgG4 in vitro. Since its discovery, PA has been received
attention by various researchers and has a widespread application
in immunology and molecular biology experiments (4).
Similar to protein A, protein G (PG) is an protein
of the Streptococcus cell wall; however, it is more useful
in IgG separation and purification (5). As protein A, protein G can bind to
the Fc region of immunoglobulin and numerous subtypes of IgG,
including IgG1, IgG2, IgG3 and IgG4 (6). Although the 4th spatial structure of
protein A is similar to that of protein G, they differ in their
amino acid composition, and therefore, protein G but not protein A
can be used for separating monoclonal antibodies (7). Compared with protein A, protein G has
a stronger IgG-binding capacity, but it does not bind to IgM, IgD
or IgA. The affinity of protein G to Ig is more advanced than that
of PA and of greater commercial value (3). During antibody separation and
purification, albumin is the main contamination source; therefore,
the albumin binding site on protein G was removed in preparation of
the present study. For convenient purification, a 6xHis label was
added to the C terminal of protein G in our prelimary experiments,
as described previously (8). E.
coli high-density fermentation is widely applied in recombinant
protein production and efficiency can be enhanced by improving
parameters of these methods (9,10).
At present, there is only a limited number of studies on PG
fermentation production as well as separation and purification. In
the present study, PG was produced by high-density fermentation of
recombinant gene engineered bacteria, and highly efficient
separation and purification techniques were implemented, providing
a theoretical and practical basis for further studies and PG
production.
Materials and methods
Strain
The E. coli strain BL21 with recombinant
vector pET32b-PG was established in our preliminary works (data not
shown) at the Gene Engineering Laboratory of Bioengineering College
of Beijing Electronic Technology Training College (Beijing, China)
(8). The E. coli strains
BL21 and pET32b were purchased from Takara Biotechnology., Co.,
Ltd. (Dalian, China).
Ragents
Yeast extracts and tryptone were purchased from
Oxoid Ltd. (Basingstoke, UK), and isopropyl
β-D-1-thiogalactopyranoside (IPTG) and kanamycin were purchased
from Takara Bio Inc. (Otsu, Japan). Buffers and solutions used were
1 mol/l pH 7.5
NaH2PO4-Na2HPO4 buffer,
5 mol/l pH 7.5 iminazole, 0.5 mol/l NaOH, 0.1 mol/l
NiSO4, 4 mol/l pH 7.5 NaCl and 20% ethanol solution.
Unless otherwise stated, all other chemical reagents were purchased
from Sinopharm Group Co., Ltd. (Beijing, China).
Instruments
The GE Image Scanner III (GE Healthcare, Little
Chalfont, UK), a NBS5LBioflo3000 fermenter (New Brunswick
Scientific Co., Edison, NJ, USA), an ultrasonicator (Ningbo Scientz
Biotechnology Co., Ltd., Ningbo, China), a high-speed centrifuge
(5810 R; Eppendorf, Hamburg, Germany), a Unico 7200
spectrophotometer (Unico Instrument Co., Shanghai, China), vertical
slab electrophoresis apparatus (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), AKTA purifier UPC10 (GE Healthcare) and a
chromatography column (diameter, 5 cm; length, 20 cm; Shanghai
BioRc Co., Ltd., Shanghai, China) were used.
Media
Primary seed medium [Luria-Bertani
(LB) medium]
The medium contained yeast extracts (5 g/l),
tryptone (10 g/l) and NaCl (10 g/l) at pH 7.3 in a total of 1 l
distilled water. The solution was sterilized at 121°C for 20 min,
and ampicillin (50 μg/m) and kanamycin (50 mg/l) were then
added.
Secondary seed medium (2xYT
medium)
The medium contained yeast extracts (10 g/l),
tryptone (16 g/l) and NaCl (5 g/l) at pH 7.3 in a total of 1 l
distilled water. The solution was sterilized at 121°C for 20 min,
and kanamycin (50 mg/l) and Amp (50 μg/ml) were added.
Fermentation medium
The medium contained yeast extracts (64 g), tryptone
(102 g), glucose (6.4 g), disodium hydrogen phosphate (48 g),
potassium dihydrogen phosphate (9.6 g), ammonium chloride (3.2 g),
CaCl2 (0.035 g), NaCl (1.6 g), MgSO4 (3.2 g)
and vitamin B1 (0.1 g), pH 7.2, with distilled water added to give
1 l of total volume. The medium was sterilized under high pressure
at 115°C for 30 min, and Amp (50 μg/ml) and kanamycin (50
mg/l) were added.
Feed supplement medium
The medium contained yeast extracts (200 g),
tryptone (20 g), glucose (10 g), MgSO4 (5 g),
(NH4)2SO4 (5 g) and vitamin B1
(0.1 g), and distilled water was added to give 0.8–1l of total
volume. The medium was sterilized under high pressure at 115°C for
30 min, and then Amp (50 μg/ml) and kanamycin (50 mg/l) were
added.
Strain activation, cultivation and
fermentation
Working seed medium preserved at −20°C was
defrosted. 10 μl seed medium was diluted with a gradient
from 10−5 to 10−6 to 10−7 and
coated on an LB plate containing Amp and kanamycin for cultivating
under 37°C for 16–18 h until single colonies were formed. For the
primary seed F1 fermentation strain, a single colony was
selected from the plate, inoculated in 50 ml LB medium containing
Amp and kanamycin in a 250-ml flask, cultivated at 37°C with
agitation at 200 rpm for 8–10 h until the optical density at 600 nm
(OD600) of the bacterial suspension reached 0.025–0.035.
For the second seed F2, the fermentation strain
F1 was inoculated in 200 ml 2xYT medium containing Amp
and kanamycin in a 1,000-ml flask at 0.5% inoculum size, cultivated
at 37°C with agitation at 200 rpm for 7–8 h until the
OD600 of the bacterial suspension reached 0.03–0.05.
500 ml grown F2 seed liquid was
inoculated in a fermenter containing Amp and kanamycin and
cultivated at 37°C until the OD600 reached 0.40–0.50.
Subsequently, 0.1–0.2 mmol/l IPTG was added for induction for
3.5–4.0 h. Compressed air was used as the air source with an air
flow of 1 vvm and 30–45% dissolved oxygen. Pure oxygen was added
2.5 h after initiation of fermentation, with the pH controlled at
7.0–7.2. Sampling was conducted once per hour to measure the
OD600 until it reached a maximum value. The feed
supplement speed was controlled using a feed supplement influx at
10–40% during the cultivating period, increased by 10% as the
fermentation time progressed; it was controlled using a feed
supplement influx at 20–40% during the induction period, with the
maximum feed supplement speed in the first hour of the induction
period. Feed supplementation was stopped half an hour prior to
opening the fermenter at the end of the process.
Measurement of the concentration of
recombinant engineered bacteria
A Unico 7200 spectrophotometer was adopted to
measure the OD600, with distilled water as the control.
The sample from fermentation broth was washed using distilled water
three times, and diluted to 0.2–0.8 at OD600.
Ultrasonic fragmentation of
recombinant E. coli BL21
The centrifuged bacterial bodies were weighed and
added into 10 mmol/l phosphate buffer (pH 7.0,
Na2HPO4-NaH2PO4) at a
ratio of 1:10. Following mixing, the suspension was cooled with ice
water and subjected to ultrasonication at 200 watt for 100–150
times (10 sec on, 10 sec off). Following ultrasonication, the
solution was transferred into centrifuge tubes, centrifuged at
15,000 ×g for >20 min with the supernatant carefully removed,
and the supernatant was filtered through double layered filter
paper and a 0.8-μm membrane in sequence to remove any
particles. To the filtered sample, NaCl (0.5 mol/l) and iminazole
(5 mmol/l) were added, and the resulting solution was loaded onto
columns for purification.
Nickel column purification
Five milliliter desalting solution (containing 1.46
mg total protein) was purified on 30 ml
diethylaminoethanol-sepharose fast flow (DEAE-FF). Balance solution
1 (10 mmol/l sodium phosphate and 0.5 mol/l NaCl; pH 7.5) was used
for balancing five column volumes at a flow speed of 1 cm/min; the
sample was slowly pumped into the nickel nitrilotriacetic acid
(Ni-NTA)-agarose column (Puribest, Shanghai, China) at a flow speed
of 0.5 cm/min; following loading of the sample, the column was
flushed with five column volumes of balance solution 1 and balance
solution 2 (10 mmol/l sodium phosphate and 0.05 mol/l NaCl; pH 7.5)
in sequence, followed by five column volumes of washing solution 1
(10 mmol/l sodium phosphate, 0.05 mol/l NaCl and 0.02 mol/l
iminazole; pH 7.5). The column was then flushed with 2–3 column
volumes of spent eluent 1 (10 mmol/l sodium phosphate, 0.05 mol/l
NaCl and 0.1 mol/l iminazole; pH 7.5) for collecting the target
protein, and then washed with regeneration solution 1 (10 mmol/l
sodium phosphate, 0.05 mol/l NaCl and 0.4 mol/l iminazole; pH 7.5)
for regeneration of the nickel column.
Gel filtration (Sephadex G-25) for
desalination
The column was washed with five column volumes of
balance solution 3 (10 mM sodium phosphate; pH 7.5) at a flow speed
of 1 cm/min. The sample was then loaded onto the column, and at a
flow speed of 0.5 cm/min, the column was flushed with five volumes
of balance solution 3 for collecting the protein sample.
Negative ion exchange column
purification DEAE-FF
Five milliliter lysate was purified on 20 ml Ni-NTA
Agarose, using a UV280 nm detector to detect the products. Five
column volumes of balance solution 3 (10 mM sodium phosphate
solution; pH 7.5) were used for balancing at a flow speed of 1
cm/min. The sample was slowly loaded onto the column (Puribest),
and at a flow speed of 0.5 cm/min, the column was flushed with five
column volumes of balance solution 3 and washing solution 2 (10
mmol/l sodium phosphate and 0.05 mol/l NaCl; pH 7.5) in sequence.
Subsequently, the column was washed with 2–3 column volumes of
spent regenerant 2 (10 mmol/l sodium phosphate and 0.1 mol/l NaCl;
pH 7.5), spent regenerant 3 (10 mmol/l sodium phosphate and 0.15
mol/l NaCl; pH 7.5) and spent regenerant 4 (10 mmol/l sodium
phosphate and 0.2 mol/l NaCl; pH 7.5) for eluting the target
protein. The column was then treated with regeneration solution 2
(10 mmol/l sodium phosphate and 0.5 mol/l NaCl; pH 7.5) for
regenerating the ion exchange solid phase.
Protein detection
The recombinant PG (rPG) protein in the samples was
identified as follows: 12% SDS-PAGE (Bio-Rad Laboratories, Inc.),
gel staining with 0.25% Commassie Blue staining solution (methanol:
acetic acid: dH2O=5:1:5), destaining with a solution
(methanol: acetic acid: dH2O=2:3:35). Following
destaining, the gel was visualized by GE Image scanner III (GE
Healthcare). The protein concentration was measured by the methods
according to Bradford or Lowry (11) with bovine serum albumin as the
standard (8).
Purity determination and
identification of rPG
The purified dried rPG was dissolved in 50 mM sodium
phosphate buffer (pH 7.2) containing 50 mM NaCl and underwent high
performance liquid chromatography (HPLC; Agilent 1200, Santa Clara,
CA, USA; TSK-GEL G2000SWXL, 5 μm, 7.8 mmx300 mm; Tosoh Co.,
Tokyo, Japan), alongside molecular weight standards bovine serum
albumin (66.4 kDa), ovalbumin (44.3 kDa), pepsin (35.0 kDa),
lysozyme (14.3 kDa), insulin (58.8 kDa) and hydroxocobalamine (1.4
kDa). All the standards were purchased from Tiangen (Tiangen
Biotech Co., Ltd, Beijing, China). The mobile phase, at a flow rate
of 0.5 ml/min, was 50 mM sodium phosphate buffer (pH 7.2)
containing 50 mM NaCl. The experiment was monitored using a UV
detector (280 nm).
The purified rPG and the commercialized PG were
separated by 12% SDS-PAGE gel, and were transferred to
polyvinylidene difluoride (PVDF) membranes using semi-dry
electro-blotting apparatus (Bio-Rad Laboratories, Inc.). The
transfer was carried out for 2 h at 18 V. Subsequently, the
membranes were blocked with TBST (20 mmol/l Tris-HCl, pH 8.0; 150
mmol/l NaCl; 0.05% Tween) containing 5% non-fat milk for 1 h, and
then incubated with a 1:10,000 dilution of goat horseradish
peroxidase-conjugated anti-Protein G antibody (cat. no. SA101;
Tiangen Biotech Co., Ltd) at room temperature for 1 h. The membrane
was then washed six times with TBST (10 min per wash), and positive
bands in the membrane were detected by Enhanced Chemiluminescent
reagent (Beyotime Institute of Biotechnology, Beijing, China).
Statistical analysis
SPSS 13.0 statistical software (SPSS Inc., Chicago,
IL, USA) was used to analyze the data. Measurement data were
compared using one-sample t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
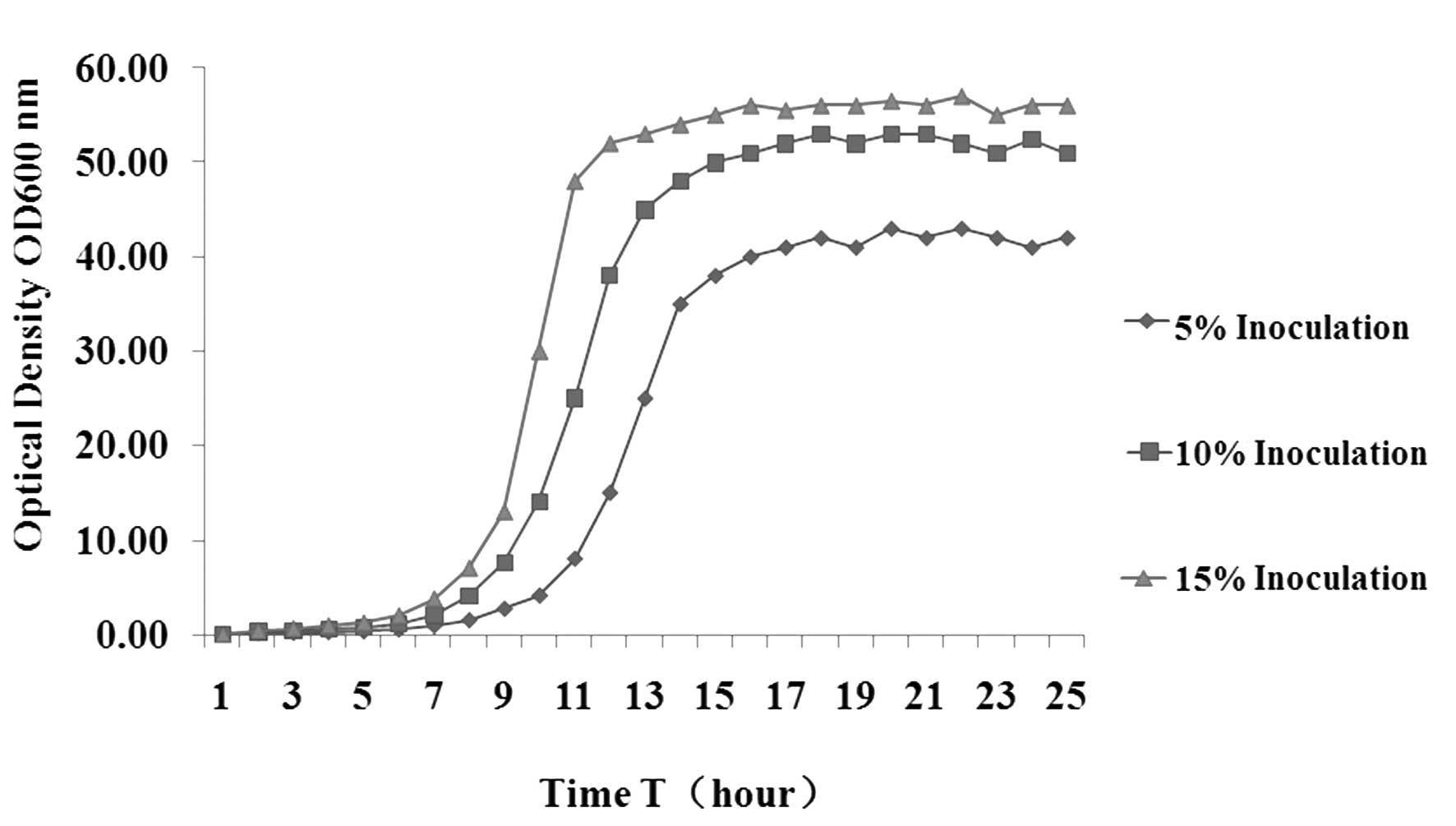
Influence of the seed inoculum size on
the productivity and time of fermentation
The productivity and time of PG fermentation were
influenced by the inoculum size in the fermenter. In the
fermentation experiments, inoculum sizes of 5, 10 and 15% were
adopted (Fig. 1) in order to
observe their influence on the mass of bacterial bodies. The
results indicated that at a relatively low inoculum size of 5%, the
lag phase was significantly longer, which may be due to strain
aging, extended growth time and a lower number of bacteria in the
stationary phase, resulting in lower protein expression. However,
at 10% inoculum size, the lag phase was significantly shortened,
and the rapid proliferation of recombinant bacteria during
fermentation made them enter the Logarithmic growth phase earlier
with more nutrition for target product synthesis, leading to a
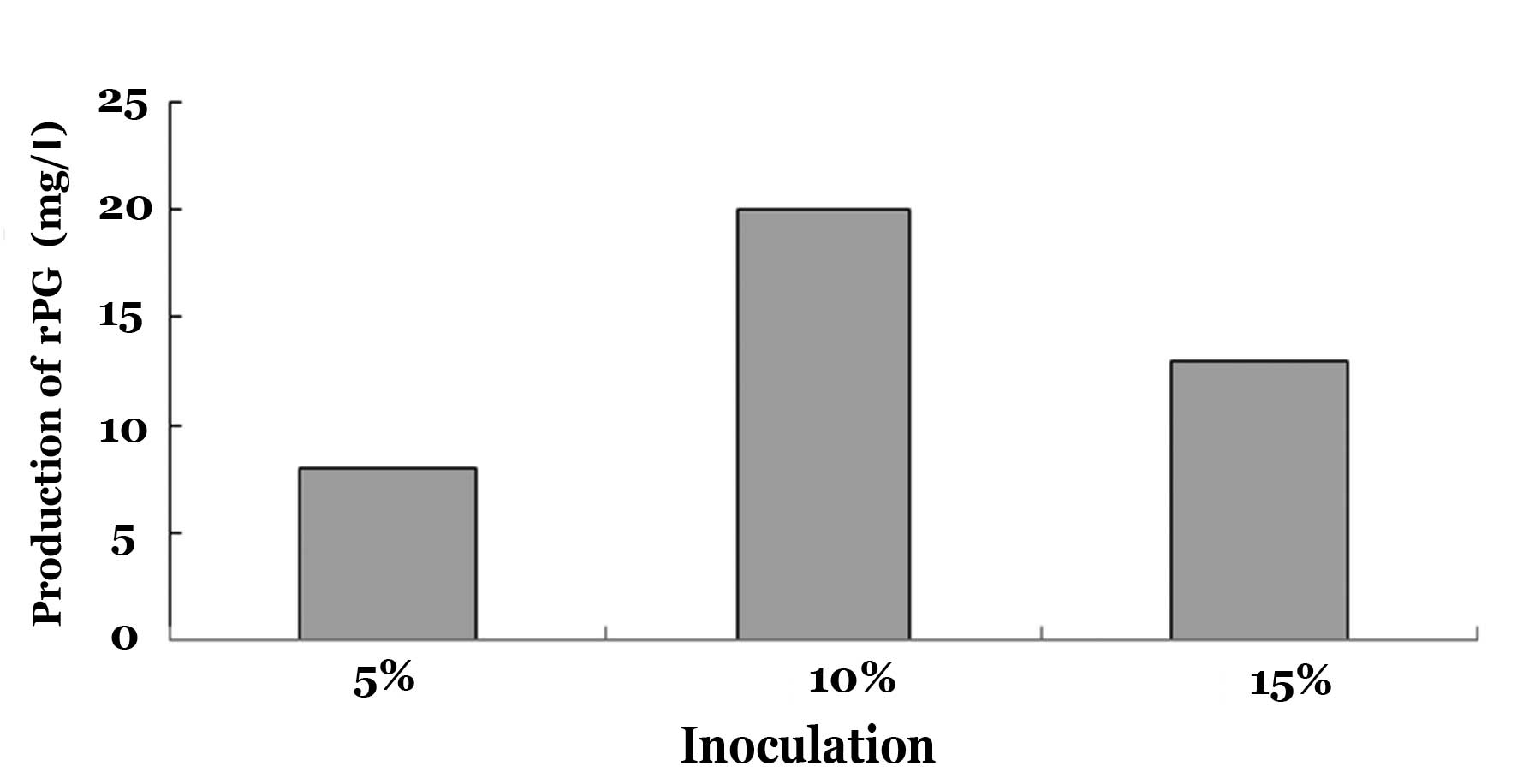
recombinant PG productivity of 20 mg/l (Fig. 2). When the inoculum size was 15%,
the large amount of bacteria exhausted the availability of
nutrition bacterial growth, resulting in a very short lag phase.
The density of bacteria in the stationary phase was >10% of the
inoculum size, but exogenous PG productivity decreased (Fig. 2); the potential cause of this is
that nutrition was absorbed by the large amount of bacteria, while
there was less nutrition for PG production; in addition, the
metabolic waste of the bacteria may have had a negative effect on
subsequent PG synthesis.
Dissolved oxygen levels affect the
productivity and time of fermentation
The dissolved oxygen concentration is another
important factor influencing bacterial growth during high-density
fermentation. The bacterial density and recombinant protein
productivity are influenced by the concentration of dissolved
oxygen. E. coli require a large amount of oxygen for
metabolism during rapid proliferation, and therefore, a timely
supply of saturated oxygen is very important. The bacterial density
was very high at the late period of high-density fermentation,
which therefore required extremely large amounts of dissolved
oxygen to maintain target protein synthesis. The compressed air
used in the present study was not sufficient to meet the oxygen
demand of the bacteria, as indicated by a DO2 value of 0
or even a negative value, high bacterial density and a bacterial
wet weight of 150 g/l, while PG expression levels remained low.
During the initial fermentation period, the required quantities of
oxygen were not high; therefore, the present study used pressured
air with levels of dissolved oxygen controlled at 30–45% during the
initial period. Following 2.5 h of fermentation, oxygen was added,
and at the late period of fermentation, high-pressure pure oxygen
was supplied to enhance the oxygen supply.
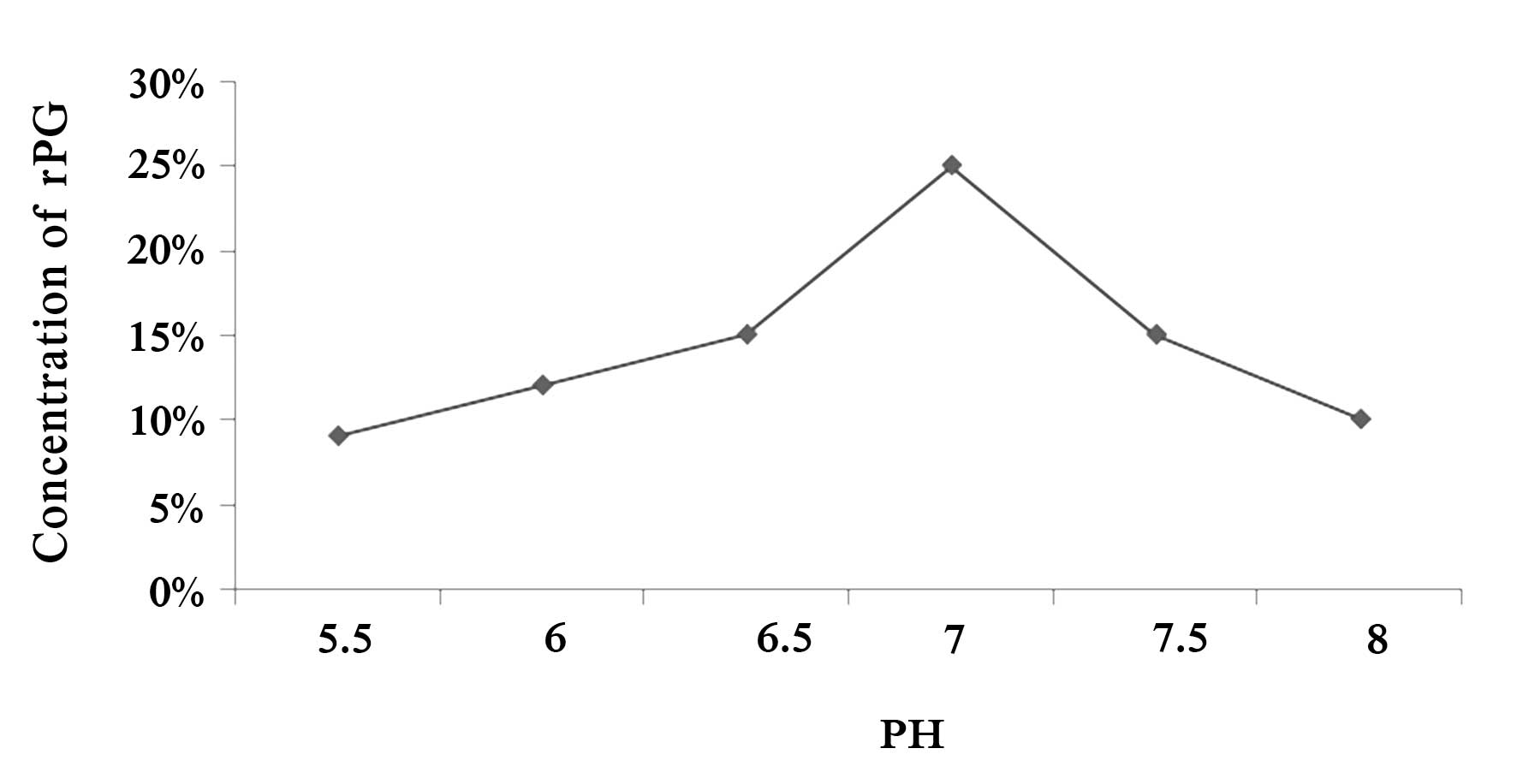
Influence of the pH on fermentation
efficiency
pH variance in the fermentation environment
influences the internal environment and variance in the cell
metabolism of bacteria; furthermore, it directly influences the
biomass of genetically engineered bacteria and the amount of gene
expression of the product. The influence of the initial pH value of
the medium on the production of PG was investigated in the present
study. A higher or lower initial pH value of the medium resulted in
reduced PG expression, while maximum production of recombinant PG
was achieved when the pH of the medium was 7.0 at the beginning of
the fermentation (Fig. 3).
IPTG influences the amount of PG
expression
Following the logarithmic phase, the bacteria were
cultivated with the inducer IPTG at 0, 0.1, 0.2, 0.3, 0.4 and 0.5
mmol/l for 4 h, and the content of recombinant PG in the soluble
protein of the bacteria was analyzed. The amount of rPG expression
increased as the IPTG concentration was enhanced, but above a
concentration of 0.2 mmol/l IPTG did not lead to any further
obvious improvement of PG expression; in fact, it decreased to a
certain extent (results not shown). Therefore, the IPTG
concentration used for induction was fixed at 0.2 mmol/l. The
induction time of IPTG influenced the levels of PG expression, as
shown in Fig. 4. The amount of rPG
protein expression gradually increased as the induction time was
increased from 1 h and reached a peak when induction was performed
for 4 h, leading to an rPG yield of 20% of total proteins. However,
further extension of the induction time decreased the yield of
recombinant PG, which may be due to degradation or denaturation of
PG over longer time periods.
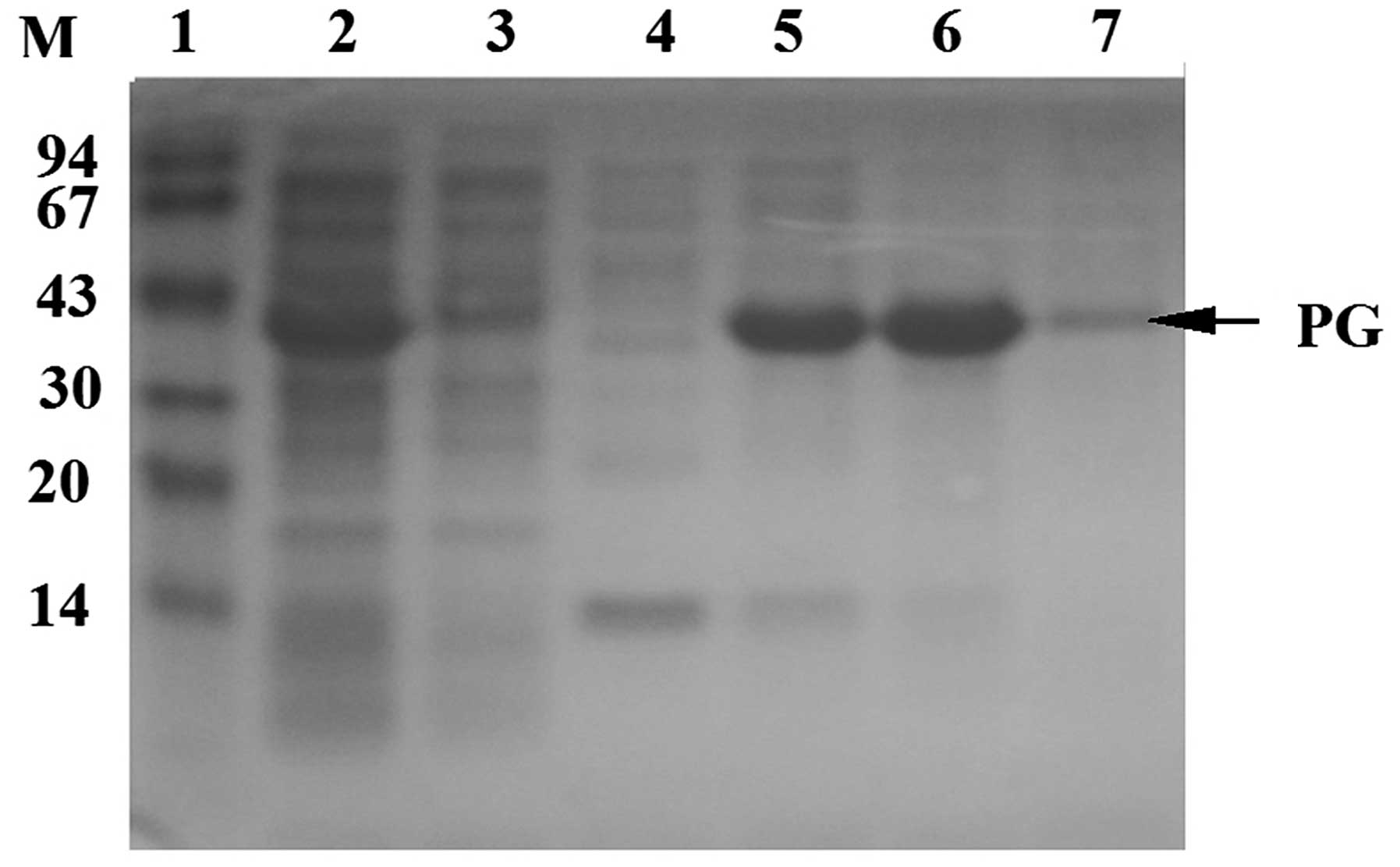
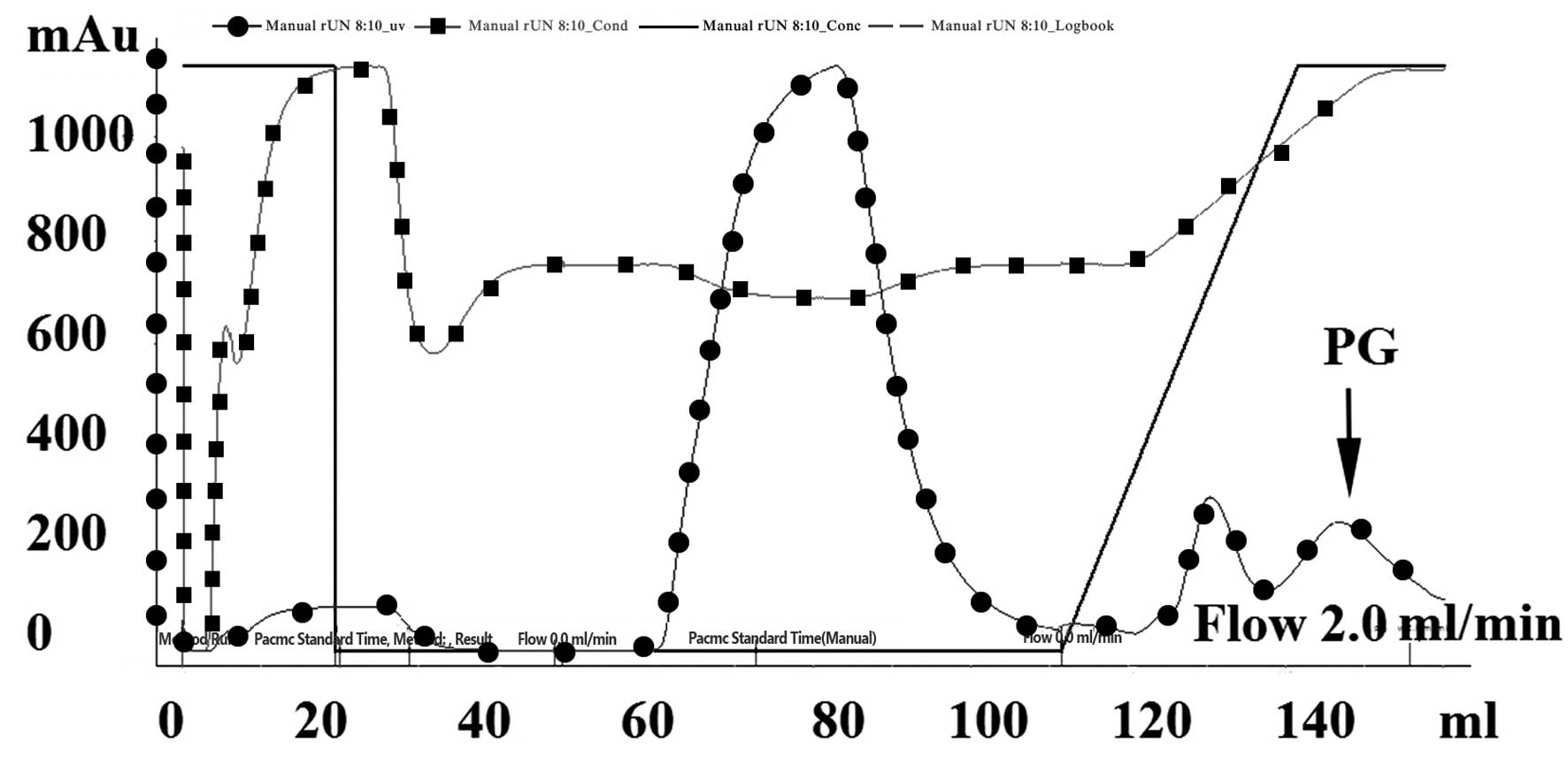
PG separation and purification
Metal chelate affinity chromatography, also known as
fixed metal ion affinity chromatography, utilizes the coordination
interaction between various metal ions, including Cu2+,
Zn2+, Ni2+, Co2+ and
Fe3+, and histidines on the protein surface as a
principle for separation and purification. Agarose gel containing
these fixed metal ions is able to selectively purify proteins
containing multiple histidines. In a previous study (12), PG with a C terminus containing six
histidines was separated from cell lysate using a Ni-NTA affinity
column, desalinated by gel filtration (Sephadex G-25) and purified
by anion exchange column (DEAE-FF). In the present study, PG was
obtained at a maximum yield following one step of nickel column
affinity chromatography (Fig. 5).
Following gel filtration and anion exchange column chromatography,
an PG purity of >95% was achieved (Fig. 4).
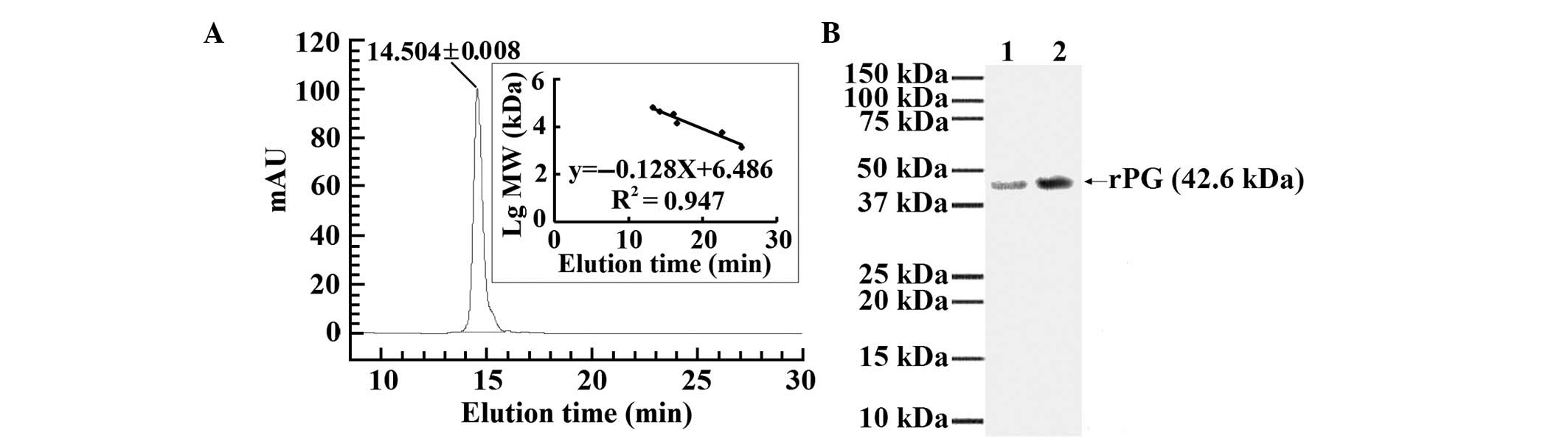
Purity and identification of rPG
The purified rPG showed a single peak at UV
adsorption 280 nm following HPLC (Fig.
6A). To detect the IgG-binding ability of the purified rPG and
compare the ability with that of the commercialized PG, each
protein was separately immobilized onto PVDF at a protein
concentration of 15 μg for the purified rPG and the
commercialized PG. The purified rPG and the commercialized PG were
shown to bind well to goat IgG (Fig.
6B).
Statistical analysis of purification
methods
The highest rPG expression level was >20% of the
total protein being expressed. The rPG was isolated and purified
with affinity Ni-NTA chromatography and Anion exchange DEAE-FF
chromatography. A final yield of 15±1.8 ml of 0.45 mg/ml rPG was
obtained, giving a cumulative yield of 86.3±6.8% for rPG (Table 1).
 | Table ISummary of the purification of rPG
expressed in E. coli BL21. |
Table I
Summary of the purification of rPG
expressed in E. coli BL21.
| Purification
step | Volume (ml) | Total protein
(mg) | rPG (mg) | Purity (%) | Cumulative
yield(%) |
|---|
| Ultra sonication | 10±1 | 29.2±0.3 | 7.8±0.5 | 26.7±2 | 100 |
| Ni-NTA
chromatography | 20±1.5 | 7.5±0.2 | 7.2±0.1 | 96.1±3.9 | 92.8±7.3 |
| Desalting column | 25±2.3 | 7.3±0.1 | 7.0±0.1 | 95.9±2.7 | 90.2±7.1 |
| DEAE-FF | 15±1.8 | 6.9±0.1 | 6.7±0.1 | 97.1±2.8 | 86.3±6.8 |
Discussion
PG is a cell wall protein separated from
Streptococcus and is able to bind to the Fc fragment of IgG
of most mammals, but it binds to human IgM, IgD and IgA. In
preliminary experiments, a recombinant E. coli strain
expressing PG was generated, with albumin and cell surface binding
sites removed in order to reduce cross reaction and non-specific
binding. Therefore, recombinant PG had higher affinity for Igs than
natural protein G and protein A; it can be applied widely in
immunochemistry by replacing secondary antibodies whilst having
high affinity and stability. PG high density fermentation
technology using the recombinant E. coli BL21 strain as well
as improved conditions of recombinant PG separation and
purification were assessed in the present study in order to
establish a basis for the development of a large-scale production
of PG.
In high-affinity fermentation, the biggest
difficulty is the large amount of acetic acid generated during the
fermentation process, which has an inhibiting effect on bacterial
growth and protein expression (13). Acetic acid synthesis is influenced
by the strain type, cultivating conditions and carbon source supply
method. To eliminate acetic acid during high-density fermentation,
genes associated with acetic acid synthesis are removed by gene
engineering method and acetic acid is removed by dialysis (13). Simply increasing the biomass does
not always increase the productivity but likely results in acetic
acid accumulation (14). During
fermentation in the present study, acetic acid synthesis was
reduced by balancing the feed supplements, which is the most common
method (15–17). In the present study, nutrition such
as a carbon source was added at the exponential growth phase in
order to ensure that nutrition and oxygen supply are maximal,
according to the methodology used in most high-density fermentation
processes (18). In the present
study, the concentration of acetic acid was reduced using
recombinant E. coli BL21 and a carbon source was added to
balance the feed supplements.
High production of target proteins depends on high
concentration of bacteria, which in turn largely depends on a
saturated oxygen supply during high-density fermentation. The
oxygen demand of the microorganisms largely exceeded the oxygen
concentration supplied in the fermentation device. In the present
study, the oxygen concentration was artificially enhanced by using
pure oxygen (13) and enhancing
the oxygen gas pressure (17,19).
Apart from being influenced by acetic acid and
oxygen, the production of PG target protein is influenced by
nutrition. In the present study, high concentrations of bacteria
and protein product were obtained by controlling the influx speed
and concentration of feed supplement (20).
Ni-NTA affinity column chromatography allowed for
simple recombinant protein separation, as it contained a 6xHis
affinity label, providing it with adsorption sites. In perliminary
experiments, a 6 His codon was added to the C-terminal region of
the PG gene to facilitate the extraction of large amounts of target
protein from bacterial lysate. PG was obtained at high purity
through desalination by gel filtration and ion exchange
chromatography.
A subsequent study will focus on further exploration
of recombinant PG activity.
Acknowledgments
The present study was funded by the Beijing
Municipal Education Commission of Science and Technology General
Program (no. SQKM201210858002).
References
|
1
|
Bjork I, Petersson BA and Sjoquist J: Some
physiochemical properties of protein A from Staphylococcus aureus.
Eur J Biochem. 29:579–584. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen YW, Wang H, Hupert M and Soper SA:
Identification of methicillin-resistant Staphylococcus aureus using
an integrated and molecular microfluidic system. Analyst.
138:1075–1083. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sheerin DJ, Menon C, Zur Oven-Krockhaus S,
Enderle B, Zhu L, Johnen P, Schleifenbaum F, Stierhof YD, Huq E and
Hiltbrunner A: Light-activated phytochrome A and B interact with
members of the SPA family to promote photomorphogenesis in
Arabidopsis by reorganizing the COP1/SPA complex. Plant Cell.
27:189–201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Graille M, Stura EA, Corper AL, Sutton BL,
Taussig MJ, Charbonnier JB and Silverman GJ: Crystal structure of a
Staphylococcus aureus protein A domain complexed with the Fab
fragment of a human IgM antibody: structural basis for recognition
of B-cell receptors and super antigen activity. Proc Natl Acad Sci
USA. 97:5399–5404. 2000. View Article : Google Scholar
|
|
5
|
Ditse Z, Adrian PV, Kuwanda L and Madhi
SA: Association of Streptococcus pneumoniae common protein antigen
(CPA) antibodies and pneumococcal nasopharyngeal colonization in
HIV-infected and HIV-uninfected African children. Vaccine.
31:4421–4427. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kmiecik S and Kolinski A: Folding pathway
of the B1 domain of protein G explored by a multiscale modeling.
Biophys J. 94:726–736. 2008. View Article : Google Scholar
|
|
7
|
Eliasson M, Andersson R, Olsson A, Wigzell
H and Uhlén M: Differential IgG-binding characteristics of
staphylococcal protein A, streptococcal protein G, and a chimeric
protein AG. J Immunol. 142:575–581. 1989.PubMed/NCBI
|
|
8
|
Green MR and Sambrook J: Expressing cloned
genes for protein production, purification, and analysis. Molecular
cloning. 4th edition. Cold Spring Harbor Laboratory Press; Long
Island, NY: pp. 1481–1611. 2012
|
|
9
|
Lee Y and Blanch H: Recombinant protein
expression in high cell density fed-batch cultures of Escherichia
coli. Biotech. 10:1550–1556. 1992. View Article : Google Scholar
|
|
10
|
Riesenberg D and Guthke R:
High-cell-density cultivation of microorganisms. Appl Microbiol
Biotechnol. 51:422–430. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lowry OH, Rosenburg NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
12
|
Hutt M, Färber-Schwarz A, Unverdorben F,
Richter F and Hontermann RE: Plasma half-life extension of small
recombinant antibodies by fusion to immunoglobulin-binding domains.
J Biol Chem. 287:4462–4469. 2012. View Article : Google Scholar :
|
|
13
|
Jenzsch M, Lange M, Bär J, Rahfeld JU and
Lubbert A: Bioreactor retrofitting to avoid aeration with oxygen in
Pichia pastoris cultivation processes for recombinant protein
production. Chem Eng Res Design. 82:1144–1152. 2004. View Article : Google Scholar
|
|
14
|
DeLisa MP, Chae HJ, Weigand WA, Valdes JJ,
Rao G and Bentley WE: Generic model control of induced protein
expression in high cell density cultivation of Escherichia coli
using on-line GFP-fusion monitoring. Bioproc Biosyst Eng. 24:83–91.
2001. View Article : Google Scholar
|
|
15
|
Lara AR, Caspeta L, Gosset G, Bolívar F
and Ramírez OT: Utility of an Escherichia coli strain engineered in
the substrate uptake system for improved culture performance at
high glucose and cell concentrations: An alternative to fed-batch
cultures. Biotechnol Bioeng. 99:893–901. 2008. View Article : Google Scholar
|
|
16
|
Matsui T, Sato H, Yamamuro H, Misawa S,
Shinzato N, Matsuda H, Takahashi J and Sato S: High cell density
cultivation of recombinant Escherichia coli for hirudin variant 1
production. J Biotechnol. 134:88–92. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Knoll A, Bartsch S, Husemann B, Engel P,
Betina S, Stockmann C, Seletzky J and Buchs S: High cell density
cultivation of recombinant yeasts and bacteria under
non-pressurized and pressurized conditions in stirred tank
bioreactors. J Biotechnol. 132:167–179. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sandén AM, Prytz I, Tubulekas I, Förberg
C, Le H, Hektor Andrea, Neubauer Peter, Pragai Z, Harwood C, Ward
A, Picon A, de Mattos JT, Postma P, Farewell A, Nyström T, Reeh S,
Pedersen S and Larsson G: Limiting factors in Escherichia coli
fed-batch production of recombinant proteins. Biotechnol Bioeng.
81:158–166. 2003. View Article : Google Scholar
|
|
19
|
Belo I, Pinheiro R and Mota M: Fed-batch
cultivation of Saccharomyces cerevisiae in a hyperbaric bioreactor.
Biotechnol Prog. 19:665–671. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eiteman MA and Altman E: Overcoming
acetate in Escherichia coli recombinant protein fermentations.
Trends Biotechnol. 24:530–536. 2006. View Article : Google Scholar : PubMed/NCBI
|