Introduction
Camurati-Engelmann disease (CED; MIM 131300), or
progressive diaphyseal dysplasia, is a rare autosomal dominant bone
disease, which is associated with molecular defects within the
transforming growth factor-β1 (TGFβ1) gene on chromosome
19q13.1-13.3 (1–3). The hallmark of CED is bilateral and
symmetrical cortical thickening of the diaphyses of the long bones,
both on the periosteal and endosteal surface, resulting in
sclerotic and expanded diaphyseal segments, and narrowed medullary
cavities. Hyperostosis is usually initiated at the diaphyses of the
femora and tibiae, and gradually spreads to involve all bones,
including the skull base, which has been reported to occur in
>50% of all patients (4–6).
CED has variable penetrance and wide expressivity
(4,7). The majority of patients exhibit
initial manifestations, which most commonly include limb pain,
waddling gait and muscle weakness, before the age of 30, and
occasionally before the age of 10 (4,7).
Other associated features include reduced subcutaneous fat, delayed
puberty and hepatosplenomegaly (4,6).
Sclerosis of the skull base is most associated with hearing and/or
vision loss, headaches and exophthalmos (6). Although biochemical measurements are
usually normal in CED, elevated erythrocyte sedimentation rate
(ESR) and abnormal bone turnover markers have been reported
(4,8,9). At
present, in the English literature, >300 cases of CED have been
reported (4,6); however, to the best of our knowledge,
no case has been reported to present with exophthalmos as the
initial manifestation.
The present study reported on a consanguineous
Chinese family with one affected individual, as confirmed by
genetic analysis, which presented with progressive proptosis as the
initial manifestation. The clinical, biochemical and radiological
findings of the proband are reported.
Subjects and methods
Subjects
The present study examined the unaffected parents
and the affected female proband in a single Chinese family. The
22-year-old female proband presented her first manifestation at the
age of 19 years and was first admitted to our clinic in August
2013. Medical history was recorded and comprehensive physical
examinations were performed, including degree of exophthalmos and
intraocular pressure measured by Non-Contact Tonometer, weight and
height, full examination of the skin, chest, abdomen and genital
organs. Skeletal deformities were also examined. The present study
was approved by the institutional review board (ethics committee)
of the Department of Scientific Research, Peking Union Medical
College Hospital (PUMCH; Beijing, China). Prior to study
participation, written informed consent was obtained from all
subjects for DNA analysis, other investigations and permission of
image publication.
Biochemical investigations
All biochemical investigations were performed at the
Central Laboratory, PUMCH. Overnight fasting blood samples were
obtained and full-day urine collection was conducted. Complete
blood count, hormone levels, thyroid function, serum levels of
phosphate (P), total calcium (Ca), ESR, high sensitivity C reactive
protein and alkaline phosphatase (ALP) were measured by standard
methods. Hormonal testing and detection of the serum levels of
25-hydroxyvitamin D, 1,25 dihydroxyvitamin D, intact parathyroid
hormone and C-terminal telopeptide of type I collagen (β-CTX) were
measured using an automated Roche electrochemiluminescence system
(E170; Roche Diagnostics, Basel, Switzerland). Urinary levels of Ca
and P were analyzed using a Urinary Chemical Analyzer (Clinitek
500; Siemens Healthcare, Malvern, PA, USA).
Radiological assessment
Radiography of the extremities and skull, computed
tomography (CT) of the head and orbital bones, and magnetic
resonance imaging (MRI) of head and bone scintigraphy were
performed on the proband. Radiological abnormalities were assessed
by experienced radiologists at PUMCH.
Genetic analysis
Blood samples were obtained from the patient and her
parents. Genomic DNA was extracted from 0.2 ml whole blood using a
commercial DNA extraction kit (QIAamp DNA Micro kit; Qiagen,
Hilden, Germany) according to the manufacturer's protocol. Using
polymerase chain reaction, the seven exons and flanking intron
sequences of TGFβ1 were amplified using seven pairs of
primers (Table I), which were
designed using Primer Premier 6.0 software (PREMIER Biosoft, Palo
Alto, CA, USA) and synthesized by TsingKe Biological Technology,
Beijing, China. The total volume of the reaction was 30 µl,
including 15 µl Taq DNA polymerase (Takara Bio, Inc.,
Otsu, Japan), 2.6 µl DNA templates, 1.2 µl forward
primer, 1.2 µl reverse primer and 10 µl double
distilled water. Taq DNA polymerase and its standard buffer
were used in all reactions under the following conditions: Initial
denaturation at 94°C for 5 min, followed by 30 cycles at 94°C for
30 sec, 54–64°C for 30 sec and 72°C for 50 sec. Direct DNA sequence
analysis was performed by automated DNA sequencing using an ABI DNA
sequencer (Model 377; Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA).
 | Table IPrimer sequences used for polymerase
chain reaction of transforming growth factor-β1. |
Table I
Primer sequences used for polymerase
chain reaction of transforming growth factor-β1.
| Exon | Direction | Primer sequence | Fragment length
(bp) |
|---|
| 1 | Sense | 5′
ATCCCCTATTCAAGACCACCCAC 3′ | 906 |
| Antisense | 5′
TCCCCCTATTGCTTGTCTCCCTCT 3′ | |
| 2 | Sense | 5′
CTGTCAGCTCCAAAACTCC 3′ | 345 |
| Antisense | 5′ ACCTTGTAACCAGCCGAC
3′ | |
| 3 | Sense | 5′
TGGGTACTGTTGGGGAGGAT 3′ | 337 |
| Antisense | 5′ GGGAGAAACAGGGGTGGG
3′ | |
| 4 | Sense | 5′
TGGGGTTTGCTCCTTCCTTC 3′ | 292 |
| Antisense | 5′
TGTGGGAGTCAGGGGATAGG 3′ | |
| 5 | Sense | 5′
CGCCCCACTTATCTATCCCTC 3′ | 379 |
| Antisense | 5′
TCTTACACCCAGACCTCATCCC 3′ | |
| 6 | Sense | 5′
GTTATTTTGTATGTTCCAGG 3′ | 704 |
| Antisense | 5′
CTCTGTGGGTCTTCATAGC 3′ | |
| 7 | Sense | 5′
TAGAAGATAAGAGAGACCG 3′ | 691 |
| Antisense | 5′
TGCTATGGTGACTGAATG 3′ | |
Other investigations
Bone mineral density (BMD) of the proband was
measured by Dual-energy X-ray absorptiometry (Lunar DPX; Lunar
Corporation, Madison, WI, USA) at the posteroanterior spine (L1-4),
lateral spine (L2-4), total hip, femoral neck and trochanter.
Prodigy enCORE version 6.70 software (GE Healthcare, Madison, WI,
USA) was used to analyze the data using the standard-array mode.
Ultrasonic investigations of the thyroid, abdominal and urogenital
system were also performed.
Results
The proband was a 22-year-old Chinese woman who had
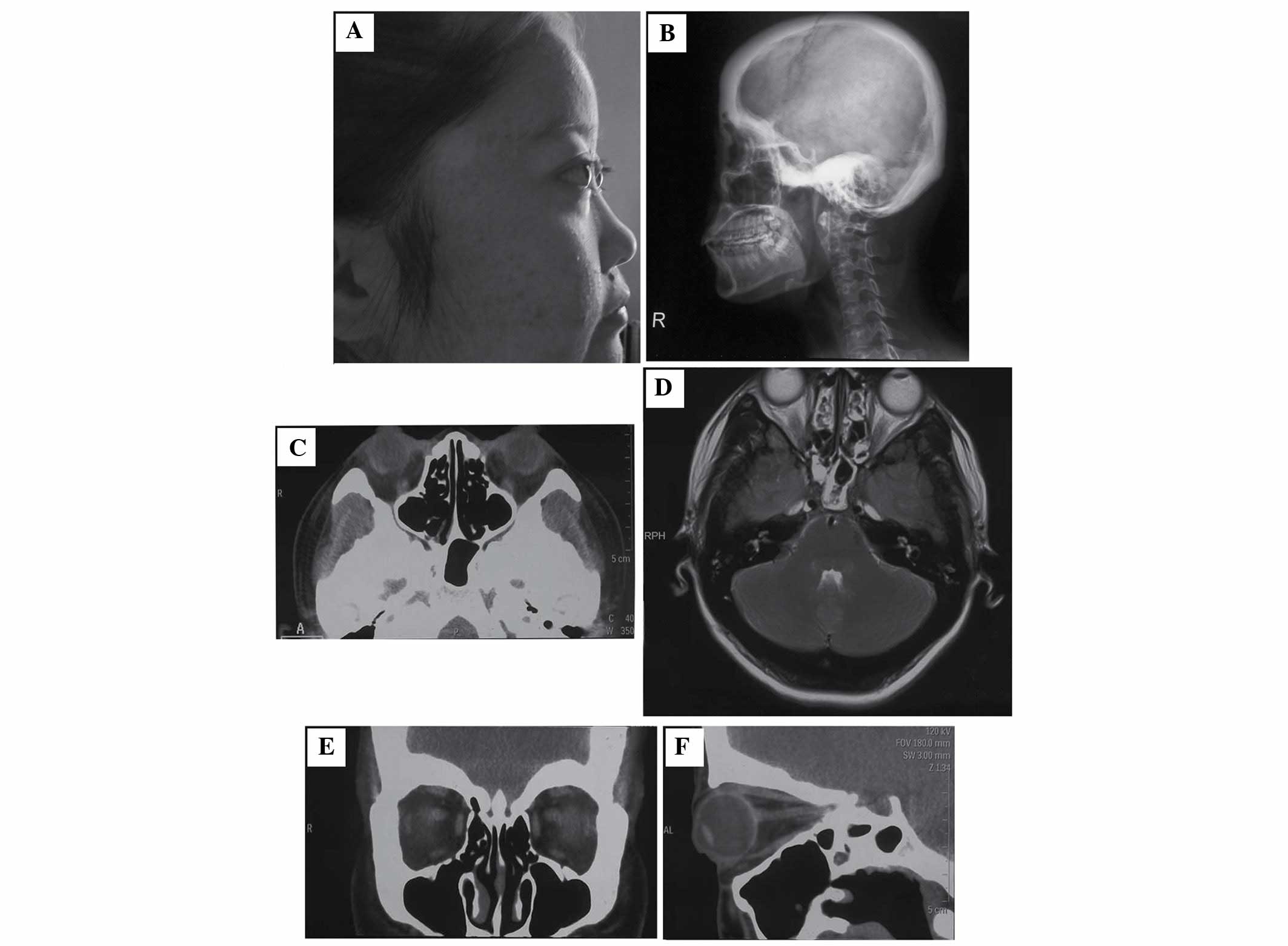
been complaining of bilateral and progressive proptosis (Fig. 1A) for the past 1 year. The patient
experienced mild eye pain, discomfort accompanied by eye movement,
and easy fatigability. Nonspecific bone pain occasionally occurred
at the knees following certain activities or during cold weather,
but was mostly unnoticeable. The patient had never exhibited a
waddling gait, muscle tenderness or headaches, and had good
mobility, hearing, vision and appetite. Menstruation began at the
age of 13, and the patient experienced regular periods. She had
previously been well with no relevant medical history. Family
history was unremarkable, neither parents nor any consanguinity
exhibited the features of CED. Physical examinations revealed the
degree of exophthalmos (left, 20 mm; right, 19 mm); high
intraocular pressure (measured by non-contact tonometer; left, 25.3
mmHg; right, 23.5 mmHg); normal body mass index (BMI; weight, 52
kg; height, 1.58 m; BMI 20.8 kg/m2); scattered acne on
the face and back; remarkable vellus hair on the back, forehead and
back of neck; an enlarged mandible; Tanner stage 5 with regards to
breast development and pubic hair; no hepatosplenomegaly; no muscle
weakness in the extremities; and no evident skeletal deformities,
such as cubitus valgus, genu valgum, scoliosis and elongation of
the long bones.
No specific biochemical abnormalities were detected
in the patient (Table II), except
for increased levels of the bone formation marker ALP and the bone
resorption marker β-CTX.
| Table IIBiochemical measurements of the
proband. |
Table II
Biochemical measurements of the
proband.
A, Assessment of
complete blood count, thyroid and gonadal function
|
|---|
| Biochemical
parameters | Complete blood
count
| Thyroid function
test
| Hormonal
testa
|
|---|
WBC
(×109/l) | Hb
(g/l) | PLT
(×109/l) | FT3
(pg/ml) | FT4
(ng/dl) | TSH
(µIU/ml) | FSH
(mIU/ml) | LH
(mIU/ml) | E
(pg/ml) | TSTO
(ng/dl) | PRL
(ng/ml) |
|---|
| Value | 6.57 | 133 | 194 | 3.18 | 1.446 | 1.776 | 4.9 | 5.88 | 53.8 | 16.1 | 20.1 |
| Reference | 4.0–10.0 | 110–150 | 100–300 | 1.8–4.1 | 0.81–1.89 | 0.38–4.34 | 5.1–7.0 | 4.4–6.1 | 50–154.5 | 25.6–42.6 | 7.2–9.2 |
B, Assessment of
bone metabolism
|
|---|
| Biochemical
parameters | ESR
(mm/h) | hsCRP
(mg/l) | 25(OH)D
(ng/ml) |
1,25(OH)2D
(pg/ml) | i-PTH
(pg/ml) | Serum Ca
(mmol/l) | Serum P
(mmol/l) | ALP
(U/l) | β-CTX
(ng/ml) | 24 h UCa
(mg) | 24 h UPb
(mg) |
|---|
| Value | 7 | 0.78 | 25.4 | 59.39 | 36.1 | 2.27 | 1.24 | 133 | 0.9 | 132.8 | 310 |
| Reference | 0–20 | 0–3 | 8–40 | 19.6–54.3 | 12–65 | 2.13–2.7 | 0.87–1.52 | 27.0–107.0 | 0.21–0.44 | 75–225 | - |
Skull radiography and CT detected prominent
hyperostosis and sclerosis of the calvarium (Fig. 1B) and skull base (Fig. 1B and C). Coronal (Fig. 1E) and sagittal (Fig. 1F) CT of the orbital bones revealed
markedly thickened and sclerotic orbits, associated with decreased
orbital volume shown in the sagittal view, which may 'push out' the
eyeballs and may have caused the obvious exophthalmos presented by
the patient. No obvious abnormalities, including compression of the
optic nerves or narrowed auditory canals, were detected by MRI
(Fig. 1D) of the head. X-rays of
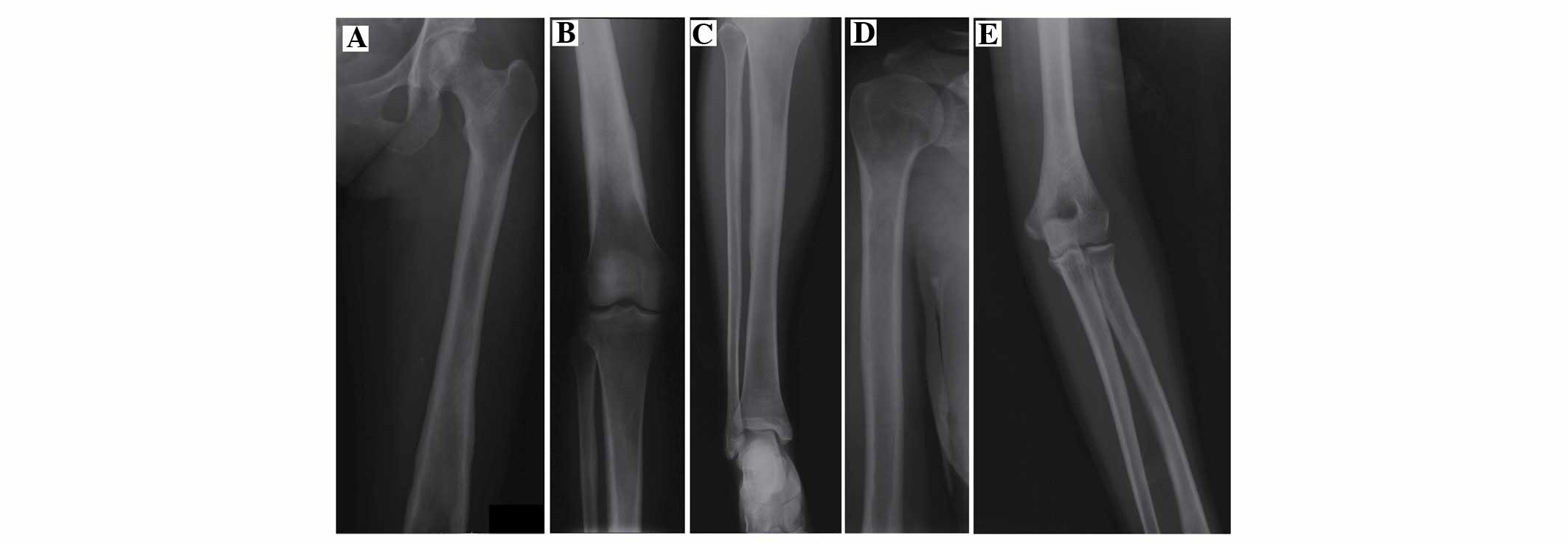
the long bones (Fig. 2) detected
cortical sclerosis, and thickness occurred in descending severity
in both femora, the upper end of both tibiae and both humeri.
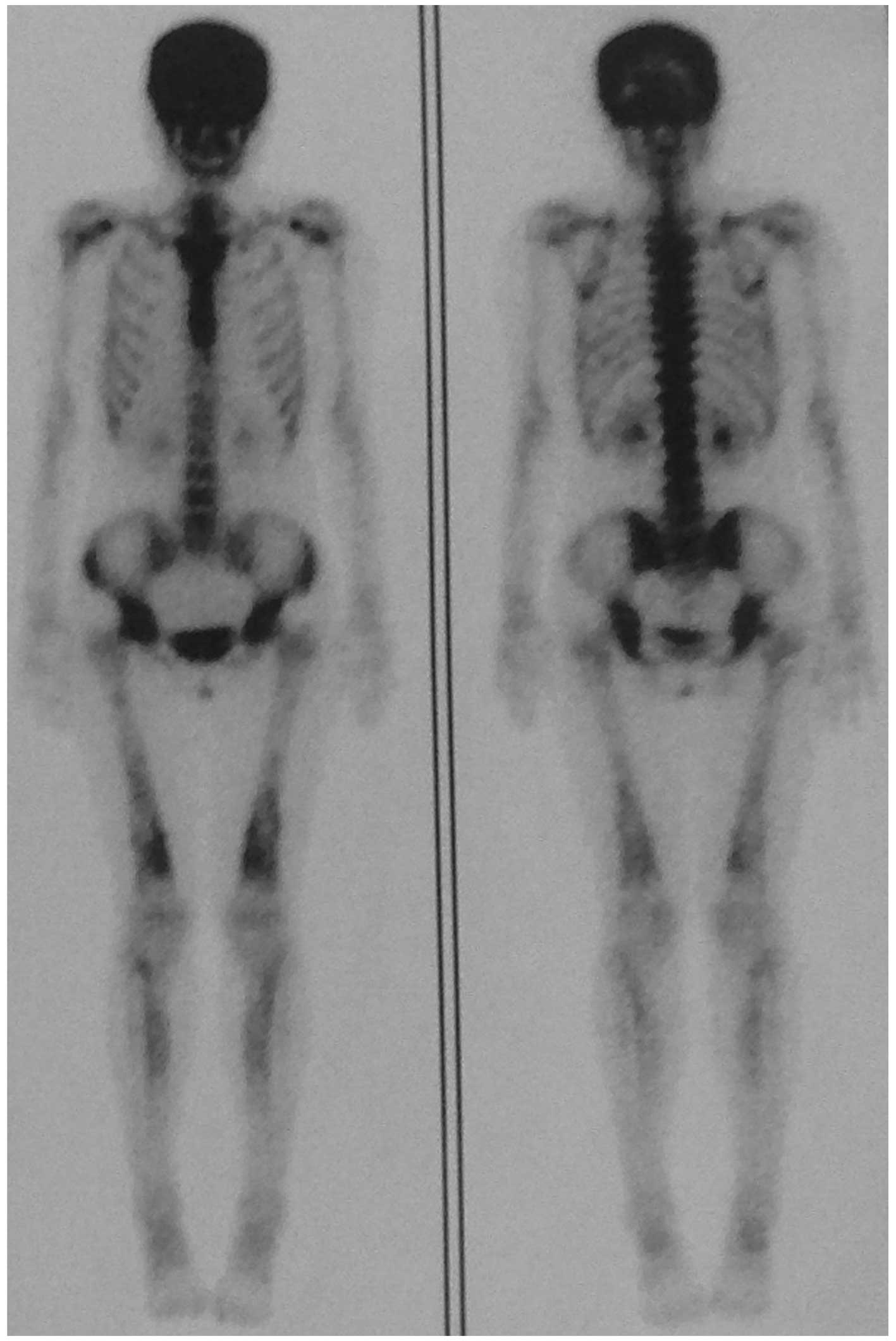
Narrowed medullary cavities were also detected. Bone scintigraphy
(Fig. 3) revealed markedly
increased tracer uptake in the skull and moderately increased
uptake in the bilateral lower half of the femora, upper end of
tibiae and shoulder joints. The rest of the skeleton was relatively
unaffected.
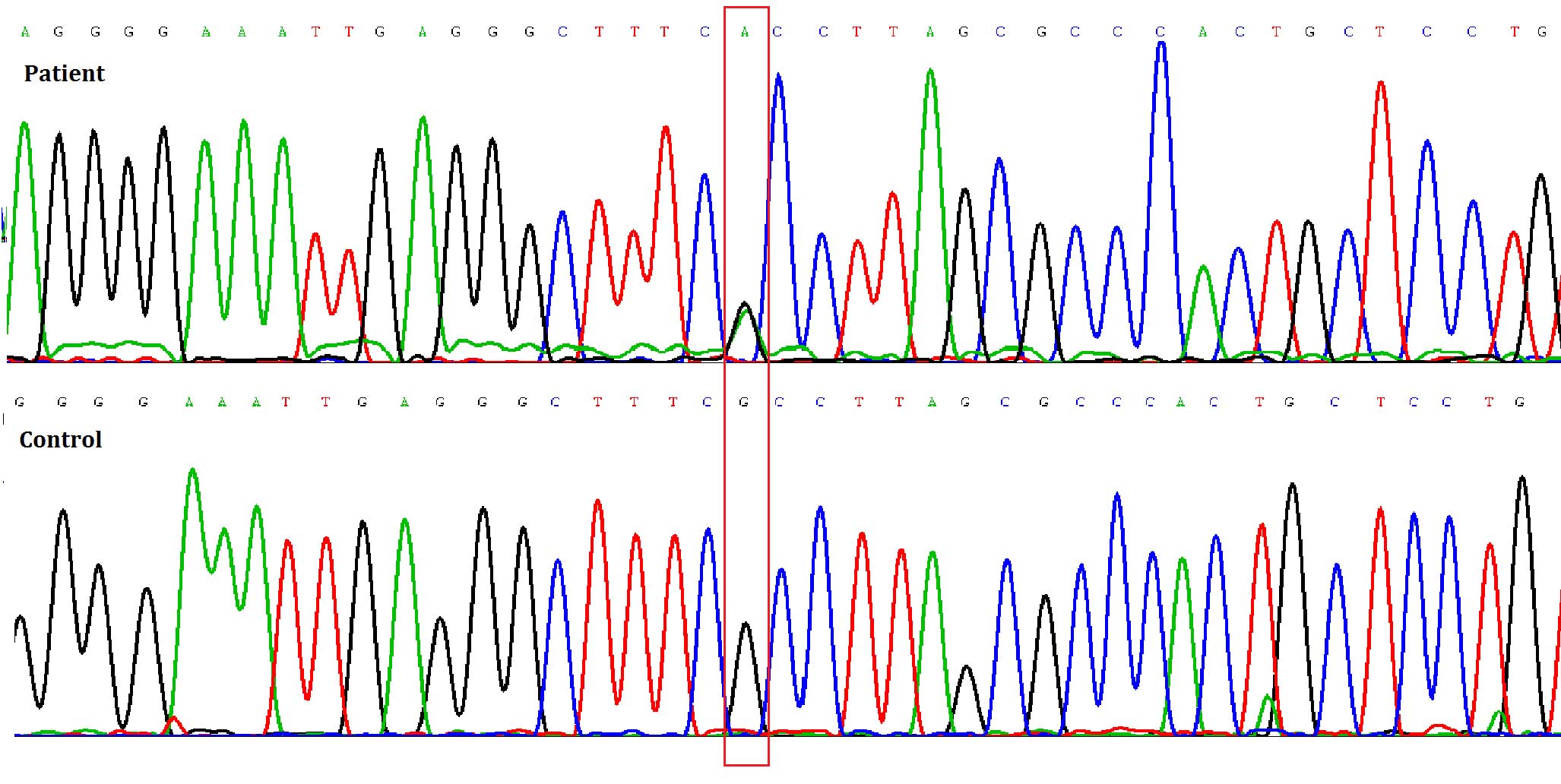
A heterozygous mutation involving a G to A
transition at the cDNA position +653 of TGFβ1 (Fig. 4) was detected only in the patient,
but not in her parents, thus resulting in an arginine to histidine
substitution at amino acid 218 (R218H) near the carboxy-terminus of
the latency associated peptide (LAP). Such mutation was not
detected in any of her parents, including a de novo mutation
in the proband.
BMD values at L1-4, L2-4, total hip, femoral neck
and trochanter were 1.245 (Z=1.3), 1.246 (Z=1.1), 1.063 (Z=0.9),
0.988 (Z=0.7) and 0.817 (Z=0.8), respectively. All BMD values of
the proband were markedly increased, especially in the spine,
compared with the site-, age- and gender-matched mean reference
values (10). Ultrasonic
investigations of the thyroid, abdominal and urogenital systems
detected no obvious abnormalities.
Discussion
The TGFβ1 gene, on chromosome 19q13.1-13.3,
has been well identified as the causative gene of CED (1–3). The
full-length precursor form of TGFβ1 consists of the signal peptide,
the LAP and the mature peptide. Post-translational processing by
proteolytic cleavage and dimerization leads to release of the
signal peptide and dimerization of the LAP, due to the formation of
a disulphide bridge (11).
Dimerization of the LAP maintains TGFβ1 in a latent form, either
alone as a small latent complex, or in conjunction with a latent
TGFβ1-binding protein as a large latent complex, which can not be
activated to bind the TGFβ1 receptor unless subjected to specific
activation conditions (11–14).
TGFβ1 gene mutations, all of which have been reported to
increase TGFβ1 protein activity (11), are associated with these activation
conditions. At present, 13 mutations have been reported in the
literature (2,4,11,15,16).
Over 80% of all mutations reported thus far are missense mutations
clustered in exon 4, and around the residues responsible for
homodimerzation of LAP (Cys223 and Cys225) (4). An arginine residue at position 218 is
the most common mutation (4),
which was detected in the proband in the present study. Mutations
in exon 4 destabilize the disulphide bridge, disrupt the LAP-TGFβ1
association, and result in subsequent release of the mature
activated TGFβ1 (4,17).
Almost all manifestations of CED can be explained by
enhanced TGFβ1 activity. Although TGFβ1 stimulates bone formation
and suppresses bone resorption under physiological conditions
(18), it appears to stimulate
entire bone turnover once mutated. Previous in vivo studies
(16,19), as with the patient in the present
study, have detected increased levels of bone formation and bone
resorption markers. In vitro, Saito et al (17) reported that CED fibroblasts with
mutant (R218H) TGFβ1 promoted the proliferation of co-cultured
osteoblast cells. McGowan et al (20) demonstrated that CED peripheral
blood mononuclear cells with mutant (R218C) TGFβ1 markedly
increased osteoclast formation and bone resorption. Increased bone
formation leads to typical hyperostosis in CED. The majority of
clinical features are secondary to hyperostosis and sclerosis of
the skeleton, including bone pain due to periosteal stretching;
skeletal deformities (such as genu valgum and enlarged mandible)
due to inappropriately increased bone growth; hearing and/or vision
loss, headaches and exophthalmos due to sclerosis of the skull
base; and systemic manifestations (such as anemia, leucopenia and
hepatosplenomegaly) due to hyperostosis encroaching on marrow
cavities, with secondary extramedullary haemopoiesis in the spleen
and liver (8,21–23).
TGFβ1 also has a crucial role in the inhibition of myogenesis
(24) and adipogenesis (25), which may explain the reduced
subcutaneous fat and easy fatigability associated with CED.
However, TGFβ1 mutation alone is insufficient
to explain the extremely variable penetrance and wide-range
expressivity associated with CED, both between families sharing the
same mutation, and even within families with genetic anticipation
in successive generations (4,5,26).
The present study provides strong evidence of variable penetrance
for CED. The majority of typical patients with CED, including
several cases with the same R218H mutation (4,7,27–30),
initially present with limb pain and a waddling gait. The patient
described in the present study is the first case, to the best of
our knowledge, to initially present with only exophthalmos.
Alongside occasional fatigability, nonspecific limb pain, and mild
or moderate hyperostosis of long bones, the present patient
appeared to suffer from mild CED. Furthermore, some unaffected
patients have been reported within the investigated cases of the
R218 mutation (4,7,28–30),
further indicating the extreme phenotypic variability in CED.
Single nucleotide polymorphisms (SNPs) in
TGFβ1 were once considered the cause of variability in CED;
however, no association between promoter SNPs or coding SNPs and
disease severity have been reported (7,30).
Whyte et al (16) detected
a receptor activator of nuclear factor κ-B ligand (RANKL) variant
and an allele dosage effect in a family of two patients with
different disease severity, thus suggesting that RANKL may be a
potential gene, other than TGFβ1, which has the ability of
modulating the outcome of the principal TGFβ1 mutation,
resulting in CED variability. Janssens et al (4) proposed another theory, that the
latent TGFβ1-binding protein may control the degree of TGFβ1
activation and thus the variable penetrance of CED. In addition, it
was suggested that the capacity of a mutation to alter the
conformation structure needed for premature activation of the
mature peptide depended on the presence of the latent TGFβ1-binding
protein. Different from the majority of other tissues, which
produce large latent complexes (LAP-TGFβ1-latent TGFβ1-binding
protein), bone predominantly produces small latent complexes
(LAP-TGFβ1), which is readily available and easily activated by
TGFβ1 mutations. As a result, patients with CED
predominantly exhibit bones abnormalities, whereas the majority of
other tissues are unaffected. Further studies are required to
confirm or reject this theory.
In conclusion, the present study was the first, to
the best of our knowledge, to describe a patient with exophthalmos
as the initial manifestation of mild CED with the heterozygous
missense mutation R218C in the TGFβ1 gene. CED is a
sclerosing bone disease with extreme variability, the potential
modulatory factors of which require further investigation.
Regarding the results of the present study, it may be recommended
that CED should be considered in the differential diagnosis of
exophthalmos, easy fatigability and nonspecific limb pain in young
individuals. Clinical examination, biochemical evaluation, bone
scintigraphic and radiological investigations, and genetic analysis
are all helpful in confirming diagnosis.
Acknowledgments
The authors of the present study would like to thank
the patients and their families for participating in this
study.
The present study was supported by grants from the
Ministry of Science and Technology of the People's Republic of
China (National Science and Technology Major Projects for 'Major
New Drugs Innovation and Development'; grant no. 2008ZX09312-016),
the National Natural Science Foundation of China (grant nos.
81471088 and 81170805), the Beijing Natural Science Foundation
(grant no. 7121012) and the National Key Program of Clinical
Science (grant no. WBYZ2011-873).
References
|
1
|
Janssens K, Gershoni-Baruch R, Van Hul E,
Brik R, Guañabens N, Migone N, Verbruggen LA, Ralston SH, Bonduelle
M, Van Maldergem L, et al: Localisation of the gene causing
diaphyseal dysplasia Camurati-Engelmann to chromosome 19q13. J Med
Genet. 37:245–249. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Janssens K, Gershoni-Baruch R, Guañabens
N, Migone N, Ralston S, Bonduelle M, Lissens W, Van Maldergem L,
Vanhoenacker F, Verbruggen L and Van Hul W: Mutations in the gene
encoding the latency-associated peptide of TGF-beta 1 cause
Camurati-Engelmann disease. Nat Genet. 26:273–275. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kinoshita A, Saito T, Tomita H, Makita Y,
Yoshida K, Ghadami M, Yamada K, Kondo S, Ikegawa S, Nishimura G, et
al: Domain-specific mutations in TGFB1 result in Camurati-Engelmann
disease. Nat Genet. 26:19–20. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Janssens K, Vanhoenacker F, Bonduelle M,
Verbruggen L, Van Maldergem L, Ralston S, Guañabens N, Migone N,
Wientroub S, Divizia MT, et al: Camurati-Engelmann disease: Review
of the clinical, radiological, and molecular data of 24 families
and implications for diagnosis and treatment. J Med Genet. 43:1–11.
2006. View Article : Google Scholar
|
|
5
|
Sparkes RS and Graham CB:
Camurati-Engelmann disease. Genetics and clinical manifestations
with a review of the literature. J Med Genet. 9:73–85. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carlson ML, Beatty CW, Neff BA, Link MJ
and Driscoll CL: Skull base manifestations of Camurati-Engelmann
disease. Arch Otolaryngol Head Neck Surg. 136:566–575. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Campos-Xavier B, Saraiva JM, Savarirayan
R, Verloes A, Feingold J, Faivre L, Munnich A, Le Merrer M and
Cormier-Daire V: Phenotypic variability at the TGF-beta1 locus in
Camurati-Engelmann disease. Hum Genet. 109:653–658. 2001.
View Article : Google Scholar
|
|
8
|
Smith R, Walton RJ, Corner BD and Gordon
IR: Clinical and biochemical studies in Engelmann's disease
(progressive diaphyseal dysplasia). Q J Med. 46:273–294.
1977.PubMed/NCBI
|
|
9
|
Hernández MV, Peris P, Guañabens N,
Alvarez L, Monegal A, Pons F, Ponce A and Muñoz-Gómez J:
Biochemical markers of bone turnover in Camurati-Engelmann disease:
A report on four cases in one family. Calcif Tissue Int. 61:48–51.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu XP, Yang YH, Zhang H, Yuan LQ, Luo XH,
Cao XZ and Liao EY: Gender differences in bone density at different
skeletal sites of acquisition with age in Chinese children and
adolescents. J Bone Miner Metab. 23:253–260. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Janssens K, ten Dijke P, Ralston SH,
Bergmann C and Van Hul W: Transforming growth factor-beta 1
mutations in Camurati-Engelmann disease lead to increased signaling
by altering either activation or secretion of the mutant protein. J
Biol Chem. 278:7718–7724. 2003. View Article : Google Scholar
|
|
12
|
Oklü R and Hesketh R: The latent
transforming growth factor beta binding protein (LTBP) family.
Biochem J. 352(Pt 3): 601–610. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pedrozo HA, Schwartz Z, Mokeyev T, Ornoy
A, Xin-Sheng W, Bonewald LF, Dean DD and Boyan BD: Vitamin D3
metabolites regulate LTBP1 and latent TGF-beta1 expression and
latent TGF-beta1 incorporation in the extracellular matrix of
chondrocytes. J Cell Biochem. 72:151–165. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bonewald LF, Wakefield L, Oreffo RO,
Escobedo A, Twardzik DR and Mundy GR: Latent forms of transforming
growth factor-beta (TGF beta) derived from bone cultures:
Identification of a naturally occurring 100-kDa complex with
similarity to recombinant latent TGF beta. Mol Endocrinol.
5:741–751. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu S, Liang S, Yan Y, Wang Y, Li F, Deng
Y, Huang W, Yuan W, Luo N, Zhu C, et al: A novel mutation of TGF
beta1 in a Chinese family with Camurati-Engelmann disease. Bone.
40:1630–1634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Whyte MP, Totty WG, Novack DV, Zhang X,
Wenkert D and Mumm S: Camurati-Engelmann disease: Unique variant
featuring a novel mutation in TGFβ1 encoding transforming growth
factor beta 1 and a missense change in TNFSF11 encoding RANK
ligand. J Bone Miner Res. 26:920–933. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Saito T, Kinoshita A, Yoshiura Ki, Makita
Y, Wakui K, Honke K, Niikawa N and Taniguchi N: Domain-specific
mutations of a transforming growth factor (TGF)-beta 1
latency-associated peptide cause Camurati-Engelmann disease because
of the formation of a constitutively active form of TGF-beta 1. J
Biol Chem. 276:11469–11472. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Janssens K, ten Dijke P, Janssens S and
Van Hul W: Transforming growth factor-beta1 to the bone. Endocr
Rev. 26:743–774. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang C, Zhang BH, Liu YJ, Hu YQ, He JW and
Zhang ZL: Transforming growth factor-β1 gene mutations and
phenotypes in pediatric patients with Camurati-Engelmann disease.
Mol Med Rep. 7:1695–1699. 2013.PubMed/NCBI
|
|
20
|
McGowan NW, MacPherson H, Janssens K, Van
Hul W, Frith JC, Fraser WD, Ralston SH and Helfrich MH: A mutation
affecting the latency-associated peptide of TGFbeta1 in
Camurati-Engelmann disease enhances osteoclast formation in vitro.
J Clin Endocrinol Metab. 88:3321–3326. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Crisp AJ and Brenton DP: Engelmann's
disease of bone - a systemic disorder? Ann Rheum Dis. 41:183–188.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gupta S and Cheikh IE: Camurati-Engelmann
disease in conjunction with hypogonadism. Endocr Pract. 11:399–407.
2005. View Article : Google Scholar
|
|
23
|
Meczekalski B, Czyzyk A, Podfigurna-Stopa
A, Rydzewski B, Sroczynski J, Lipinska M, Sokalski J, Krawczynski
M, Jamsheer A, Katulski K and Genazzani A: Hypothalamic amenorrhea
in a Camurati-Engelmann disease - a case report. Gynecol
Endocrinol. 29:511–514. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Massagué J, Cheifetz S, Endo T and
Nadal-Ginard B: Type beta transforming growth factor is an
inhibitor of myogenic differentiation. Proc Natl Acad Sci USA.
83:8206–8210. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ignotz RA and Massagué J: Type beta
transforming growth factor controls the adipogenic differentiation
of 3T3 fibroblasts. Proc Natl Acad Sci USA. 82:8530–8534. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Saraiva JM: Progressive diaphyseal
dysplasia: A three-generation family with markedly variable
expressivity. Am J Med Genet. 71:348–352. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liang YH, Li W, Li LY, Ye YY and Lu GX: A
mutation in TGF beta1 gene encoding the latency-associated peptide
in a Chinese patient with Camurati-Engelmann disease. Zhonghua Yi
Xue Yi Chuan Xue Za Zhi. 23:502–504. 2006.PubMed/NCBI
|
|
28
|
Park SJ, Yoon CS, Park HW, Choi JR, Chung
JS and Lee KA: The first Korean case of Camurati-Engelmann disease
(progressive diaphyseal dysplasia) confirmed by TGFB1 gene mutation
analysis. J Korean Med Sci. 24:737–740. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhadada SK, Sridhar S, Steenackers E,
Dhiman V, Mortier G, Bhansali A and Van Hul W: Camurati-Engelmann
disease (progressive diaphyseal dysplasia): Reports of an Indian
kindred. Calcif Tissue Int. 94:240–247. 2014. View Article : Google Scholar
|
|
30
|
Wallace SE, Lachman RS, Mekikian PB, Bui
KK and Wilcox WR: Marked phenotypic variability in progressive
diaphyseal dysplasia (Camurati-Engelmann disease): Report of a
four-generation pedigree, identification of a mutation in TGFB1,
and review. Am J Med Genet A. 129A:235–247. 2004. View Article : Google Scholar : PubMed/NCBI
|