Introduction
Osteosarcoma (OS) is the most common type of primary
bone malignancy, and the eighth most common type of cancer in
children, comprising 2.4% of all malignancies in pediatric patients
and ~35% of all bone cancers worldwide (1). The five-year survival rate of
patients with OS has significantly improved over recent decades to
60–70%, since the advent of combinatorial chemotherapy (2). However, survival has since leveled,
despite advances in therapeutic strategies (3). Standard chemotherapy of OS is based
on a combination of several drugs: Neoadjuvant therapy with
methotrexate, cisplatin and doxorubicin, followed by surgery and
post-operative chemotherapy with methotrexate, cisplatin,
doxorubicin, cyclophosphamide and vincristine (4). The use of cisplatin, which is an
effective anti-tumor agent with a wide spectrum of activity against
human solid tumors (5), has been
shown to be a useful chemotherapeutic strategy for pre-operative
induction therapy of OS. Patients who responded well to
pre-operative cisplatin chemotherapy were also shown to have an
improved survival rate (6).
Furthermore, the inclusion of cisplatin has been shown to be
associated with a better outcome for high-grade OS (7). Intrinsic or acquired chemoresistance
is the predominant reason for poor survival and disease relapse in
patients with OS (8). Recently,
novel molecular-targeted drugs have emerged; however, these drugs
have not been well established for the treatment of OS (9). In addition, the molecular mechanisms
underlying OS chemoresistance remain to be elucidated. Therefore,
identification of factors that contribute to OS chemoresistance and
elucidation of the underlying mechanisms is essential for the
development of novel therapeutic strategies.
Podocalyxin (PCX) is a highly glycosylated and
sialylated transmembrane protein. PCX is a CD34 ortholog that is
usually expressed on hematopoietic stem cells, hemangioblasts,
vascular endothelial cells, podocytes and a subset of neural
progenitors (10). Aberrant PCX
expression has been reported in leukemia (11,12),
undifferentiated thyroid carcinoma (13) and renal cell carcinoma (14). High protein expression levels of
PCX have been shown to be correlated with poor outcome in a subset
of breast carcinoma, and have also been associated with increased
aggressiveness of breast and prostate cancer cells (15,16).
Recently, it has been demonstrated that PCX promotes astrocytoma
cell survival against temozolomide-induced apoptotic stress
(17), thus suggesting that PCX
may also contribute to cancer chemoresistance. The present study
aimed to explore the role of PCX in OS by determining its effect on
cisplatin chemoresistance in OS cells.
Materials and methods
Cell lines, plasmids and reagents
MG-63 (CRL-1427) and U2OS (HTB-96) human OS cell
lines were purchased from the American Tissue Culture Collection
(Manassas, VA, USA). The cells were grown in Dulbecco's modified
Eagle's medium (Invitrogen Life Technologies, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (Invitrogen Life
Technologies) and 100 U/ml penicillin-streptomycin (Sigma-Aldrich,
Beijing, China) in an incubator with a humidified atmosphere of 95%
air and 5% CO2 at 37°C. Human full-length PCX cDNA
(SC302189; Origene Technologies, Inc., Beijing China) was
sub-cloned into a pcDNA 3.1 expression vector (Invitrogen Life
Technologies) at the KpnI and NotI sites. A human PCX short hairpin
(sh)RNA plasmid (RHS3979-98487921; 5′-AGTTCATCCCATTTGTCCT-3′) was
purchased from Open Biosystems (Huntsville, AL, USA). Mouse
anti-human anti-PCX (3D3) monoclonal (cat. no. 39-3800) antibody
and Lipofectamine® 2000 transfection reagent were
purchased from Invitrogen Life Technologies (Carlsbad, CA, USA).
Selective phosphatidylinositide 3-kinase (PI3K) inhibitor BKM120
(cat. no. sc-364437A), and rabbit anti-human anti-Akt serine 473
(ser473) (cat. no. sc-1618) and rabbit anti-human
anti-phosphorylated (p)-Akt (ser473) (cat. no. sc-101629)
polyclonal antibodies were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). All primary antibodies were incubated with
the membrane at a dilution of 1:500 for 1 h in Tris-buffered saline
with Tween 20 containing 5% skimmed milk powder for blocking at
room temperature. All secondary antibodies were purchased from
Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA).
The TiterTACS in situ Apoptosis Detection kit (cat. no.
4822-96-K) was purchased from R&D Systems (Minneapolis, MN,
USA). The PI3K Activity ELISA kit (cat. no. K-1000s) was purchased
from Echelon Biosciences, Inc. (Salt Lake City, UT, USA).
Cisplatin, puromycin, G418 and all chemicals of reagent grade were
purchased from Sigma-Aldrich (St. Louis, MO, USA).
Transfection and lentiviral
transduction
The PCX expression construct was transfected into
the MG-63 and U2OS cells using Lipofectamine® 2000
transfection reagent, according to the manufacturer's instructions.
Pools of stable transductants were generated via selection with
G418 (700 µg/ml), according to the manufacturer's
instructions. Lentiviral transduction was performed in the MG-63
and U2OS cells. Lentiviral particles were packaged with vector
psPAX2 and vector pMD2.G, according to the manufacturer's
instructions (Open Biosystems). A control virus containing a
scrambled shRNA sequence, which did not lead to the specific
degradation of any cellular mRNA, was used as a negative control
for PCX-shRNA lentiviral particles. Pools of stable transductants
were generated via selection with puromycin (5 µg/ml).
Western blot analysis
The cells were dissolved in 250 µl 2X SDS
loading buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 25% glycerol, 5%
2-mercaptoethanol; Sigma-Aldrich) and incubated at 95°C for 10 min
The supernatant collected following centrifugation at 2,000 × g for
15 min at 4°C was used for protein determination using the
Coomassie blue method. Equal amounts of protein for each sample
were separated by 10% SDS-PAGE and blotted onto polyvinylidene
difluoride microporous membranes (EMD Millipore, Billerica, MA,
USA). The membranes were then incubated for 1 h with the primary
antibodies at a 1:500 dilution and then washed. The membranes were
subsequently incubated with the horseradish peroxidase-conjugated
secondary antibodies (1:4,000) for 1 h. The blots were visualized
using a GE Healthcare Enhanced Chemiluminescence kit (GE
Healthcare, Little Chalfont, UK). Three independent experiments
were performed for each western blot analysis. Western blots were
quantified using ImageJ software version 1.42 (National Institutes
of Health, Bethesda, MD, USA).
Cisplatin
chemosensitivity/chemoresistance assay
The cells were plated in duplicates in 96-well
plates at a density of 5×103 cells/well. Following a
24-h incubation, the medium was replaced with fresh medium with or
without various concentrations of cisplatin (0.1, 0.25, 0.5, 1.0,
1.5, 3.0, 6.0, 15.0, 30.0 and 55.0 mM). For BKM120 treatment, cells
were treated with BKM120 (50 µM) for 48 h. Cell viability
was assayed six days later, using a modified MTT assay
(Sigma-Aldrich) as previously described (18). The 96-well plate used was purchased
from Thermo Fisher Scientific (Beijing, China) and the microplate
reader (SpectraMax Plus 384) was purchased from Molecular Devices,
LLC (Beijing, China). The half maximal inhibitory concentration
(IC50) was defined as the concentration resulting in a
50% reduction in growth, as compared with that of the control
cells.
Cell colony formation assay
Colony formation assays were performed as previously
described (19). Briefly, the
cells were treated with cisplatin (5 mM) at 37°C for 48 h, then
trypsinized (Sigma-Aldrich), re-seeded in 100-mm plates
(1×103 cells/plate) and cultured at 37°C for 2 weeks.
For BKM120 treatment, cells were treated with BKM120 (50 µM)
for 48 h. The plates were subsequently fixed with cold methanol
(Sigma-Aldrich) at −20°C for 30 min, stained with crystal violet
(0.1%) (Sigma-Aldrich) and finally washed with water in order to
remove background staining. Images were captured using a scanner
(InGenius3; Syngene Inc., Frederick, MD, USA), and all colonies
>1.5 mm in diameter were counted.
Cell apoptosis assay
The cells were cultured at 9×104
cells/well in 96-well tissue culture plates and treated with
cisplatin (5 mM) at 37°C for 48 h. For BKM120 treatment, cells were
treated with BKM120 (50 µM) for 48 h. The rate of cell
apoptosis was measured at 48 h using the microplate reader-based
TiterTACS in situ Apoptosis Detection kit, according to the
manufacturer's instructions. The SpectraMax Plus 384 microplate
reader was used to measure optical density. Each experiment was
repeated three times in duplicates.
PI3K activity assay
PI3K activity was determined using the PI3K Activity
ELISA kit, according to the manufacturer's instructions (20,21).
For BKM120 treatment, cells were treated with BKM120 (50 µM)
for 48 h. For direct functional assessment of PI3K activity, PI3K
was isolated by immuno-precipitation using an anti-PI3K antibody
(cat. no. 06–195; EMD Millipore) to the p85 adapter subunit. The
ability of the co-precipitated p110 catalytic subunit to convert
standard phosphatidylinositol 4,5-bisphosphate to
phosphatidylinositol 3,4,5-trisphosphate (PIP3) in a kinase
reaction was assessed by measuring the generated PIP3 by ELISA. The
SpectraMax Plus 384 microplate reader was used to measure optical
density. Each experiment was repeated three times in
duplicates.
Statistical analysis
Statistical analyses were performed using SPSS for
Windows 19.0 (International Business Machines, Armonk, NY, USA).
Values are expressed as the mean ± standard deviation. Comparisons
of the means among multiple groups were performed by one-way
analysis of variance followed by post hoc pairwise comparisons
using Tukey's tests. A two-tailed P<0.05 was considered to
indicate a statistically significant difference.
Results
Overexpression and knockdown of PCX in
human OS cells
The MG-63 and U2OS human OS cell lines were used as
cell models in the present study. As shown in Fig. 1, PCX was constitutively expressed
in the two cell lines. PCX was overexpressed or knocked down in the
cells by stable transfection with a PCX expression vector or
lentiviral transduction of PCX-shRNA, respectively. PCX was
overexpressed by 9.6- and 7.3-fold in the MG-63 and U2OS cells
transfected with the PCX expression vector, respectively, as
compared with that in the control cells. Conversely, the endogenous
expression levels of PCX were knocked down by ~80 and 75% in the
MG-63 and U2OS cells transduced with PCX-shRNA, respectively
(Fig. 1). A preliminary study
using the selective PI3K inhibitor BKM120 had suggested that PCX
may regulate OS cell chemoresistance predominantly through a
PI3K-dependent mechanism (data not shown); therefore, the present
study used the selective PI3K inhibitor BKM120 (50 µM) in
all experiments. As shown in Fig.
1, treatment with BKM120 had no significant effect on the
expression levels of PCX in the two cell lines.
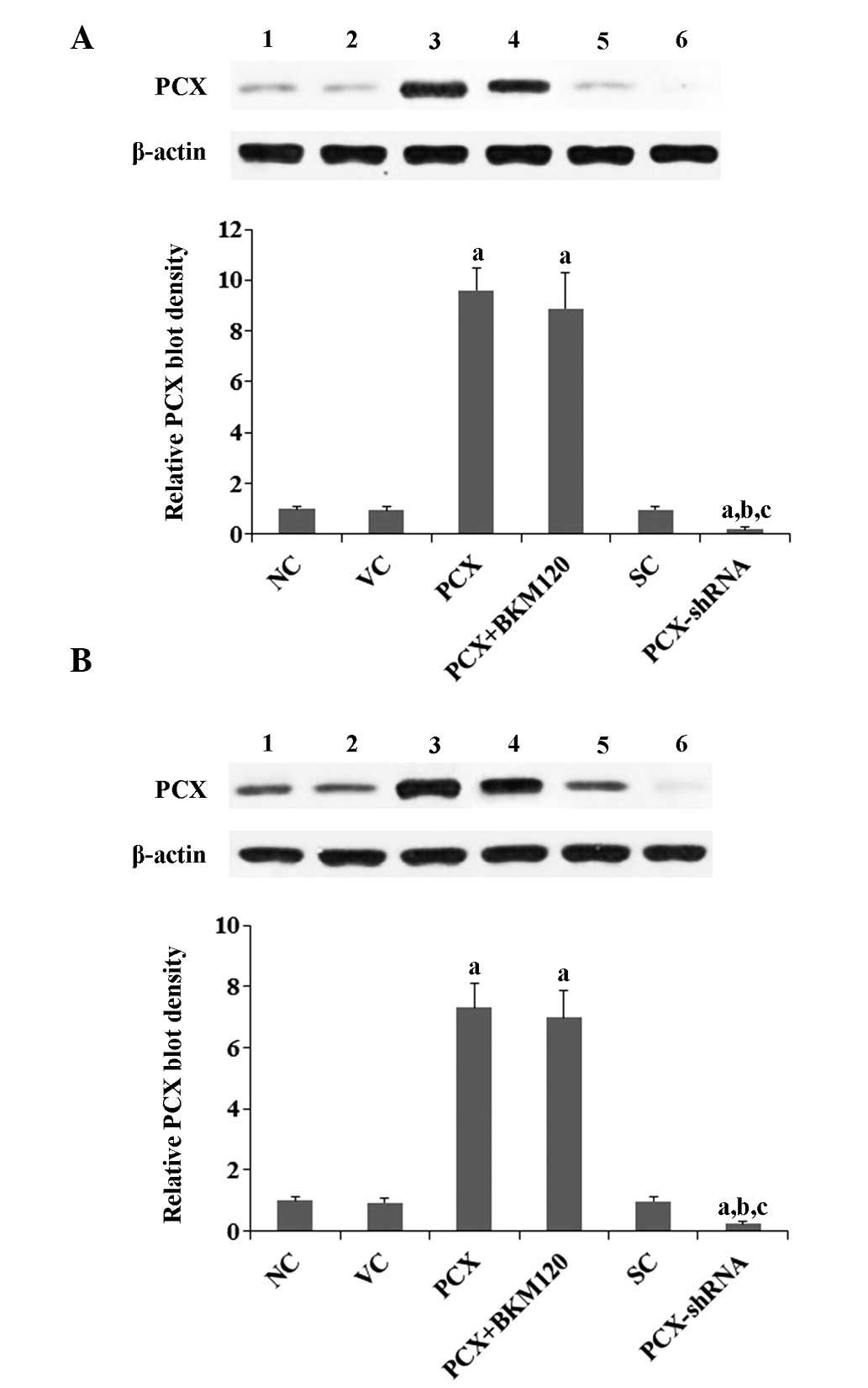 | Figure 1Expression of PCX in OS cells
following overexpression or knockdown of PCX. Expression of PCX was
detected in (A) MG-63 and (B) U2OS human OS cells by western blot
analysis. Lanes: 1, NC; 2, VC; 3, PCX; 4, PCX + BKM120; 5, SC; and
6, PCX-shRNA. β-actin was used as a loading control. The density of
the PCX blot was normalized against that of the β-actin blot in
order to obtain the relative PCX blot density, which was expressed
as a fold change relative to that of the NC group (designated as
1). Three independent experiments were performed for each western
blot analysis. Values are expressed as the mean + standard
deviation. aP<0.05, vs. the controls (NC, VC and SC);
bP<0.05, vs. the PCX group; cP<0.05,
vs. the PCX + BKM120 group. Groups: NC, normal control cells; VC,
cells stably transfected with empty pcDNA3.1 vector; PCX, cells
stably transfected with PCX; PCX + BKM120, cells stably transfected
with PCX and treated with the phosphatidylinositide 3-kinase
inhibitor BKM120 (50 µM) for 48 h; SC, cells stably
transduced with scramble control shRNA; and PCX-shRNA, cells stably
transduced with PCX-shRNA. PCX, podocalyxin; OS, osteosar-coma;
shRNA, small hairpin RNA. |
Effects of PCX on cisplatin
chemoresistance in OS cells
To explore the effects of PCX on OS chemoresistance,
the present study examined the IC50 values of cisplatin
on OS cells. As shown in Fig. 2,
following six days of cisplatin treatment, the cisplatin
IC50 values on MG-63 and U2OS cells were 1.2 and 6.2
µM, respectively. Overexpression of PCX significantly
increased the IC50 values to 3.9 and 17.8 µM,
respectively, which was reduced by ~50% following treatment with
BKM120 (50 µM) (Fig. 2).
Conversely, knockdown of PCX expression markedly decreased the
IC50 values of cisplatin on MG-63 and U2OS cells to 0.5
and 2.1 µM, respectively (Fig.
2).
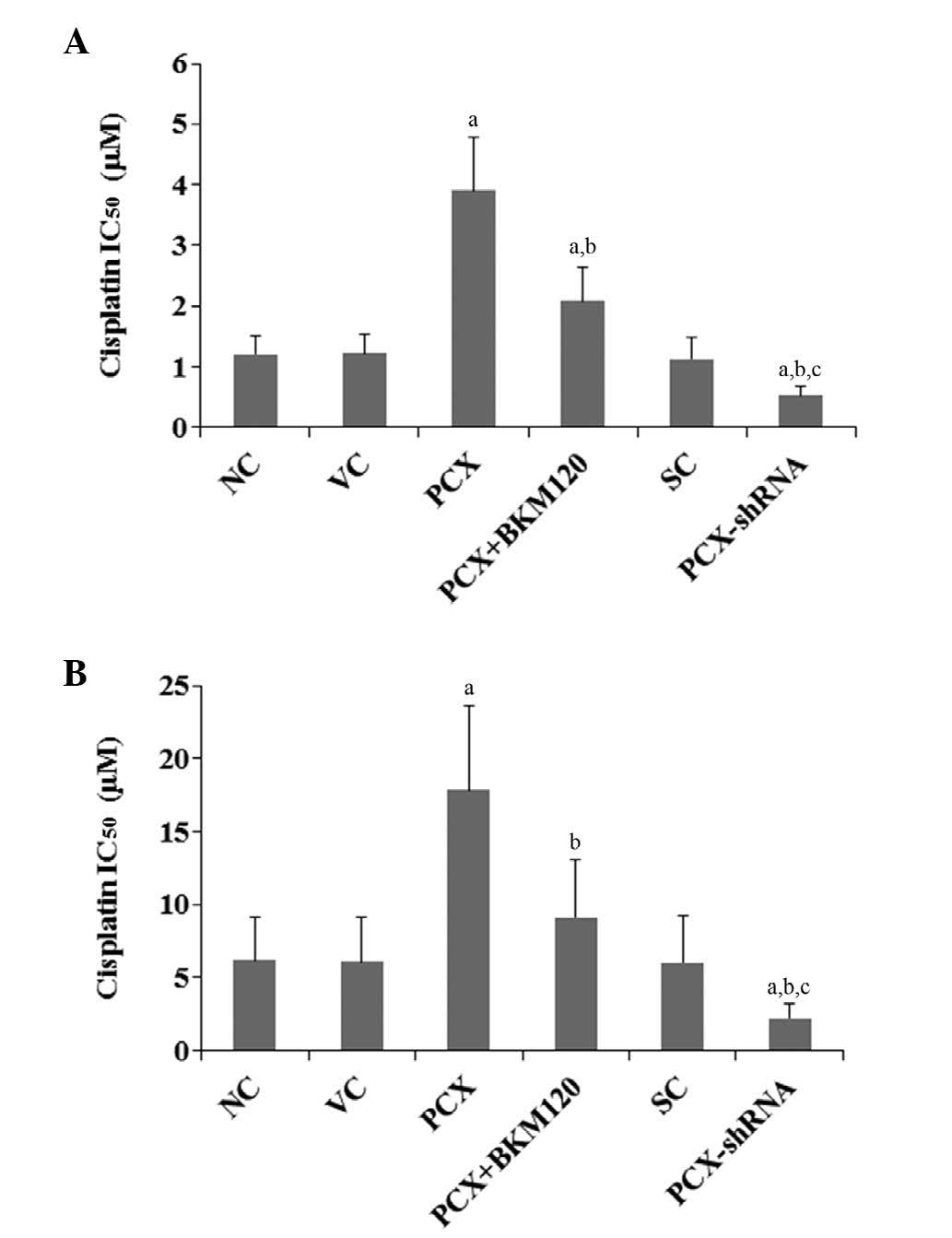 | Figure 2Effects of PCX on the IC50
of cisplatin in (A) MG-63 and (B) U2OS human OS cells. The cells
were treated with or without various concentrations of cisplatin
for six days, and the IC50 values were determined in the
following groups: NC, normal control cells; VC, cells stably
transfected with empty pcDNA3.1 vector; PCX, cells stably
transfected with PCX; PCX + BKM120, cells stably transfected with
PCX and treated with the phosphatidylinositide 3-kinase inhibitor
BKM120 (50 µM) for 48 h; SC, cells stably transduced with
scramble control shRNA; and PCX-shRNA, cells stably transduced with
PCX-shRNA. Each experiment was repeated three times in duplicate.
Values are expressed as the mean + standard deviation.
aP<0.05, vs. the controls (NC, VC and SC);
bP<0.05, vs. the PCX group; cP<0.05,
vs. the PCX + BKM120 group. PCX, podocalyxin; OS, osteosarcoma;
shRNA, small hairpin RNA; IC50, half maximal inhibitory
concentration. |
Effects of PCX on OS cell colony
formation following cisplatin treatment
To determine the effects of PCX on the long-term
survival of OS cells, colony formation assays were performed in
MG-63 and U2OS cells treated with 5 mM cisplatin for 48 h. After
two weeks, the cells were fixed and stained to visualize the
colonies. As shown in Fig. 3, the
number of cell colonies in the MG-63 and U2OS cells was 20 and 48,
respectively. Overexpression of PCX significantly increased the
number of colonies to 147 and 259, respectively (P<0.05), which
was reduced by ~50% following treatment with BKM120 (50 µM)
(Fig. 3). Conversely, knockdown of
PCX expression markedly decreased the number of colonies in the
MG-63 and U2OS cells to 7 and 10, respectively (Fig. 3).
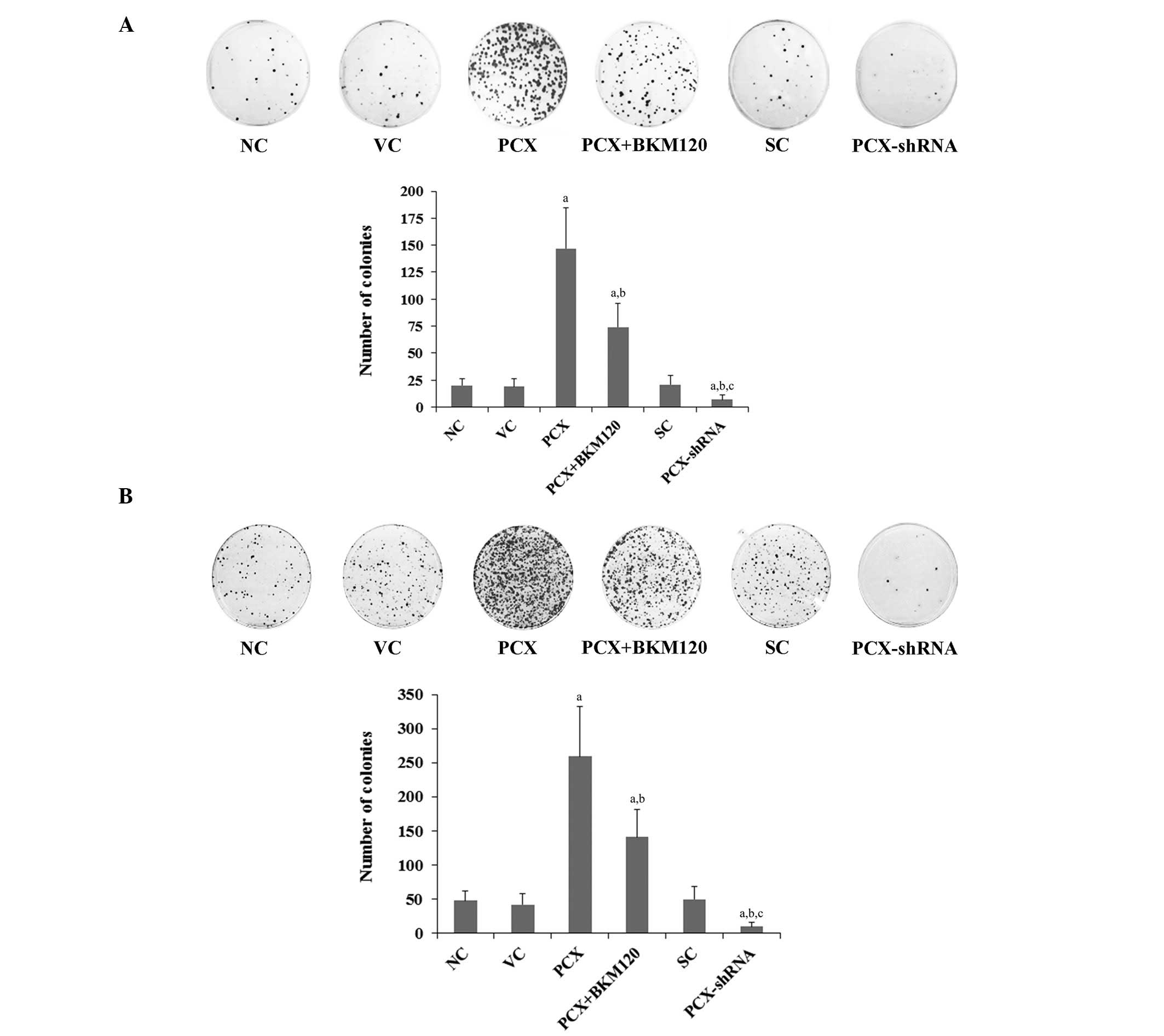 | Figure 3Effects of PCX on cell colony
formation in OS cells treated with cisplatin. (A) MG-63 and (B)
U2OS human OS cells were treated with 5 µM cisplatin for 48
h and then cultured for two weeks. The colonies were stained with
crystal violet and counted in the following groups: NC, normal
control cells; VC, cells stably transfected with empty pcDNA3.1
vector; PCX, cells stably transfected with PCX; PCX + BKM120, cells
stably transfected with PCX and treated with the
phosphatidylinositide 3-kinase inhibitor BKM120 (50 µM) for
48 h; SC, cells stably transduced with scramble control shRNA; and
PCX-shRNA, cells stably transduced with PCX-shRNA. Each experiment
was repeated three times in duplicate. Values are expressed as the
mean + standard deviation. aP<0.05, vs. the controls
(NC, VC and SC); bP<0.05, vs. the PCX group;
cP<0.05, vs. the PCX + BKM120 group. Magnification,
×5. PCX, podocalyxin; OS, osteosarcoma; shRNA, small hairpin
RNA. |
Effects of PCX on cisplatin-induced OS
cell apoptosis
The present study also examined the effects of PCX
on cisplatin-induced apoptosis in OS cells. Under normal culture
conditions, overexpression and knockdown of PCX had no significant
effects on OS cell apoptosis (P>0.05), as compared with that of
the control cells (data not shown). Following 48 h of cisplatin
treatment (5 mM), the percentages of apoptotic MG-63 and U2OS cells
were 27.3 and 21.5%, respectively (Fig. 4). Overexpression of PCX markedly
decreased the percentage of apoptotic cells to 8.3 and 5.1%,
respectively, which was partially reversed by treatment with BKM120
(50 µM) to 16.9 and 10.2%, respectively (Fig. 4). Conversely, knockdown of PCX
markedly increased the percentage of apoptotic cells to 62.0 and
67.9%, respectively (Fig. 4;
P<0.05).
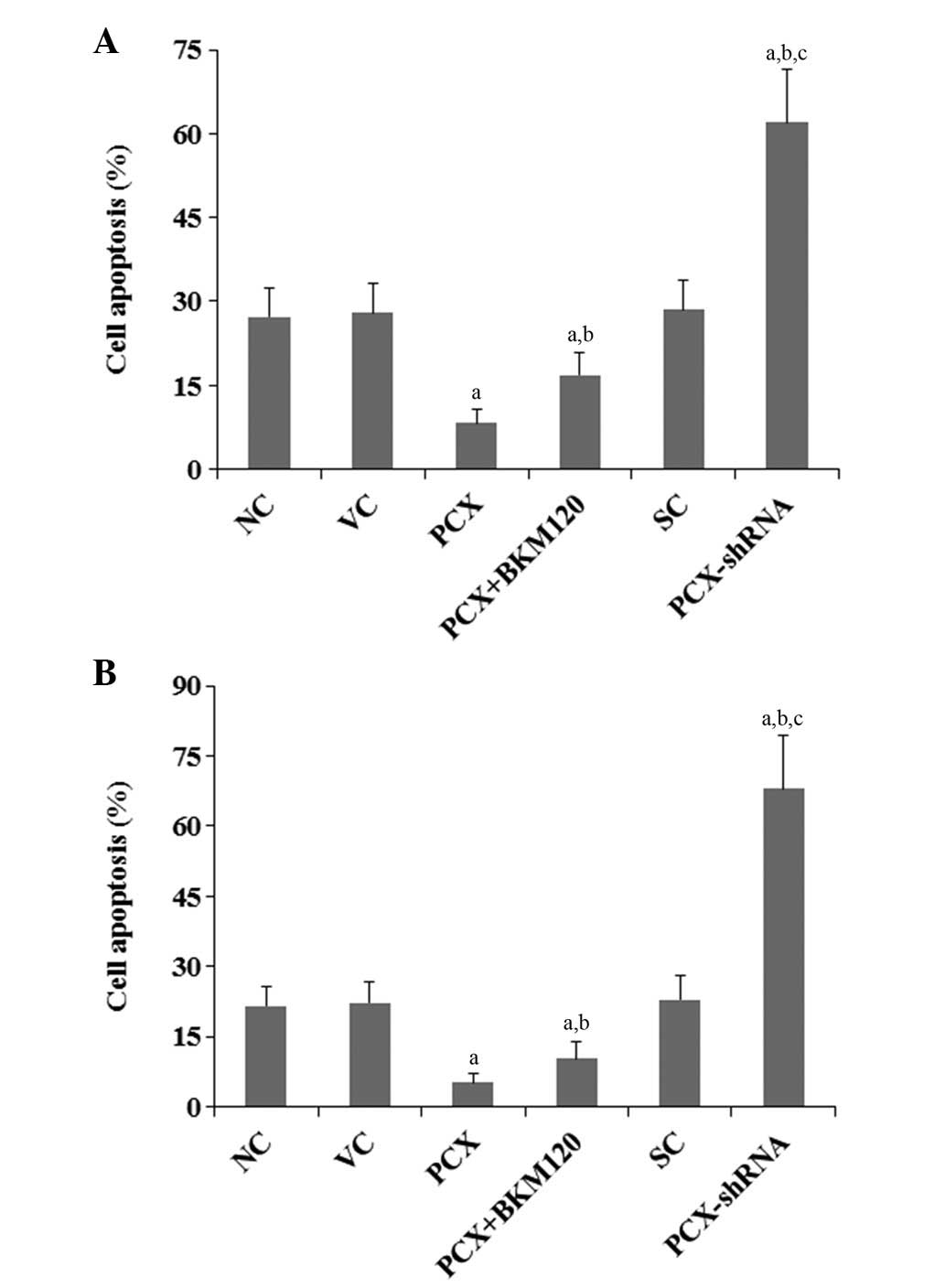 | Figure 4Effects of PCX on cell apoptosis in
OS cells treated with cisplatin. (A) MG-63 and (B) U2OS human OS
cells were treated with 5 µM cisplatin for 48 h. Cell
apoptosis was measured using a microplate reader-based TiterTACS
in situ Apoptosis Detection kit in the following groups: NC,
normal control cells; VC, cells stably transfected with empty
pcDNA3.1 vector; PCX, cells stably transfected with PCX; PCX +
BKM120, cells stably transfected with PCX and treated with the
phosphatidylinositide 3-kinase inhibitor BKM120 (50 µM) for
48 h; SC, cells stably transduced with scramble control shRNA; and
PCX-shRNA, cells stably transduced with PCX-shRNA. Each experiment
was repeated three times in duplicate. Values are expressed as the
mean + standard deviation. aP<0.05, vs. the controls
(NC, VC and SC); bP<0.05, vs. the PCX group;
cP<0.05, vs. the PCX + BKM120 group; PCX,
podocalyxin; OS, osteosarcoma; shRNA, small hairpin RNA. |
Effects of PCX on PI3K activity and
phosphorylation of Akt in OS cells
The results of the present study suggested that PCX
promotes cisplatin chemoresistance in OS cells, predominantly
through a PI3K-dependent mechanism. Previous studies have
demonstrated that the PI3K/Akt pathway is important for cancer cell
chemoresistance (22–25). Therefore, the present study
examined the effects of PCX on the activity of PI3K and
phosphorylation of Akt in OS cells. As shown in Fig. 5, overexpression of PCX increased
PI3K activity by 5.3-fold in MG-63 cells and by 4.2-fold in U2OS
cells, as compared with that in the control cells, which was
attenuated by BKM120 treatment (50 µM). Conversely,
knockdown of PCX expression decreased PI3K activity by ~50% in the
two cell lines (Fig. 5;
P<0.05). A similar trend was observed for the phosphorylation at
serine 473 of Akt (Fig. 6;
P<0.05), which is required for full activation of Akt by PI3K.
Overexpression of PCX was observed to increas Akt phosphorylation
by approximately 4.5-fold in MG-63 cells and by approximately
3.6-fold in U2OS cells, as compared with that of the control cells,
the phosphorylation of which was abolished by BKM120 treatment (50
µM) (Fig. 6). Conversely,
knockdown of PCX expres sion reduced Akt phosphorylation by
approximately 55% in the two cell lines (Fig. 6).
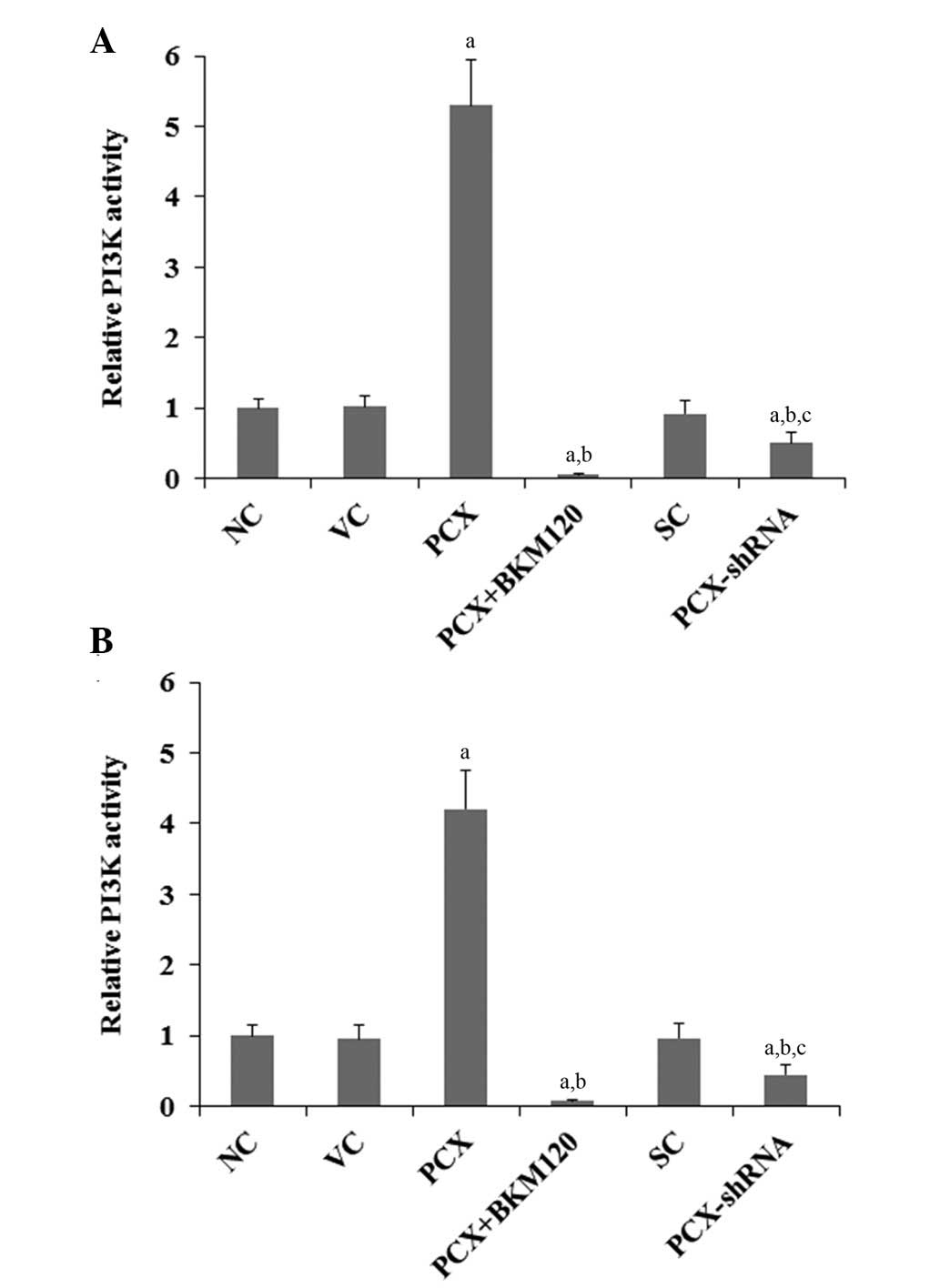 | Figure 5Effects of PCX on PI3K activity in OS
cells. In (A) MG-63 and (B) U2OS human OS cells, PI3K activities
were determined using a PI3K Activity ELISA kit in the following
groups: NC, normal control cells; VC, cells stably transfected with
empty pcDNA3.1 vector; PCX, cells stably trans-fected with PCX; PCX
+ BKM120, cells stably transfected with PCX and treated with the
PI3K inhibitor BKM120 (50 µM) for 48 h; SC, cells stably
transduced with scramble control shRNA; and PCX-shRNA, cells stably
transduced with PCX-shRNA. Each experiment was repeated three times
in duplicate. Values are expressed as the mean + standard
deviation. aP<0.05, vs. the controls (NC, VC and SC);
bP<0.05, vs. the PCX group; cP<0.05,
vs. the PCX+BKM120 group. PCX, podocalyxin; OS, osteosarcoma;
shRNA, small hairpin RNA; PI3K, phosphatidylinositide 3-kinase. |
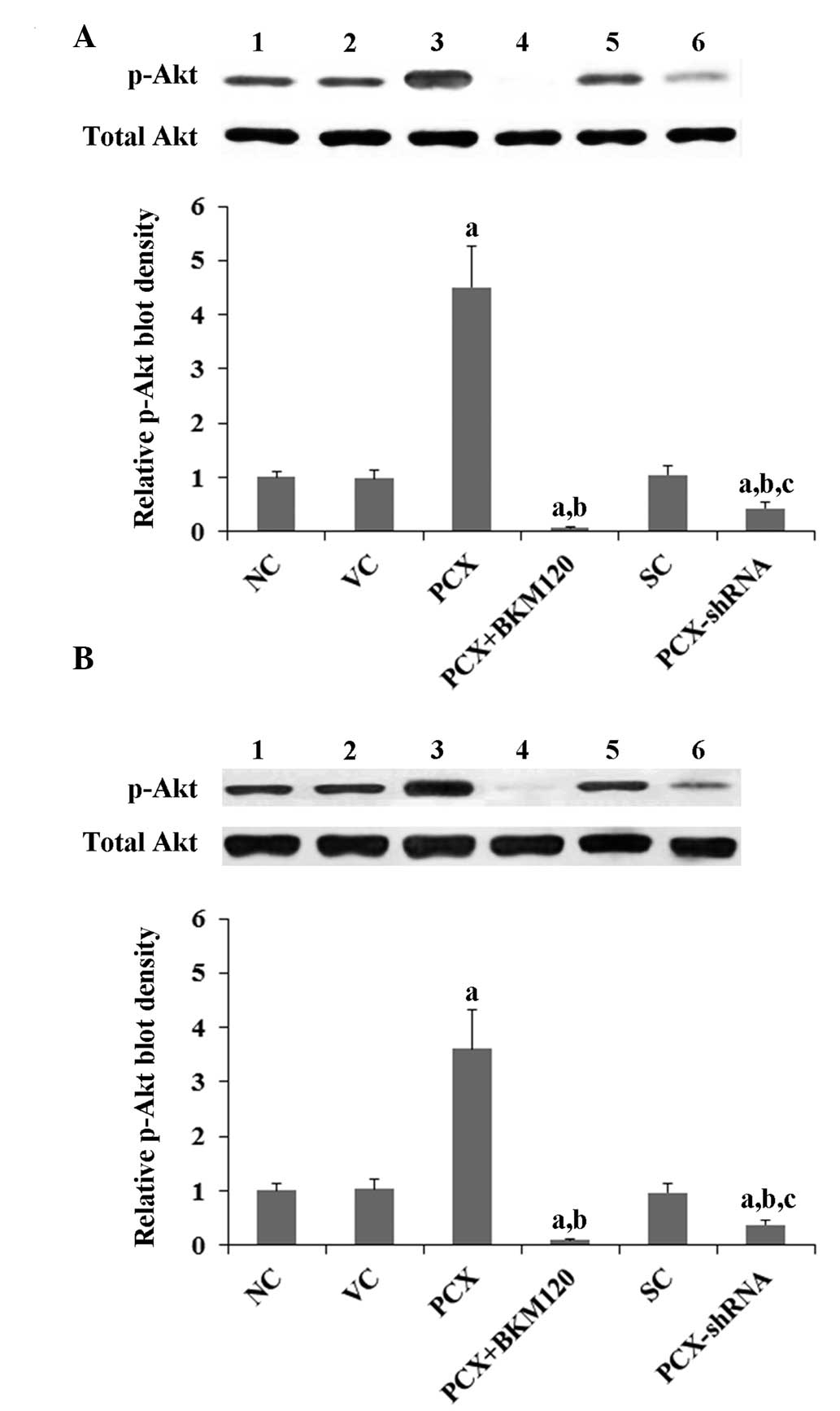 | Figure 6Effects of PCX on p-Akt expression
levels in OS cells. In (A) MG-63 and (B) U2OS human OS cells,
expression levels of total Akt and p-Akt at ser473 were determined
by western blot analyses. Lanes: 1, NC; 2, VC; 3, PCX; 4, PCX +
BKM120; 5, SC; and 6, PCX-shRNA. Total Akt expression levels were
not significantly altered by PCX in MG-63 and U2OS cells. The
density of the p-Akt (ser473) blot was normalized against that of
total Akt in order to obtain the relative p-Akt blot density, which
was expressed as a fold changes of that of the NC (designated as
1). Three independent experiments were performed for each western
blot analysis. Values are expressed as the mean + standard
deviation. aP<0.05, vs. the controls (NC, VC and SC);
bP<0.05, vs. the PCX group; cP<0.05,
vs. the PCX + BKM120 group. Groups: NC, normal control cells; VC,
cells stably transfected with empty pcDNA3.1 vector; PCX, cells
stably transfected with PCX; PCX + BKM120, cells stably transfected
with PCX and treated with the phosphatidylinositide 3-kinase
inhibitor BKM120 (50 µM) for 48 h; SC, cells stably
transduced with scramble control shRNA; and PCX-shRNA, cells stably
transduced with PCX-shRNA. p-Akt, phosphorylated Akt; PCX,
podocalyxin; OS, osteosarcoma; shRNA, small hairpin RNA; ser,
serine. |
Discussion
The use of multiagent, intensive chemotherapy has
markedly improved the long-term survival rate of patients with OS
(5). However, chemoresistance
continues to be the principal reason for poor survival and disease
recurrence in patients with OS (26). Innate or acquired resistance to
cisplatin, one of the most effective drugs against OS (27), is common (28). Therefore, understanding the
molecular basis for cisplatin chemoresistance in OS cells may serve
as a basis for identification of novel therapeutic targets and
biomarkers. A previous study suggested that PCX, a transmembrane
protein that is critical for the malignant progression of various
types of cancer (11–16), may also contribute to cancer
chemoresistance (17). The present
study was the first, to the best of our knowledge, to provide
evidence supporting an important role of PCX in promoting cisplatin
chemoresistance in OS cells.
The clinical significance of PCX in cancer
progression has been investigated in numerous types of tumor,
including uterine (29), colon
(30) and breast (31) carcinomas. In uterine endometrioid
adenocarcinoma, PCX expression has been shown to be correlated with
tumor grade (29). Furthermore,
overex-pression of PCX is an independent indicator of poor outcome
in breast and colorectal carcinoma (30,31).
Specifically in colorectal cancer, PCX expression was observed
predominantly on the invasive tumor front, thus suggesting its
importance in the metastatic spread of this type of cancer
(30). A previous study
demonstrated that PCX promotes astrocytoma cell survival against
temozolomide-induced apoptotic stress (17), suggesting that PCX may also promote
cancer chemoresistance. Therefore, in the present study, the role
of PCX was investigated in OS cell chemoresistance, in order to
provide novel insight into the functional role of PCX in
cancer.
The present study used two OS cell lines with
relatively large differences in their genetic background as cell
models to demonstrate a generalizable role of PCX in OS cell
chemoresistance to cisplatin. The IC50 of cisplatin was
used as a measure of cisplatin chemoresistance, with an increased
IC50 value reflecting clinical chemoresistance to
cisplatin (32). In addition, cell
colony formation and apoptosis assays were used to measure
long-term and early cell survival against cisplatin treatment,
respectively. In the two cell lines, overexpression of PCX
significantly increased the IC50 of cisplatin and cell
colony formation, and decreased cisplatin-induced cell apoptosis,
whereas knockdown of PCX expression markedly decreased the
IC50 of cisplatin and cell colony formation, and
enhanced cisplatin-induced cell apoptosis. Potential OS cell
chemoresistance mechanisms include dysfunctional membrane
transport, resistance to apoptosis and the persistence of stem
cell-like tumor cells (33). The
results of the present study suggested that PCX may promote
cisplatin chemoresistance in OS cells predominantly through
inhibition of cisplatin-induced apoptosis and promotion of cell
survival.
The promoting effect of PCX on the IC50
of cisplatin and cell survival of OS cells was markedly attenuated
by treatment with a selective PI3K inhibitor, which did not affect
the expression of PCX. These results suggested that PCX is able to
promote cisplatin chemoresistance in OS cells, predominantly
through a PI3K-dependent mechanism, which was corroborated by the
results showing that knockdown of PCX expression decreased PI3K
activity and PI3K-mediated Akt phosphorylation in OS cells. These
findings are also concordant with those of previous studies, which
indicated that the PI3K/Akt pathway has a critical role in cancer
chemoresistance (22–25). Of note, the selective PI3K
inhibitor only blocked ~50% of the effect of PCX on cisplatin
chemoresistance and cell survival in OS cells, suggesting the
involvement of other signaling pathways, which may be identified in
further studies.
Neoadjuvant therapy usually consists of
methotrexate, cisplatin and doxorubicin. Post-operative
chemotherapy for OS usually consists of methotrexate, cisplatin,
doxorubicin, cyclophosphamide and vincristine. As one of the most
common first-line chemotherapy drugs for OS, cisplatin elicits DNA
repair mechanisms by crosslinking DNA, which in turn activates
apoptosis when DNA repair proves impossible (34). Given the important role of
cisplatin in OS chemotherapy, and the significant promoting effect
of PCX on cisplatin chemoresistance in OS cells, PCX may be a
potential target for overcoming chemoresistance in OS. Future
studies by our group are aimed at investigating whether PCX may
impact OS cell resistance to other chemotherapy agents used in
neoadjuvant therapy and post-operative chemotherapy.
In conclusion, the present study was the first, to
the best of our knowledge, to provide evidence suggesting that PCX
promotes cisplatin chemoresistance in OS cells through a
PI3K-dependent mechanism. The results of the present study provided
novel insight not only into the functional role of PCX in cancer,
but also into the molecular mechanisms underlying OS
chemoresistance.
Acknowledgments
The present study was supported by the National
Natural Science Foundation, China (grant no. 81272947).
References
|
1
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar
|
|
2
|
Subbiah V and Kurzrock R: Phase 1 clinical
trials for sarcomas: The cutting edge. Curr Opin Oncol. 23:352–360.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fellenber J, Bernd L, Delling G, Witte D
and Zahlten-Hinguranage A: Prognostic significance of
drug-regulated genes in high-grade osteosarcoma. Mod Pathol.
20:1085–1094. 2007. View Article : Google Scholar
|
|
4
|
Chou AJ and Gorlick R: Chemotherapy
resistance in osteo-sarcoma: Current challenges and future
directions. Expert Rev Anticancer Ther. 6:1075–1085. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang L, Jin F, Qin A, et al: Targeting
Notch1 signaling pathway positively affects the sensitivity of
osteosarcoma to cisplatin by regulating the expression and/or
activity of Caspase family. Mol Cancer. 13:1392014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abe S, Nishimoto Y, Isu K, Ishii T and
Goto T; Japanese Musculoskeletal Oncology Group: Preoperative
cisplatin for initial treatment of limb osteosarcoma: Its local
effect and impact on prognosis. Cancer Chemother Pharmacol.
50:320–324. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anninga JK, Gelderblom H, Fiocco M, et al:
Chemotherapeutic adjuvant treatment for osteosarcoma: Where do we
stand? Eur J Cancer. 47:2431–2445. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Uribe-Botero G, Russell WO, Sutow WW and
Martin RG: Primary osteosarcoma of bone. Clinicopathologic
investigation of 243 cases, with necropsy studies in 54. Am J Clin
Pathol. 67:427–435. 1977.PubMed/NCBI
|
|
9
|
Geller DS and Gorlick R: Osteosarcoma: A
review of diagnosis, management and treatment strategies. Clin Adv
Hematol Oncol. 8:705–718. 2010.
|
|
10
|
Nielsen JS and McNagny KM: The role of
podocalyxin in health and disease. J Am Soc Nephrol. 20:1669–1676.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Riccioni R, Calzolari A, Biffoni M, et al:
Podocalyxin is expressed in normal and leukemic monocytes. Blood
Cells Mol Dis. 37:218–225. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kelley TW, Huntsman D, McNagny KM,
Roskelley CD and Hsi ED: Podocalyxin: A marker of blasts in acute
leukemia. Am J Clin Pathol. 124:134–142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yasuoka H, Tsujimoto M, Hirokawa M, et al:
Podocalyxin expression in undifferentiated thyroid carcinomas. J
Clin Pathol. 61:1228–1229. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hsu YH, Lin WL, Hou YT, et al: Podocalyxin
EBP50 ezrin molecular complex enhances the metastatic potential of
renal cell carcinoma through recruiting Rac1 guanine nucleotide
exchange factor ARHGEF7. Am J Pathol. 176:3050–3061. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sizemore S, Cicek M, Sizemore N, Ng KP and
Casey G: Podocalyxin increases the aggressive phenotype of breast
and prostate cancer cells in vitro through its interaction with
ezrin. Cancer Res. 67:6183–6191. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Somasiri A, Nielsen JS, Makretsov N, et
al: Overexpression of the anti-adhesin podocalyxin is an
independent predictor of breast cancer progression. Cancer Res.
64:5068–5073. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu H, Yang L, Liao D, Chen Y, Wang W and
Fang J: Podocalyxin regulates astrocytoma cell invasion and
survival against temozolomide. Exp Ther Med. 5:1025–1029.
2013.PubMed/NCBI
|
|
18
|
Ding X, Zhang Z, Li S and Wang A:
Combretastatin A4 phosphate induces programmed cell death in
vascular endothelial cells. Oncol Res. 19:303–309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gu Y, Fan S, Xiong Y, et al: Cloning and
functional characterization of TCRP1, a novel gene mediating
resistance to cisplatin in an oral squamous cell carcinoma cell
line. FEBS Lett. 585:881–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cao CM, Zhang Y, Weisleder N, et al: MG53
constitutes a primary determinant of cardiac ischemic
preconditioning. Circulation. 121:2565–2574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fos C, Salles A, Lang V, et al: ICOS
ligation recruits the p50alpha PI3K regulatory subunit to the
immunological synapse. J Immunol. 181:1969–1977. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li B, Yang Y, Jiang S, Ni B, Chen K and
Jiang L: Adenovirus-mediated overexpression of BMP-9 inhibits human
osteosarcoma cell growth and migration through downregulation of
the PI3K/AKT pathway. Int J Oncol. 41:1809–1819. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu ZL, Mao JH, Peng AF, et al: Inhibition
of fatty acid synthase suppresses osteosarcoma cell invasion and
migration via down-regulation of the PI3K/Akt signaling pathway in
vitro. Mol Med Rep. 7:608–612. 2013.
|
|
24
|
Zhao G, Cai C, Yang T, et al: MicroRNA-221
induces cell survival and cisplatin resistance through PI3K/Akt
pathway in human osteosarcoma. PLoS One. 8:e539062013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang TF, Wang H, Peng AF, et al:
Inhibition of fatty acid synthase suppresses U-2 OS cell invasion
and migration via downregulating the activity of HER2/PI3K/AKT
signaling pathway in vitro. Biochem Biophys Res Commun.
440:229–234. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fellenberg J, Kunz P, Sähr H and Depeweg
D: Overexpression of inosine 5′-monophosphate dehydrogenase type II
mediates chemo-resistance to human osteosarcoma cells. PLoS One.
5:e121792010. View Article : Google Scholar
|
|
27
|
Janeway KA and Grier HE: Sequelae of
osteosarcoma medical therapy: A review of rare acute toxicities and
late effects. Lancet Oncol. 11:670–678. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bruheim S, Xi Y, Ju J and Fodstad O: Gene
expression profiles classify human osteosarcoma xenografts
according to sensitivity to doxorubicin, cisplatin and ifosfamide.
Clin Cancer Res. 15:7161–7169. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yasuoka H, Tsujimoto M, Inagaki M, et al:
Clinicopathological significance of podocalyxin and phosphorylated
ezrin in uterine endometrioid adenocarcinoma. J Clin Pathol.
65:399–402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Larsson A, Johansson ME, Wangefjord S, et
al: Overexpression of podocalyxin-like protein is an independent
factor of poor prognosis in colorectal cancer. Br J Cancer.
105:666–672. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Somasiri A, Nielsen JS, Makretsov N, et
al: Overexpression of the anti-adhesin podocalyxin is an
independent predictor of breast cancer progression. Cancer Res.
64:5068–5073. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu M, Wang J, Huang H, et al:
miR-181a-Twist1 pathway in the chemoresistance of tongue squamous
cell carcinoma. Biochem Biophys Res Commun. 441:364–370. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tsai HC, Huang CY, Su HL and Tang CH: CCN2
enhances resistance to cisplatin-mediating cell apoptosis in human
osteo-sarcoma. PLoS One. 9:e901592014. View Article : Google Scholar
|
|
34
|
Rosenberg B, Vancamp L, Trosko JE and
Mansour VH: Platinum compounds: A new class of potent antitumour
agents. Nature. 222:385–386. 1969. View
Article : Google Scholar : PubMed/NCBI
|














