Introduction
Pulmonary arterial hypertension (PAH) is a fatal and
progressive disease, which frequently results in the elevation of
pulmonary artery pressure, right ventricular failure and mortality
(1–3). Vascular remodeling is the
pathological hallmark of PAH, leading to narrowing and obstruction
of small pulmonary arteries. The increased proliferation of
pulmonary artery smooth muscle cells (PASMCs) is a pivotal
contributor to vascular wall hypertrophy, vascular remodeling and
the resulting forms of PAH (4,5).
It is generally understood that smoking is a
significant risk factor in the occurrence and development of
chronic obstructive pulmonary disease (COPD) and pulmonary
hypertension, by directly resulting in pulmonary vascular
remodeling at the onset of the disease (6,7). In
addition, it has been reported that cigarette smoke may be involved
in the mitogenic pathway of vascular smooth muscle cells (SMCs) in
the bovine thoracic aorta, human greater saphenous vein, and aortic
and iliac arteries (8–11). In addition, a previous study
demonstrated that the expression levels of vasoactive mediators,
including endothelin-1 and vascular endothelial growth factor, were
increased in guinea pig lung vessels following chronic smoke
exposure, and these mediators were reported to be closely
associated with pulmonary vascular remodeling (12). However, the molecular mechanism
underlying this process remains to be elucidated.
FHL1 is a 30 kDa protein of the FHL subfamily, which
is structurally characterized by an N-terminal half LIM domain,
followed by four complete LIM domains (13). The LIM domain is a double-zinc
finger protein-binding motif, a term used following earlier
findings in the Lin11, Isl-1 and Mec-3 proteins (14). Previous studies have confirmed that
FHL1 is highly expressed in skeletal muscle and in the heart
(13,15), and is suggested to contribute to
sarcomere synthesis, assembly and biomechanical stress sensing
(16,17). Although considerably lower
expression levels have been detected in several other tissue types,
including colon, small intestine and prostate tissues (15), a previous study revealed that
increased expression of FHL1 may be involved in the prognosis of
Hirschsprung's disease (18). FHL1
deficiency was reported to significantly suppress proliferation,
but exhibit no effect on the apoptosis of rat aortic SMCs (19). Additionally, Kwapiszewska et
al demonstrated that knockdown of FHL1 markedly inhibited
hypoxia-induced PASMC migration and proliferation (20). These results indicated that FHL1 is
critical in the remodeling process during PAH. However, the effects
of FHL1 on cigarette smoke-induced PASMC proliferation and its
precise molecular mechanism remain to be elucidated.
Accumulating evidence suggests that cell cycle
progression is governed precisely at various biological checkpoints
by cyclin proteins, cyclin-dependent kinases (CDK) and CDK
inhibitors (CDKIs) (21). Among
the cyclin families, cyclin D1 is a critical regulator of the
progression of the cell cycle and has been demonstrated to be
important in the proliferation of SMCs from vessels, airways and
intestine (22–24). Additionally, the G1 to S
phase transition checkpoint is regulated by CDK4 and CDK6, the
activities of which are facilitated by cyclin D1, but are
attenuated by p27 (25).
The present study used small interfering (si)RNA and
adenovirus transfection methods to investigate the effects of FHL1
on CSE-induced PASMC proliferation and on the pathogenesis of PAH
induced by cigarette smoke.
Materials and methods
Materials and reagents
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS), penicillin and streptomycin were purchased from
Gibco Life Technologies (Carlsbad, CA, USA). All other reagents
were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless
otherwise specified.
Cell isolation and culture
Primary rat PASMCs were isolated and cultured, as
previously described (26).
Briefly, 20 male Sprague-Dawley rats were obtained from the Animal
Experiment Center of Wenzhou Medical University (Wenzhou, China)
and were housed alone with a 12:12 h light/dark cycle with free
access to water and food at room temperature. All animal
experiments were approved by the Committee on the Ethics of Animal
Experiments of Wenzhou Medical University and were performed in
accordance with the Wenzhou Medical University Guidelines for the
Ethical Care of Animals (WYDW2012-0032). At the age of 12 weeks,
the rats were anesthetized with 100 mg/kg intraperitoneal ketamine
and 5 mg/kg intraperitoneal xylazine. Subsequently, the distal
pulmonary arteries were isolated and immersed immediately in Hanks'
solution containing collagenase (1.5 mg/ml) for 20 min. Following
incubation at 37°C, a thin layer of adventitia and endothelium were
carefully stripped off. The PASMCs were then harvested by digesting
the remaining smooth muscle with collagenase (2.0 mg/ml) and
elastase (0.5 mg/ml) for 40 min, and were cultured in DMEM,
containing 10% FBS, 100 U/ml penicillin and 100 U/ml streptomycin
at 37°C in a 5% CO2 atmosphere for 5~7 days. The cells
were identified using immunochemistry and immunofluorescent
staining the of α-SM-actin antibody (AA132; 1:100; Beyotime
Institiute of Biotechnology, Jiangsu, China). For all experiments,
PASMCs between passages three and six were used, which were made
quiescent by replacing the medium with FBS-free DMEM for 24 h prior
to treatments.
Preparation of CSE solution
CSE was prepared freshly for each experiment, as
described previously (27).
Briefly, the smoke derived from one Double Happiness cigarette
(Shanghai Tobacco Corporation, Shanghai, China) was drawn slowly
into a 50 ml syringe, passed through 30 ml DMEM and was repeated
for five draws of 50 ml of the syringe. The resulting solution,
which was expressed as '100%' final concentration, was then
adjusted to pH 7.4 using concentrated NaOH, filtered (0.25
µm size) for sterilization and diluted in DMEM to the
required concentration (1, 2, 5, 10 and 20%) for the
experiment.
Cell proliferation and assessment of DNA
synthesis
Cell proliferation was assessed using a Cell
Counting kit-8 (CCK-8; Dojindo, Tokyo, Japan), according to the
manufacturer's instructions. The PASMCs were plated into 96-well
plates at a density of 1×104 cells/ml/well and 10
µl CCK-8 reagent was added and incubated for 2 h at 37°C.
The absorbance was measured at 450 nm using a microplate reader
(Multiskan Spectrum; Thermo Fisher Scientific, Pittsburgh, PA,
USA).
The present study also examined the BrdU
incorporation to measure DNA synthesis. The cells
(2×105) were incubated with 50 mM BrdU for 4 h at 37°C
and subsequently fixed with 4% paraformaldehyde in 0.01 M cold
phosphate buffer saline (PBS; pH 7.4). The cells were permeabilized
with 2% HCI containing 0.4% Triton X-100 for 15 min. Following
incubation with the monoclonal mouse anti-BrdU monoclonal antibody
(1:50; ab12219; Abcam, Cambridge, MA, USA) at 4°C overnight, the
samples were treated with biotinylated goat anti-mouse
immunoglobulin G antibody (1:100; #7056; Cell Signaling Technology,
Inc., Danvers, MA, USA) for 60 min and subsequently stained with
diaminobenzidene. PBS was used as a negative control by replacing
the primary antibodies. The percentage of positively stained cells
was determined by counting the numbers of stained cells and the
total cells using a Nikon Eclipse E600FN microscope (Nikon, Tokyo,
Japan).
Cell cycle analysis
The PASMCs were harvested by centrifugation at 200 ×
g for 5 min at 4°C. The pellets were washed with PBS and fixed in
70% ethanol overnight at −20°C. The samples were subsequently
resuspended in PBS, containing propidium iodide (50 µg/ml),
DNase-free RNase (10 µg1ml), 0.1% sodium citrate and 0.1%
TritonX-100. The DNA content was analyzed using a Beckman EPICS XL
flow cytometer (Beckman Coulter, Miami, FL, USA).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL)
DNA fragmentation, a marker of apoptosis, was
assessed using a TUNEL assay, as previously described (28). The cells (2×105) were
fixed with 4% paraformaldehyde and permeabilized for 30 min using
70% ethanol on ice. Cell apoptosis was measured using an In
situ Cell Death Detection kit® (Roche, Basel,
Switzerland), according to the manufacturer's instructions. The
positive cells were visualized with the Nikon Eclipse E600FN
microscope and the percentage of positive cells was determined by
counting the numbers of TUNEL positive cells and the total
cells.
Transfection of PASMCs with stealth
siRNA
The sequence of the stealth siRNA duplex
oligoribonucleotides against the rat FHL1 gene (GenBank accession
no. NM_001033926.2) was 5′-UGCCAAGCAUUGCGUGAAA-3′, and its
corresponding complementary strand was 5′-CUAAGGAGGUGGACAUAA-3′
(Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The siRNA were
transiently transfected using Lipofectamine RNAi max reagent
(Applied Biosystems, Foster City, CA, USA), according to the
manufacturer's instructions, and a negative stealth siRNA sequence
was used as a control. Briefly, the siRNA and Lipofectamine RNAi
max reagent were mixed and diluted in FBS-free DMEM. Following
coincubation for 15 min at room temperature, the mixture was added
to the PASMCs in a quiescent state and gently agitated to ensure
uniform distribution. The transfection mixture was removed
following incubation for 6 h at 37°C and complete medium was added
for further incubation at 37°C for 48 h.
Adenovirus expressing FHL1 infection
Full-length FHL1 cDNA was amplified and cloned into
a pAdTrack-CMV plasmid (Invitrogen Life Technologies, Carlsbad, CA,
USA), according to the manufacturer's instructions. The recombinant
pAdTrack-CMV shuttle plasmids containing the FHL1 gene were
linearized by PacI digestion and were transfected into 293A
cells using Lipofectamine RNAi max reagent to produce a recombinant
adenovirus. Viruses were packaged and amplified in 293A cells and
purified using CsCl (Sigma-Aldrich) banding followed by dialysis
against 10 mmol/l Tris-buffered saline with 10% glycerol. The
titers of virus were assayed using a p24 ELISA kit (Cell Biolabs,
San Diego, CA, USA). An adenovirus bearing LacZ was obtained from
Clontech (Mountain View, CA, USA) and was used as the negative
control. For overexpression of FHL1, 2×105 PASMCs were
cultured in FBS-free DMEM, containing the appropriate multiplicity
of infection of adenovirus vectors for 6 h at 37°C. The cells were
then transferred into complete medium and cultured for 48 h.
Western blot analysis
The total protein was harvested for western blot
analysis, as described previously (29). Briefly, the cells were washed with
cold PBS three times and subsequently lysed in
radioimmunoprecipitation buffer (Beyotime Institute of
Biotechnology), containing 1% protease inhibitor cocktail (Merck,
Darmstadt, Germany). Following determination of the protein content
using a Bradford assay (Bio-Rad Laboratories, Inc., Hercules, USA),
the proteins were resolved on 8–10% sodium dodecyl
sulfate-polyacrylamide gels and transferred onto nitrocellulose
membranes (Millipore, Billerica, MA, USA). The membranes were
incubated with blocking buffer for 1 h and subsequently with the
following primary antibodies at 4°C overnight: Mouse-anti-FHL-1
monoclonal antibody (sc-374246), mouse-anti-PCNA monoclonal
antibody (sc-25280) and mouse-anti-β-actin monoclonal antibody
(sc-8432) (1:1,000; Santa Cruz Biotechnology, Inc.);
rabbit-anti-cyclin D1 polyclonal antibody (#2922), rabbit-anti-Ki67
monoclonal antibody (#9129) and rabbit-anti-p27 monoclonal antibody
(#3686) (1:500; Cell Signaling Technology, Inc.). Following washing
and incubation with horseradish peroxidase (HRP)-labeled goat
anti-mouse IgG (A0216) or HRP-labeled goat anti-rabbit IgG (A0208)
secondary antibodies (1:1,000; Beyotime Institute of Biotechnology)
for 1 h at room temperature, the membranes were detected by a
chemiluminescence system (Cell Signaling Technology, Inc.). Image
quantification was performed using ImageJ software, version 1.37
(NIH, Bethesda, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was isolated from the PASMCs using
TRIzol reagent (Invitrogen Life Technologies), according to the
manufacturer's instructions. A total of 1 µg RNA was reverse
transcribed using the SuperScript III First-Strand Synthesis system
(Invitrogen Life Technologies). A Fast SYBR® Green
Master Mix kit (Applied Biosystems) was used for qPCR on a Fast
RT-PCR system (ABI 7300; Applied Biosystems). A total of 40 cycles
were performed as follows: 95°C for 15 sec, 60°C for 1 min and 72°C
for 30 sec. Relative expression was determined using GAPDH as an
internal control and was reported using the 2−ΔΔCT
method. The specific primer sequences used for amplification were
as follows: FHL1, forward 5′-GTGCCCTTGTACTCCACGTT-3′ and reverse
5′-GTGTCCAAGGATGGCAAGAT-3′ and GAPDH, forward
5′-ATGAGCCCCAGCCTTCTCCAT-3′ and reverse
5′-GGTCGGAGTCAACGGATTTG-3′.
Statistical analysis
All data are expressed as the mean ± standard error
of mean. A one-way analysis of variance or an unpaired two-tailed
Student t-test was used to analyze the differences between groups.
P<0.05 was considered to indicate a statistically significant
difference. All statistical analyses were performed using SPSS16.0
software. (SPSS, Inc., Chicago, USA).
Results
Effects of CSE on cell proliferation and
apoptosis
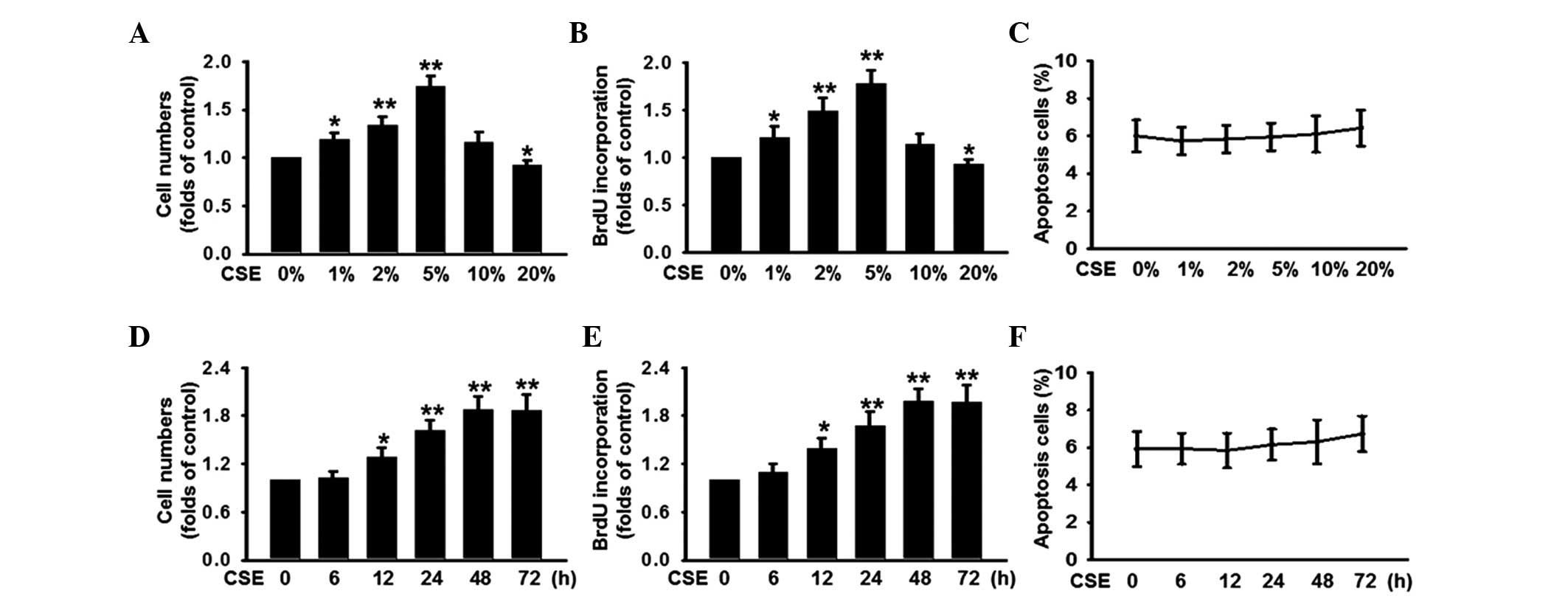
To investigate the effects of CSE on the
proliferation of PASMCs, PASMCs were treated with 0, 1, 2, 5, 10 or
20% CSE for 48 h and cell proliferation was determined using CCK-8
and BrdU incorporation assays. As shown in Fig. 1A and B, CSE at low concentrations
of 1% significantly increased the number of cells by 19% and BrdU
incorporation by 21%, compared with the control (0%). The peak
increase in cell number (69%) and BrdU incorporation (78%) were
observed at a concentration of 5%. Notably, CSE at high
concentrations (20%) exhibited a significant inhibition on cell
proliferation. Furthermore, no significant effect on cell apoptosis
was observed at any of the CSE concentrations (Fig. 1C). In addition, the PASMCs were
treated with 5% CSE for different durations, and cell proliferation
was examined. The results from the CCK-8 and BrdU incorporation
assays revealed that cell proliferation was triggered from 12 h and
reached a peak at 48 h (Fig. 1D and
E). Although incubation of CSE for 72 h marginally increased
the percentage of apoptotic cells, no significant difference was
observed compared with the control (0 h; Fig. 1F), indicating that CSE has no toxic
effect on PASMCs. These data suggested that CSE induced the PASMC
proliferation.
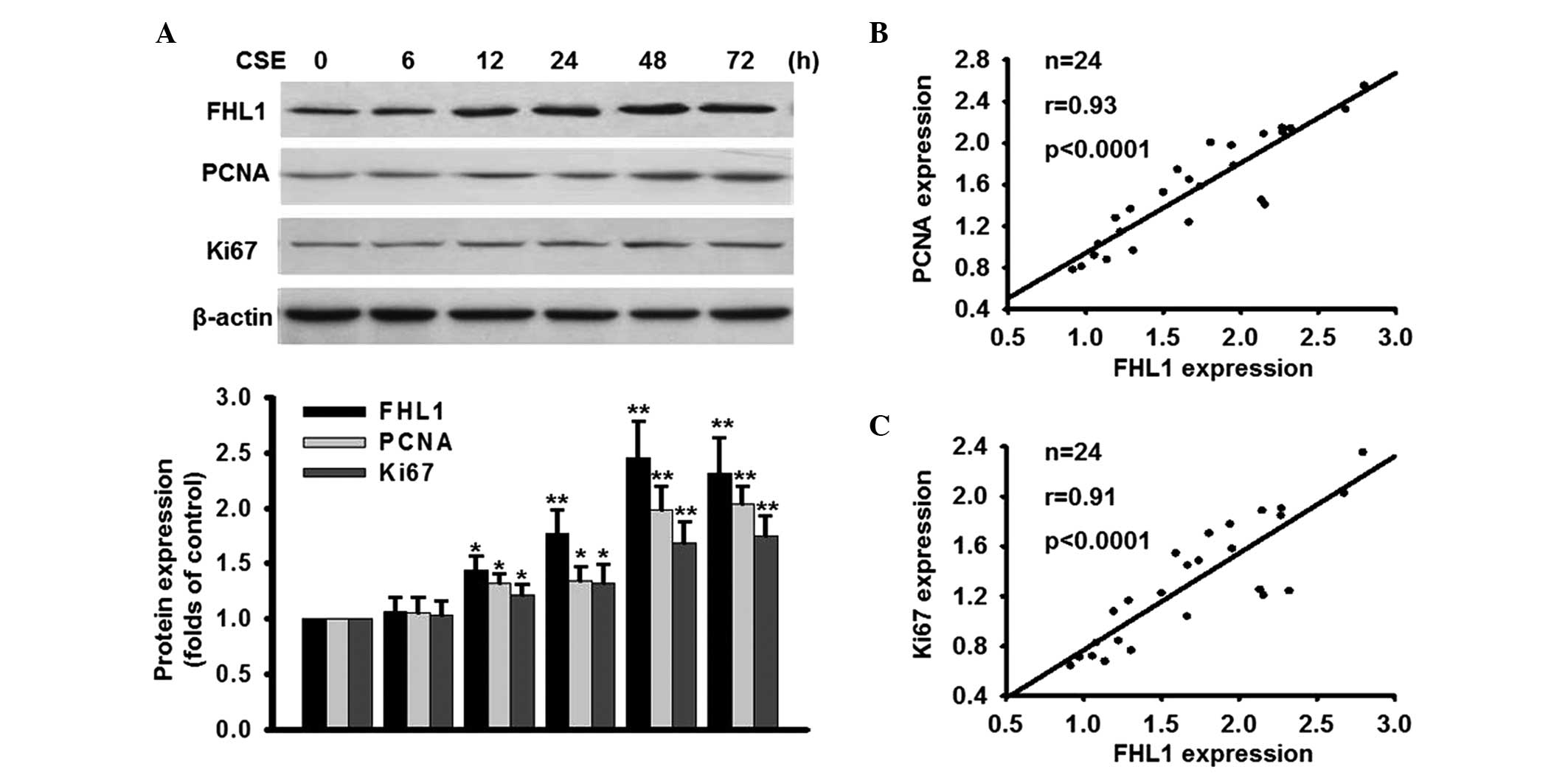
Protein expression of FHL1 parallels with
the CSE-induced proliferation of PASMCs
To investigate whether the expression of FHL1
correlated with the rate of cell proliferation, the effect of CSE
on the expression of FHL1 was determined. As shown in Fig. 2A, 5% CSE induced the protein expression of
FHL1 in a time-dependent manner. Compared with the control (0 h),
the protein expression of FHL1 at 12, 24, 48 72 h was increased
1.24±0.13, 1.57±0.21, 2.45±0.34 and 2.32±0.32-fold, respectively.
The protein expression of FHL1 reached the maximal expression level
at 48 h, which paralleled with CSE-induced cell proliferation. To
further confirm the effect of CSE on cell proliferation, the
expression levels of two proliferation markers, PCNA and Ki67, were
assessed. As expected, the expression levels of PCNA and Ki67 were
increased following treatment with CSE in a time-dependent manner.
Notably, the protein expression of FHL1 was positively correlated
with the protein expression levels of PCNA and Ki67 (Fig. 2B and C). These data suggested that
increased expression of FHL1 may be involved in the CSE-induced
proliferation of PASMCs.
FHL1 accelerates the proliferation of
PASMCs
To determine whether FHL1 is involved in the
CSE-induced proliferation of PASMCs, the effect of FHL1 knockdown
on PASMC proliferation was determined. The silencing efficiency of
the siRNA was detected using western blot and RT-qPCR analyses.
Compared with the control, 20 nM FHL1 siRNA significantly decreased
the protein expression of FHL1 by 81% and its mRNA expression by
75%, while the negative siRNA (Neg) caused no change in the mRNA or
protein expression levels of FHL1 (Fig. 3A and B). FHL1 deficiency
significantly decreased the cell number at the basal level and
inhibited the increase of cell number induced by CSE (Fig. 3C). The result of BrdU incorporation
revealed a similar tendency (Fig.
3D). The effect of FHL1 overexpression on cell proliferation
was assessed to confirm the role of FHL1 in proliferation.
Following the successful overexpression of FHL1, confirmed using
western blotand RT-qPCR analyses (Fig.
3E and F), the proliferation of the PASMCs was markedly
increased at the basal level and following CSE stimulation
(Fig. 3G and H). Negative siRNA
and Lacz transfection caused no significant alteration to the rate
of PASMCs proliferation at the basal level or following treatment
with CSE.
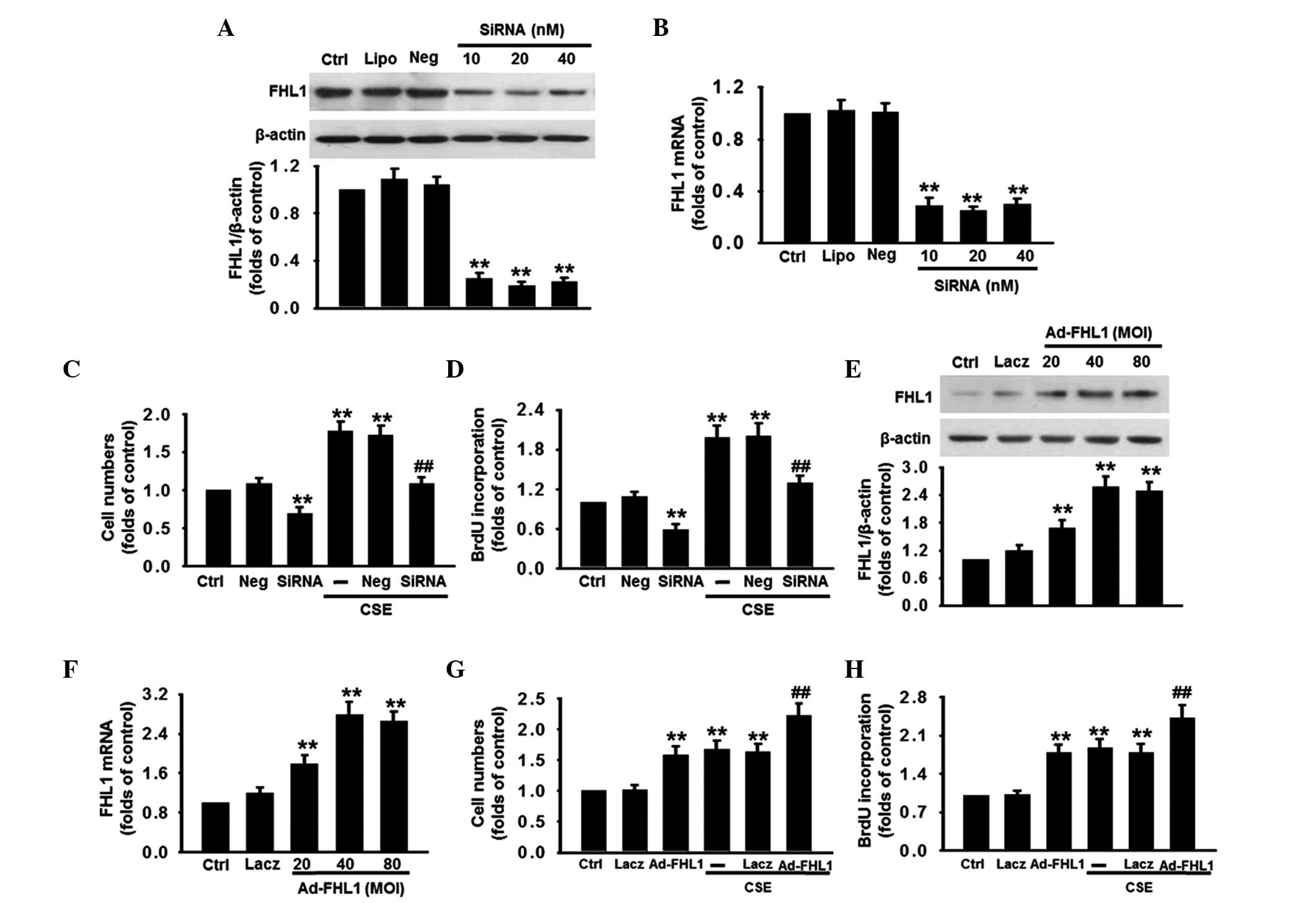 | Figure 3Effects of FHL1 on CSE-induced
proliferation of PASMCs. (A and B) Silencing efficiency of siRNA
was examined. The cells were transfected with different
concentrations of FHL1-targeting siRNA (10, 20 or 40 nM) for 48 h.
The expression of FHL1 was determined using (A) western blot and
(B) RT-qPCR analyses. Negative siRNA was used as a negative
control. (C and D) FHL1 was knocked down using siRNA, as described
for A and B, and the cell proliferation was assessed using (C)
CCK-8 and (D) BrdU incorporation assays. (E and F) PASMCs were
infected with adenovirus packaging an expression vector of FHL1
(Ad-FHL1; MOI, 20, 40 and 80) for 48 h. The expression of FHL1 was
detected using (E) western blot and (F) RT-qPCR analyses. Lacz was
used as a negative control. (G and H) FHL1 was overexpressed, as
described fot E and F, and the cell proliferation was assessed
using (G) CCK-8 and (H) BrdU incorporation assays. All data are
expressed as the mean ± standard error of the mean
(*P<0.05 and **P<0.01, vs. Ctrl;
#P<0.05 and ##P<0.01, vs. CSE alone;
n=6). siRNA, small interfering RNA; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; BrdU,
bromodeoxyuridine; CCK-8, cell counting kit-8; MOI, multiplicity of
infection; CSE, cigarette smoke extract; PASMCs, pulmonary artery
smooth muscle cells; Ctrl, control; Neg, negative; Lipo,
lipofectamine RNAi max reagent. |
Inhibition of FHL1 inhibits the
G1/S cell cycle transition
The effect of FHL1 on cell cycle status was also
investigated in the present study. According to flow cytometric
analysis, 5% CSE reduced the proportion of cells in the
G0/G1 phase between 69.3 and 48.9%, and
simultaneously increased the proportion of cells in the S phase
between 21.3 and 38.7%, compared with the control (Fig. 4A), which suggested that CSE
promoted cell cycle progression and an increase in cells entry into
the S phase. However, inhibition of FHL1 significantly decreased
the percentage of cells in S phase and resulted in
G0/G1 cell cycle arrest at the basal level
and following CSE stimulation, suggesting that the deficiency of
FHL1 arrested the cell cycle in G0/G1 phase
by preventing entrance into the S phase in the PASMCs. By contrast,
FHL1 overexpression decreased the percentage of cells in the
G1 phase and increased the percentage in the S phase,
compared with the corresponding control (Fig. 4B). To investigate the molecular
mechanisms by which FHL1 knockdown arrests cells at the
G1/S transition, the expression levels of cyclin D1 and
p27, which are involved in the regulation of the G1 to S
phase transition checkpoint were investigated. The results
demonstrated that silencing of FHL1 markedly decreased the
expression of cyclin D1 at the basal level and inhibited the
CSE-induced increase of the expression of cyclin D1. By contrast,
the expression of p27 was increased at the basal level and restored
following CSE stimulation (Fig.
4C). The inverse results were obtained in the group
overexpressing FHL1 (Fig. 4D).
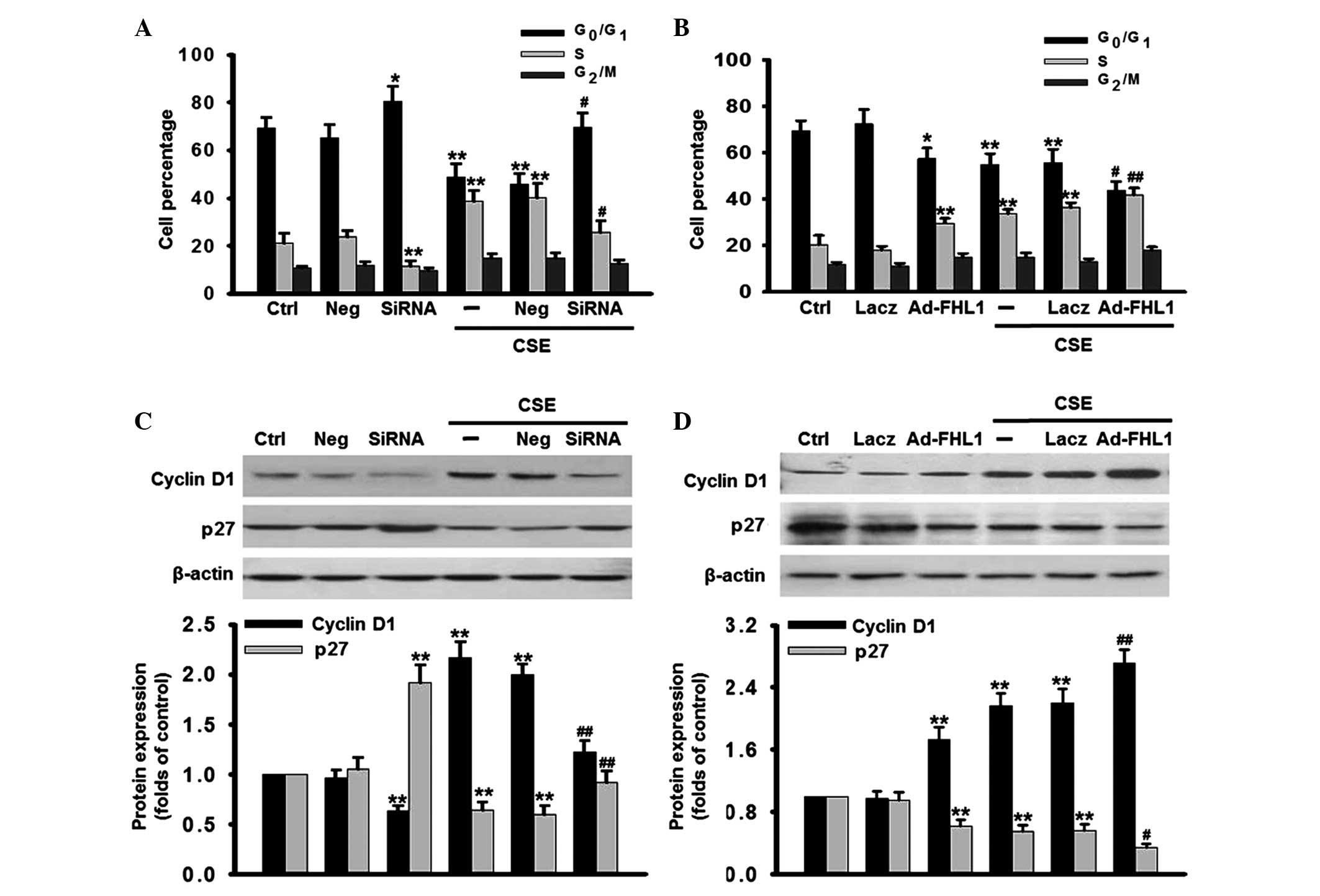 | Figure 4Effects of FHL1 on the cell cycle
phases. FHL1 was (A) knocked down using siRNA or (B) overexpressed
by adenovirus in the presence or absence of CSE for 48 h. The cell
cycle phases were determined by flow cytometry. The percentage of
cells in G0/G1, S, G2/M phases
were statistically analyzed. (C and D) Following treatment, the
expression levels of cyclin D1 and p27 were detected by western
blotting, using β-actin as an internal control. The data are
expressed as the mean ± standard error of the mean
(*P<0.05, **P<0.01, vs. control;
#P<0.05, ##P<0.01, vs. CSE alone; n=6).
siRNA, small interfering RNA; CSE, cigarette smoke extract; PASMCs,
pulmonary artery smooth muscle cells. |
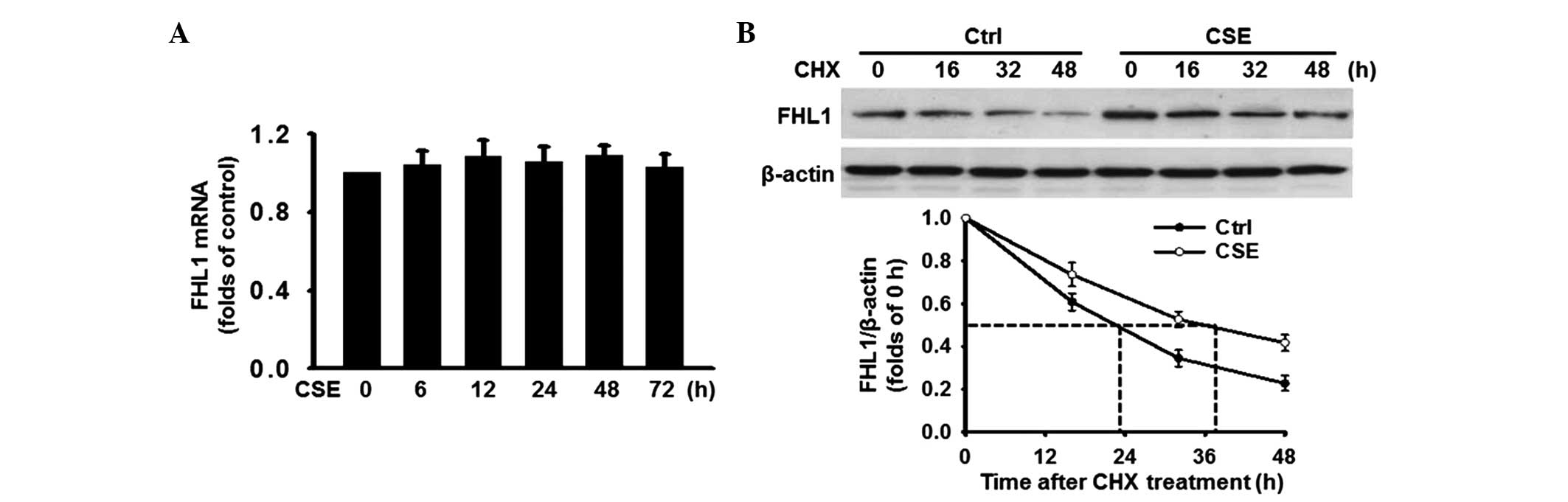
CSE induces the expression of FHL1 by
increasing protein stability
To further investigate the mechanism by which CSE
induced the expression of FHL1, the mRNA expression level of FHL1
was determined. RT-qPCR analysis revealed that the mRNA expression
of FHL1 remained unchanged following treatment with CSE (Fig. 5A), indicating that a
post-transcriptional mechanism may be involved. Since the
predominant mechanism involved in the modulation of protein
expression is degradation, the present study determined whether CSE
altered the protein stability of FHL1. The PASMCs were treated with
or without 5% CSE for 48 h and cycloheximide (CHX; 10
µg/ml), a protein synthesis inhibitor, was added at various
time points (0, 16, 32 and 48 h). The results revealed that
treatment of the PASMCs with CHX led to a time-dependent decrease
in the protein expression of FHL1 in the two groups. Notably, the
combination of CHX and CSE significantly changed the rate of FHL1
degradation, increasing the FHL1 half-life by >12 h compared
with the control (Fig. 5B). These
results suggested that CSE upregulates FHL1, at least in part, at
the post-translational level, rather than the transcriptional
level.
Discussion
The results of the present study provided compelling
evidence that the expression of FHL1 is associated with CSE-induced
proliferation of PASMCs. The present study demonstrated that
CSE-induced cell proliferation was accompanied by an increase in
the protein expression of FHL1 in a time-dependent manner,
determined using western blot analysis and a proliferation assay.
In addition, it was revealed that FHL1 accelerated cell
proliferation by promoting cell cycle transition between the G1 and
S phase. Finally, CSE was found to be involved in maintaining the
stability of the FHL1 protein. These data are the first, to the
best of our knowledge, to confirm the key role of FHL1 in cell
proliferation induced by cigarette smoke.
Cigarette smoke is a major risk factor in various
types of disease, including PAH. As the predominant cause of
hypertrophy and vascular remodeling, the increase of PASMC
proliferation contributes to PAH (5,30).
Substantial evidence from animals and humans has suggested that
smoking induces pulmonary vascular remodeling in patients with
mild-to-moderate COPD in smokers with normal lung function
(31–33). The present study demonstrated a
biphasic nature of the effect of CSE on the proliferation of
PASMCs. Low concentrations of CSE (1, 2 and 5%) increased cell
proliferation, while high concentrations exhibited a significant
inhibition on cell proliferation. These results were in accordance
with previous studies (34–36).
FHL1 is the most widely expressed member of the FHL
subfamily, containing four complete and one half, highly conserved,
LIM domains (37). LIM domains
function as important mediators of protein-protein interactions in
the cytoplasm and nucleus, and their functions are largely
dependent upon their associated binding partners (14,38,39),
including α5β1 and α7β1 integrins (40,41),
muscle-specific RING finger proteins MuRF1 and MuRF2 (42), and other protein kinases, including
ERK2 (17,43). Additionally, LIM domain proteins
have been suggested to be involved in biomechanical stress
responses, as structural adapters and as subcellular localizers
(16,44,45).
A previous study demonstrated that silencing of FHL1 significantly
decreased the migration and proliferation of PASMCs in a
hypoxia-induced rat hypertension model, and defined a novel
interaction between FHL1 and Talin1, which may be significant in
the proliferation and migration of PASMCs (20). However, the potential effects of
FHL1 on the CSE-induced proliferation of PASMCs remains to be
elucidated. In the present study, CCK-8 and BrdU incorporation
assays demonstrated that knockdown of FHL1 significantly decreased
the proliferation of PASMCs, whereas overexpression of FHL1 induced
the opposite effect. These findings are consistent with data from a
previous study (20), but are
novel, to the best of our knowledge, for CSE-induced cell
proliferation. To further confirm the role of FHL1 in cell
proliferation, the effects of FHL1 on cell-cycle distribution were
assessed. Flow cytometry demonstrated that the upregulation of FHL1
promoted cell cycle progression between the G1 and S
phase. Conversely, deficiency of FHL1 caused cell cycle arrest at
the G1 phase. Cell cycle progression is a tightly
controlled event, which is positively regulated by cyclins and
CDKs, and negatively regulated by CDKIs (46). The present study demonstrated that
loss of FHL1 almost eliminated CSE-induced expression of cyclin D1,
which promotes transition between the G1 and S phase
(21). In addition, FHL1 knockdown
also reversed the inhibitory effect of CSE on the expression of
p27, which is widely reported to restrict the G1/S
transition in the cell cycle by suppressing several cyclin-CDK
complexes (47). The inverse
results were observed in FHL1 overexpressing cells. Together, these
results demonstrated that FHL1 accelerated CSE-induced cell
proliferation by regulating the expression levels of cyclin D1 and
p27, which facilitated the cell cycle transition between the
G1 and S phase.
As CSE induced the proliferation of PASMCs and the
expression of FHL1, the present study investigated the correlation
between the expression of FHL1 and cell proliferation. Western blot
analysis and a proliferation assay revealed that the induction of
cell proliferation by CSE was accompanied by an increase in the
protein expression of FHL1 in the same time-dependent manner.
Detection of the expression levels of Ki67 and PCNA, which are
essential for DNA replication (48), further confirmed that the
expression of FHL1 correlated with the rate of PASMC proliferation.
Notably, CSE exerted no effect on the mRNA expression of FHL1,
indicating that transcriptional regulation was not involved. A
previous study demonstrated that hypoxia reduced the protein
expression levels of FHLl in the lung, pulmonary artery and
alveolar septae, but exhibited different effects on the mRNA
expression in these tissues (20).
Therefore, awareness of tissue- and stimulation-specific responses
is required. It is well known that the predominant mechanism
involved in the regulation of protein levels is degradation. The
present study demonstrated that CSE significantly decreased the
rate of FHL1 degradation, suggesting that protein stabilization at
translational levels was involved, at least partially, in the
CSE-induced increase in the expression of FHL1.
In conclusion, the results of the present study
suggested that FHL1 is an important regulator in PASMC
proliferation induced by cigarette smoke. Therefore, inhibition of
FHL1 may offer a potential therapeutic strategy for the treatment
of smokers with PAH.
Acknowledgments
This study was supported by the Zhejiang Natural
Science Foundation (no. LQ13H010002) and the Science and Technology
Planning Project of Wenzhou, China (no. Y20100292).
References
|
1
|
Humbert Marc, Sitbon Olivier and Simonneau
Gérald: Treatment of Pulmonary Arterial Hypertension. N Engl J Med.
351:1425–1436. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pidgeon GP, Tamosiuniene R, Chen G,
Leonard I, Belton O, Bradford A and Fitzgerald DJ: Intravascular
thrombosis after hypoxia-induced pulmonary hypertension: regulation
by cyclooxygenase-2. Circulation. 110:2701–2707. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mandegar M, Fung YC, Huang W, Remillard
CV, Rubin LJ and Yuan JX: Cellular and molecular mechanisms of
pulmonary vascular remodeling: role in the development of pulmonary
hypertension. Microvasc Res. 68:75–103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barberà JA, Peinado VI and Santos S:
Pulmonary hypertension in chronic obstructive pulmonary disease.
Eur Respir J. 21:892–905. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pietra GG, Capron F, Stewart S, et al:
Pathologic assessment of vasculopathies in pulmonary hypertension.
J Am Coll Cardiol. 43(Suppl 12): 25–32. 2004. View Article : Google Scholar
|
|
6
|
Wright JL, Cosio M and Churg A: Animal
models of chronic obstructive pulmonary disease. Am J Physiol Lung
Cell Mol Physiol. 295:L1–L15. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mandegar M, Fung YC, Huang W, Remillard
CV, Rubin LJ and Yuan JX: Cellular and molecular mechanisms of
pulmonary vascular remodeling: role in the development of pulmonary
hypertension. Microvasc Res. 68:75–103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carty CS, Huribal M, Marsan BU, Ricotta JJ
and Dryjski M: Nicotine and its metabolite cotinine are mitogenic
for human vascular smooth muscle cells. J Vasc Surg. 25:682–628.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nishio E and Watanabe Y: Cigarette smoke
extract is a modulator of mitogenic action in vascular smooth
muscle cells. Life Sci. 62:1339–1347. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li JM, Cui TX, Shiuchi T, Liu HW, Min LJ,
Okumura M, Jinno T, Wu L, Iwai M and Horiuchi M: Nicotine enhances
angiotensin II-induced mitogenic response in vascular smooth muscle
cells and fibroblasts. Arterioscler Thromb Vasc Biol. 24:80–84.
2004. View Article : Google Scholar
|
|
11
|
Jacob T, Clouden N, Hingorani A and Ascher
E: The effect of cotinine on telomerase activity in human vascular
smooth muscle cells. J Cardiovasc Surg (Torino). 50:345–349.
2009.
|
|
12
|
Wright JL, Tai H and Churg A: Vasoactive
mediators and pulmonary hypertension after cigarette smoke exposure
in the guinea pig. J Appl Physiol (1985). 100:672–678. 2006.
View Article : Google Scholar
|
|
13
|
Lee SM, Tsui SK, Chan KK, Garcia-Barcelo
M, Waye MM, Fung KP, Liew CC and Lee CY: Chromosomal mapping,
tissue distribution and cDNA sequence of four-and-a-half LIM domain
protein 1 (FHL1). Gene. 216:163–170. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bach I: The LIM domain: regulation by
association. Mech Dev. 91:5–17. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Greene WK, Baker E, Rabbitts TH and Kees
UR: Genomic structure, tissue expression and chromosomal location
of the LIM-only gene, SLIM1. Gene. 232:203–207. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chu PH, Ruiz-Lozano P, Zhou Q, Cai C and
Chen J: Expression patterns of FHL/SLIM family members suggest
important functional roles in skeletal muscle and cardiovascular
system. Mech Dev. 95:259–265. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sheikh F, Raskin A, Chu PH, Lange S,
Domenighetti AA, Zheng M, Liang X, Zhang T, Yajima T, Gu Y, Dalton
ND, et al: An FHL1-containing complex within the cardiomyocyte
sarcomere mediates hypertrophic biomechanical stress responses in
mice. J Clin Invest. 118:3870–3880. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang LL, Gu H, Fan Y, Zhang Y, Wu D, Miao
JN, Huang TC, Li H and Yuan ZW: Up-regulated FHL1 expression maybe
involved in the prognosis of Hirschsprung's disease. Int J Med Sci.
11:262–267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weng J, Liao M, Zou S, Bao J, Zhou J, Qu
L, Feng R, Feng X, Zhao Z and Jing Z: Downregulation of FHL1
expression in thoracic aortic dissection: implications in aortic
wall remodeling and pathogenesis of thoracic aortic dissection. Ann
Vasc Surg. 25:240–247. 2011. View Article : Google Scholar
|
|
20
|
Kwapiszewska G, Wygrecka M, Marsh LM,
Schmitt S, Trösser R, Wilhelm J, Helmus K, Eul B, Zakrzewicz A,
Ghofrani HA, Schermuly RT, et al: Fhl-1, a new key protein in
pulmonary hypertension. Circulation. 118:1183–1194. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morgan DO: Principles of CDK regulation.
Nature. 374:131–134. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zeng DX, Liu XS, Xu YJ, Wang R, Xiang M,
Xiong WN, Ni W and Chen SX: Plasmid-based short hairpin RNA against
cyclin D1 attenuated pulmonary vascular remodeling in smoking rats.
Microvasc Res. 80:116–122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pera T, Gosens R, Lesterhuis AH, Sami R,
van der Toorn M, Zaagsma J and Meurs H: Cigarette smoke and
lipopolysaccharide induce a proliferative airway smooth muscle
phenotype. Respir Res. 11:482010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kuemmerle JF, Zhou H and Bowers JG: IGF-I
stimulates human intestinal smooth muscle cell growth by regulation
of G1 phase cell cycle proteins. Am J Physiol Gastrointest Liver
Physiol. 286:G412–G419. 2004. View Article : Google Scholar
|
|
25
|
Nakayama K, Ishida N, Shirane M, Inomata
A, Inoue T, Shishido N, Horii I, Loh DY and Nakayama K: Mice
lacking p27(Kip1) display increased body size, multiple organ
hyperplasia, retinal dysplasia and pituitary tumors. Cell.
85:707–720. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Golovina VA and Blaustein MP: Preparation
of primary cultured mesenteric artery smooth muscle cells for
fluorescent imaging and physiological studies. Nat Protoc.
1:2681–2687. 2006. View Article : Google Scholar
|
|
27
|
Oltmanns U, Chung KF, Walters M, John M
and Mitchell JA: Cigarette smoke induces IL-8, but inhibits eotaxin
and RANTES release from airway smooth muscle. Respir Res. 6:742005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Q, Li X, Chen Y, Wang F, Yang Q, Chen
S, Min Y, Li X and Xiong L: Activation of epsilon protein kinase
C-mediated anti-apoptosis is involved in rapid tolerance induced by
electroacupuncture pretreatment through cannabinoid receptor type
1. Stroke. 42:389–396. 2011. View Article : Google Scholar
|
|
29
|
Leopold JA, Dam A, Maron BA, Scribner AW,
Liao R, Handy DE, Stanton RC, Pitt B and Loscalzo J: Aldosterone
impairs vascular reactivity by decreasing glucose-6-phosphate
dehydrogenase activity. Nat Med. 13:189–197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rabinovitch M: The mouse through the
looking glass: a new door into the pathophysiology of pulmonary
hypertension. Circ Res. 94:1001–1004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu SQ and Fung YC: Changes in the
structure and mechanical properties of pulmonary arteries of rats
exposed to cigarette smoke. Am Rev Respir Dis. 148:768–777. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Peinado VI, Barbera JA, Ramirez J, Gomez
FP, Roca J, Jover L, Gimferrer JM and Rodriguez-Roisin R:
Endothelial dysfunction in pulmonary arteries of patients with mild
COPD. Am J Physiol. 274:L908–L913. 1998.PubMed/NCBI
|
|
33
|
Washko GR: Diagnostic imaging in COPD.
Semin Respir Crit Care Med. 31:276–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Luppi F, Aarbiou J, van Wetering S, Rahman
I, de Boer WI, Rabe KF and Hiemstra PS: Effects of cigarette smoke
condensate on proliferation and wound closure of bronchial
epithelial cells in vitro: role of glutathione. Respir Res.
6:1402005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu K, Liu XS, Yu MQ and Xu YJ: Change of
extracellular signal-regulated kinase expression in pulmonary
arteries from smokers with and without chronic obstructive
pulmonary disease. Exp Lung Res. 39:162–172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zeng DX, Xu YJ, Liu XS, Wang R and Xiang
M: Cigarette smoke extract induced rat pulmonary artery smooth
muscle cells proliferation via PKCα-mediated cyclin D1 expression.
J Cell Biochem. 112:2082–2088. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shathasivam T, Kislinger T and Gramolini
AO: Genes, proteins and complexes: the multifaceted nature of FHL
family proteins in diverse tissues. J Cell Mol Med. 14:2702–2720.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kadrmas JL and Beckerle MC: The LIM
domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell
Biol. 5:920–931. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schmeichel KL and Beckerle MC: The LIM
domain is a modular protein-binding interface. Cell. 79:211–219.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Samson T, Smyth N, Janetzky S, Wendler O,
Müller JM, Schüle R, von der Mark H, von der Mark K and Wixler V:
The LIM-only proteins FHL2 and FHL3 interact with alpha- and
beta-subunits of the muscle alpha7beta1 integrin receptor. J Biol
Chem. 279:28641–28652. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
McGrath MJ, Mitchell CA, Coghill ID,
Robinson PA and Brown S: Skeletal muscle LIM protein 1 (SLIM11FHL1)
induces alpha 5 beta 1-integrin-dependent myocyte elongation. Am J
Physiol Cell Physiol. 285:C1513–C1526. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Witt CC, Witt SH, Lerche S, Labeit D, Back
W and Labeit S: Cooperative control of striated muscle mass and
metabolism by MuRF1 and MuRF2. EMBO J. 27:350–360. 2008. View Article : Google Scholar
|
|
43
|
Purcell NH, Darwis D, Bueno OF, Müller JM,
Schüle R and Molkentin JD: Extracellular signal-regulated kinase 2
interacts with and is negatively regulated by the LIM-only protein
FHL2 in cardiomyocytes. Mol Cell Biol. 24:1081–1095. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Knöll R, Hoshijima M, Hoffman HM, Person
V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H,
Schaper W, McKenna W, et al: The cardiac mechanical stretch sensor
machinery involves a Z disc complex that is defective in a subset
of human dilated cardiomyopathy. Cell. 111:943–955. 2002.
View Article : Google Scholar
|
|
45
|
Zhou Q, Chu PH, Huang C, Cheng CF, Martone
ME, Knoll G, Shelton GD, Evans S and Chen J: Ablation of Cypher, a
PDZ-LIM domain Z-line protein, causes a severe form of congenital
myopathy. J Cell Biol. 155:605–612. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Woo RA and Poon RY: Cyclin-dependent
kinases and S phase control in mammalian cells. Cell Cycle.
2:316–324. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Barchiesi F, Jackson EK, Fingerle J,
Gillespie DG, Odermatt B and Dubey RK: 2-Methoxyestradiol, an
estradiol metabolite, inhibits neointima formation and smooth
muscle cell growth via double blockade of the cell cycle. Circ Res.
99:266–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yerushalmi R, Woods R, Ravdin PM, Hayes MM
and Gelmon KA: Ki67 in breast cancer: prognostic and predictive
potential. Lancet Oncol. 11:174–183. 2010. View Article : Google Scholar : PubMed/NCBI
|